

Summary
Background
The p53 transcription factor directs a transcriptional program that determines whether a cell lives or dies after DNA damage. Animal survival after extensive cellular damage often requires that lost tissue be replaced through compensatory growth or regeneration. In Drosophila, damaged imaginal disc cells can induce the proliferation of neighboring viable cells, but how this is controlled is not clear. Here we provide evidence that Drosophila p53 (dp53) has a previously unidentified role in coordinating the compensatory growth response to tissue damage.
Results
We find that dp53, the sole p53 ortholog in Drosophila, is required for each component of the response to cellular damage, including two separate cell-cycle arrests, changes in patterning gene expression, cell proliferation, and growth. We demonstrate that these processes are regulated by dp53 in a manner that is independent of DNA-damage sensing but that requires the initiator caspase Dronc. Our results indicate that once induced, dp53 amplifies and sustains the response through a positive feedback loop with Dronc and the apoptosis-inducing factors Hid and Reaper.
Conclusions
How cell death and cell proliferation are coordinated during development and after stress is a fundamental question that is critical for an understanding of growth regulation. Our data suggest that dp53 may carry out an ancestral function that promotes animal survival through the coordination of responses leading to compensatory growth after tissue damage.
Introduction
Much of the cellular response to DNA damage and other cellular stresses is coordinated by the p53 transcription factor [1]. In irradiated mammalian cells, p53 is stabilized and activated in response to signaling from the damage-sensing kinase ATM, ataxia telangiectasia mutated [2]. Upon activation by ATM and the downstream kinase Chk2, p53 directs a transcriptional program that results in G1 or G2 cell-cycle arrest, which facilitates repair of genomic damage, or in apoptosis [3]. By inducing these processes, p53 ensures the safety of the genome and thus also of the animal. However, survival after extensive cellular damage often requires that lost tissue be replaced through compensatory growth or regeneration. How compensatory growth is initiated in response to tissue damage is not known, but experiments in several systems have suggested that dying cells contribute by emitting autocrine and paracrine survival and growth-stimulating signals [4–9]. In Drosophila imaginal discs, the epithelial primordia for adult structures, compensatory growth is induced by many insults, including irradiation, disc fragmentation, and genetic manipulation [10–12]. Recent work indicates that compensatory proliferation is also induced in imaginal discs containing “undead” cells, damaged by irradiation or ectopic expression of proapoptotic genes but protected from death by the baculovirus caspase inhibitor P35 [5–7]. Under these conditions, the undead cells remain in the epithelium despite receiving a constant death signal; as a result, the discs undergo prolonged growth and express the pattern-organizing genes wingless (wg) and decapentaplegic (dpp) ectopically. Wg and Dpp have been implicated in the signaling events that accompany compensatory proliferation induced by undead cells and also in regeneration [7, 13–17]. In addition, it is thought that the initiator caspase Dronc, a homolog of caspase-9, has a nonapoptotic role in the undead-induced compensatory growth [5].
Drosophila has one p53 ortholog, dp53, which is necessary for apoptosis after irradiation [18–23]. To date, dp53 has not been shown to have a role in damage-induced growth arrest [18–23]. However, homozygous dp53 mutants are hypersensitive to ionizing irradiation, with reduced viability at late larval and pupal stages [20, 23]. This raises the interesting possibility that tissue recovery after cellular damage is impaired in dp53 mutant animals. p53 is not necessary for normal development in vertebrates or in flies, although its loss in mice can lead to spontaneous tumor formation due to accumulation of damaged cells [3]. In contrast, the vertebrate p53 paralogs, p63 and p73, which do not exist in Drosophila, are required for specific developmental processes unrelated to DNA damage. For example, mice lacking p63 have severe epidermal and limb abnormalities, arising in part from an epidermal stratification failure and inadequate renewal of basal progenitor cells [24–26].
The role of p53 as director of many aspects of the DNA-damage response puts it in a position to coordinate additional processes related to tissue damage. This prospect, along with the roles of its close relative p63 and the reduced survival of irradiated dp53 mutants, prompted us to ask whether dp53 plays a regulatory role in the growth response after cellular damage in Drosophila. We have used the generation of undead cells to induce cellular damage and compensatory proliferation and examined this process in detail. We report here that priortothe onset of growth, a disc-wide response is generated, with many undead cells transiently arresting cell division while other, viable cells in the disc arrest permanently. Compensatory proliferation then occurs with normal kinetics, but results in prolonged larval development. We find that the cell-cycle arrests, the subsequent compensatory growth, and the patterning gene alterations induced by undead cells all require dp53. Strikingly, dp53 activity under these conditions is not dependent upon DNA-damage sensing; rather, it requires a nonapoptotic role of the Dronc caspase. Our data indicate that dp53 activity is at the center of a feedback loop involving Dronc and the proapoptotic genes hid and reaper that amplifies and sustains compensatory proliferation. Finally, we demonstrate that in a model of disc regeneration, the formation of a blastema, a step necessary for compensatory proliferation during regeneration, is impaired in dronc and dp53 mutants.
Results
To create undead cells, we used the Gal4/UAS system to coexpress the UAS-regulated prodeath genes Hid or Reaper (Rpr) and the caspase inhibitor UAS-P35 in posterior cells, with the Hedgehog Gal4 (HhGal4) driver. In all of our experiments, the undead cells were marked by coexpression of GFP. As reported previously, expression of Hid or Rpr alone was lethal at early larval stages, but expression of either proapoptotic gene with P35 allowed survival until early pupal stages (data not shown) [5, 7]. The development of Hid or Rpr + P35-expressing animals was significantly delayed. Embryogenesis and the first two larval instars progressed with appropriate timing, but the last instar continued 3–4 days longer than controls (see Figure S1A in the Supplemental Data available with this article online). Early in the last instar (78 hr after egg laying [AEL]), wing discs containing undead cells were similar in size and morphology to 78 hr control discs (Figures 1A and 1E). However, by 96 hr AEL, the discs were significantly smaller than controls, suggesting that their growth was blocked. The posterior compartments, containing undead cells, were disproportionately small and distorted (Figures 1B and 1F; Figure S1B). However, within the next 24 hr the discs began to grow again, eventually losing their normal monolayered architecture and taking on extra folds (Figure 1G). By the end of the prolonged growth period, the discs were up to 48% larger than control discs (Figure S1B). As observed previously, expression of Hid or Rpr + P35 led to ectopic expression of the pattern regulators wingless (wg) and decapentaplegic (dpp), beginning between 87 and 96 hr AEL. Their expression was restricted to undead cells and located near the endogenous wg and dpp expression domains (Figures 1D and 1H and data not shown) [5–7]. Undead cells created with expression of Rpr + P35 gave similar phenotypes, as did the use of another posterior-specific Gal4 driver, Engrailed Gal4 (Figures S1A and S1B).
Figure 1. Undergrowth Is the First Detectable Phenotype of Undead Cells.
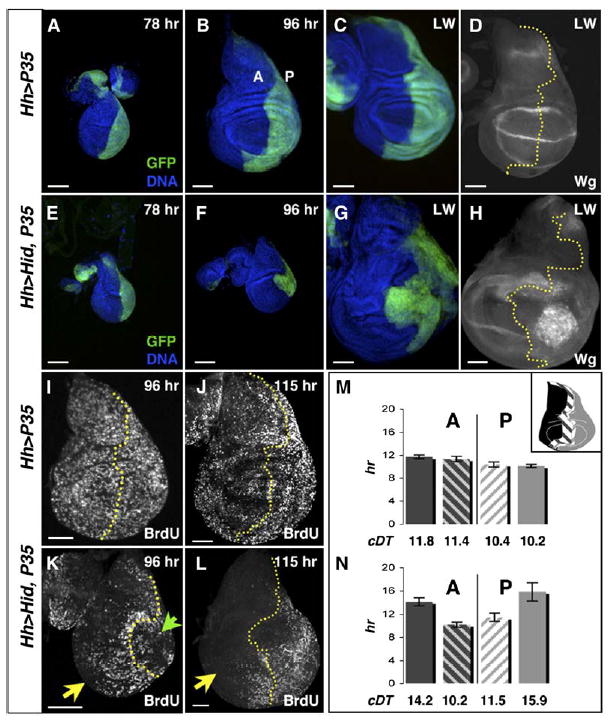
(A–D) Control wing imaginal discs expressing UAS-P35 in posterior cells (green) under Hedgehog (Hh) Gal4 control. In this and all subsequent images, anterior is left, dorsal is up. A, anterior; P, posterior.
(A) 78 hr disc.
(B) 96 hr wing imaginal disc.
(C) Late wandering stage (LW) wing disc, near the end of the disc growth period.
(D) LW wing disc stained for Wingless (Wg) protein.
(E–H) Wing discs with undead cells in the posterior.
(E) 78 hr wing disc, similar in size to the control (A).
(F) By 96 hr, the wing discs with undead cells are smaller than controls (compare with [B]), with a small, distorted posterior compartment (green).
(G) LW stage wing disc with undead cells in the posterior. This disc is 158 hr old and substantially overgrown in both the anterior (blue) and the posterior (green) compartments. Only the posterior compartment contains undead cells.
(H) LW wing disc with undead posterior cells, stained for Wg protein. Note that Wg is expressed ectopically; these Wg-expressing cells clump together near the center of the posterior compartment.
(I and J) BrdU incorporation in 96 hr (I) and 115 hr (J) control wing discs.
(K) BrdU incorporation in the 96 hr disc from (F). Note the lack of BrdU incorporation in the posterior undead cells (green arrowhead) and in the anterior (yellow arrow). Cells along the anterior-posterior (A/P) boundary (marked by dotted yellow line) continue to incorporate BrdU.
(L) 115 hr wing disc with undead cells, showing that undead cells have begun to incorporate BrdU again, along with the anterior cells along the A/P boundary. However, lateral anterior cells remain quiescent (yellow arrow). In all images, scale bars show relative size.
(M)Cell proliferation rates in control wing discs. Gray bars, posterior cells; black bars, anterior cells. Each bar represents a region of the wing disc as depicted in the inset: solid bars, lateral regions; scored bars, medial regions. Cell doubling times (cDT), below each bar, of lateral cells and medial cells are similar; posterior cells cycle moderately faster than anterior cells.
(N) Cell proliferation rates in wing discs with posterior undead cells. Undead, posterior, lateral cells cycle significantly slower than controls (p < 0.003). Anterior lateral cells also cycle slower than control anterior lateral cells (p < 0.002). The anterior and posterior lateral proliferation rates are likely to be an underestimate, given that BrdU incorporation was normal for the first 8–10 hr of the 26 hr clonal growth period. In contrast, cells in medial regions of both compartments cycle with normal kinetics.
Error bars in (M) and (N) denote standard errors of the mean.
Cell-Cycle Arrests Precede Compensatory Proliferation
We investigated the reason for the small disc size at 96 hr AEL by labeling cells with the DNA replication marker BrdU. At 78 hr AEL, when still the same size as controls, wing discs with undead cells incorporated BrdU with the same general pattern as 78 hr control discs (data not shown). By 96 hr, most undead cells no longer incorporated BrdU, and remarkably, anterior cells far from the undead cells also proliferated sparsely (Figure 1K). By contrast, cells flanking the A/P boundary in both compartments continued to incorporate BrdU.
To examine the alterations in cell proliferation in more detail, we measured proliferation rates during the time disc growth was slowed. We used flp-out clonal analysis [27, 28] to generate cell clones marked by expression of β-galactosidase randomly throughout the disc. This assay estimates the rate of cell proliferation (cell division + cell survival) that occurs in the wing disc over a defined period of time. Clones were induced while cells still proliferated normally, at 70 hr AEL, and allowed to grow until 96 hr AEL. At the end of this growth period, we counted the number of β-gal-positive cells that had accumulated in each clone to determine the rate of their proliferation. Since we had observed proliferation differences across the disc, the location of each clone in the disc was also noted (Figures 1M and 1N, inset). By these measurements, posterior cells located laterally (with respect to the A/P boundary; see inset) doubled every 10.2 hr in control discs, but when the cells expressed Hid + P35, they took 15.9 hr to double, nearly 6 hr longer (Figures 1M and 1N; p < 0.003). Likewise, anterior cells located laterally proliferated significantly slower than comparable control cells (14.2 hr versus 11.8 hr, respectively, p < 0.002; Figure 1M versus 1N). All the cells in the disc proliferated normally prior to 87 hr AEL, so these proliferation rates are likely to be an underestimate. In contrast to the slow-proliferating cells, the BrdU-incorporating cells on both sides of the A/P border divided at the same rate as controls (Figures 1M and 1N).
To determine which phases of the cell cycle were affected in the slow-growing cells, we analyzed wing disc cells via flow cytometry at three time points spanning the third instar, using GFP expression to identify the posterior, undead cells. In control wing discs, cell cycles shifted from G1 to G2 regulation between 96 hr and late wandering (LW) stage (Figure 2A) [28, 29]. In contrast, at 96 hr, undead cells had accumulated in G2 and were 70% larger than anterior cells (Figure 2B). Expression of the Cdc25 homolog string (stg) mRNA, which is rate limiting for progression into mitosis and tightly transcriptionally controlled, was reduced or absent from many undead cells, indicating that cells had arrested in G2 (Figure 2E [n = 36]) [30, 31]. Their large size suggested that although blocked in G2, the undead cells continued to grow in size. Later, at early wandering (EW) stage, undead cells re-entered the cell cycle, expressed stg again, and by LW proliferated with a G1-regulated cell cycle more appropriate of younger cells (Figures 2A, 2B, and 2F). Consistent with this, undead cell size decreased during this period and by LW was similar to controls (Figure 2B, LW).
Figure 2. Autonomous and Nonautonomous G2 Arrests Precede Compensatory Proliferation.
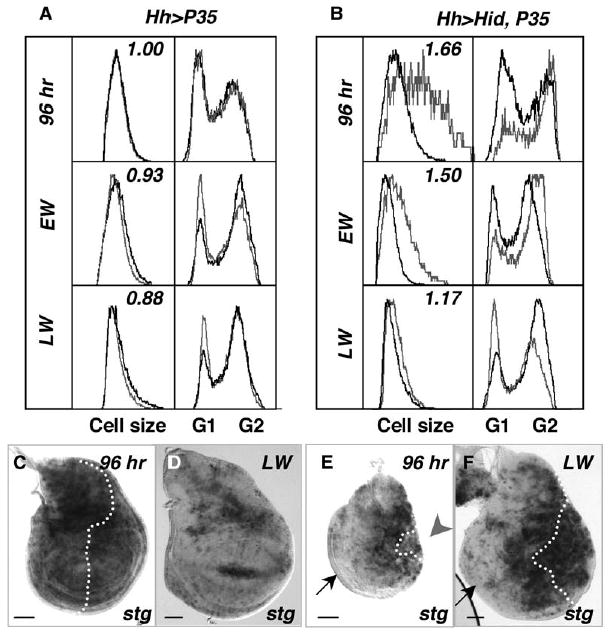
(A) Cell-cycle profiles and cell size from control wing discs at 96 hr, early wandering (EW), and late wandering (LW) stage. Posterior cells are marked with GFP and are represented by gray traces; black traces represent profiles of anterior cells. Left boxes: relative cell size, measured by forward scatter. The number in these boxes is the ratio of posterior/anterior cell size. Right boxes: cell-cycle profiles.
(B) Cell-cycle profiles and cell size from wing discs with undead cells. Undead cells (posterior), wild-type cells (anterior). Trace colors are same as controls. At 96 hr, most undead cells are in G2 and nearly 1.7× larger than anterior cells. At EW stage, the undead cells are somewhat smaller, but most still remain in G2. By LW stage, undead cells are similar in size to anterior cells, but their cell cycles have shifted to one similar to younger control wing discs (96 hr and EW). At LW stage, the wild-type anterior cells (black trace) adopt a cell cycle that is predominantly G2 regulated, the characteristic profile of LW stage wing discs (e.g., control discs in [A], LW).
(C–F) stringcdc25 (stg) mRNA expression in control wing discs at 96 hr AEL (C) and LW (D), and in discs with undead posterior cells at 96 hr AEL (E) and LW (F). Wing discs are all oriented with the posterior to the left. stg is expressed in a periodic fashion throughout the control wing disc at 96 hr (C), but by LW stage most cells in control discs do not express stg and are arrested in G2 (D). In discs with undead cells, stg is expressed in anterior cells and posterior cells surrounding the A/P boundary at 96 hr but is largely missing in lateral anterior cells and in most undead cells (E). Later, when the undead cells are proliferating, stg is expressed at high levels in these cells; however, note that it is still absent in lateral anterior cells (F). Scale bars in (C)–(F) represent 50 μn.
In contrast to the posterior, undead cells, the cell-cycle profile of anterior cells was similar to controls at 96 hr (Figure 2B versus 2A). However, we suspect that this reflected the large population of proliferating cells at the A/P boundary, since many cells at the lateral anterior edge of the wing discs lacked stg mRNA, suggesting that, like the undead cells at this stage, they had arrested in G2 (Figures 2E [n = 36] and 2F [n = 20]). This anterior cell arrest was permanent, because stg (and BrdU) was absent from most of the cells at all stages after 96 hr.
These data indicate that the earliest detectable consequence of tissue damage due to undead cells is a G2 arrest in two separate cell populations, which explains the small wing disc size at 96 hr of development. Our results indicate that tissue damage imposed by undead cells leads to global signaling that instructs cells to arrest either transiently or permanently or to proliferate. Interestingly, many undead cells were capable of proliferating after their initial arrest and apparently contributed to the eventual disc overgrowth since most of these cells incorporated BrdU despite a significant level of Hid and activated caspase-3 (Figures 3A and 3B [n = 23 and 27, respectively] and data not shown). However, cells with highest levels of Hid and caspase activity were often devoid of BrdU (Figure 3A, inset and data not shown). Unlike control wing discs, cell proliferation in discs with undead cells continued into pupal development; the cells did not differentiate and eventually disintegrated (data not shown).
Figure 3. dp53 Is Transcriptionally Induced in Undead Cells Independent of DNA-Damage Sensing.
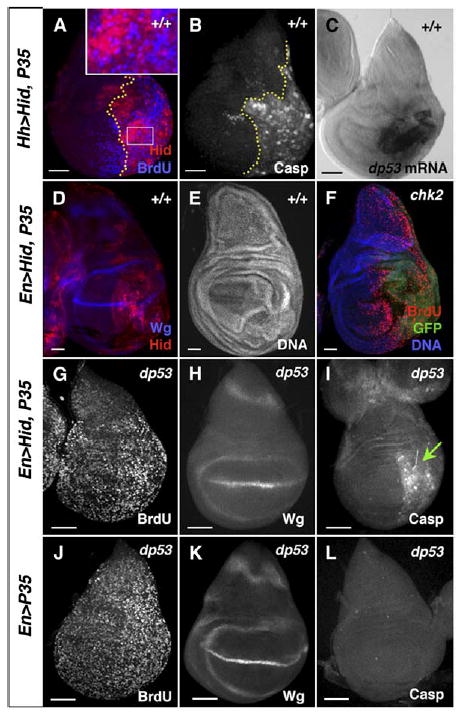
(A) 115 hr wing disc expressing Hid + P35 under HhGal4 control in the posterior. The A/P boundary is marked by a dotted yellow line. Undead cells express Hid protein (red) and, except for those with the highest level of Hid, also incorporate BrdU.
(B) Similar disc with undead posterior cells, stained with an antibody against the cleaved form of caspase-3, which marks most undead cells and also some anterior, wild-type cells. The A/P boundary is marked by a dotted yellow line.
C) dp53 mRNA is specifically upregulated in undead cells.
(D and E) Wing discs expressing Hid + P35 in posterior cells with Engrailed (En) Gal4 driver, showing ectopic Wg protein (D) and overgrowth (E).
(F) chk2 mutant wing disc with posterior undead cells (in this genotype induced by HhGal4). Loss of chk2 does not alleviate the overgrowth or the anterior cell-cycle arrest.
(G–L) Loss of dp53 completely alleviates the cell-cycle arrests and undergrowth of 96 hr wing discs caused by undead cells.
(G) BrdU incorporation in a 96 hr dp53 mutant wing disc with posterior undead cells. Neither posterior cells, expressing Hid + P35, nor anterior cells are arrested. The size of the posterior compartment and of the entire disc is normal. Despite the presence of undead cells, the discs have normal morphology.
(H) Wg protein is not ectopically expressed in these dp53 mutant discs.
(I) Caspase-3 is still present in the cleaved form, indicating that Hid still induces the apoptotic signal (green arrow).
(J–L) Control dp53 mutant wing discs, expressing P35 alone. Bars indicate relative scale.
dp53 Is Induced in Undead Cells and Is Required for the Growth Alterations
The G2 arrests in both cell populations were reminiscent of the behavior of irradiated imaginal discs and suggested that a damage response had been activated [10,18,19,23,32]. We examined flies carrying mutations in genes in the DNA-damage-sensing pathway, including atm and chk2. Drosophila atm is essential during development, and although mutant animals are very sick, they are viable until the pupal stage [33]. We examined atm6 null mutant wing discs containing undead cells and found that they expressed wg ectopically and overgrew relative to control atm6 mutant discs. chk2 null mutant wing discs behaved similarly; thus, neither atm nor chk2 were required for the compensatory proliferation or ectopic expression of Wg (Figure 3F; Figure S2). Consistent with these results, we found that very few cells in wing discs with undead cells expressed the DNA-damage marker γ-H2AX, suggesting that the DNA-damage response had not been activated (data not shown).
Nevertheless, dp53 mRNA was dramatically induced, specifically in the undead cells (Figure 3C [n = 26 discs]). To test whether dp53 was required for undead cell-induced effects, we used two different targeted null alleles of dp53 [20, 21]. Remarkably, we found that loss of dp53 completely prevented all of the phenotypes associated with the growth arrests caused by undead cells. In dp53 mutants, both the undead cells and lateral anterior cells incorporated BrdU normally throughout development (Figure 3G [n = 24 discs]). Despite the persistent expression of Hid + P35, loss of dp53 also suppressed the subsequent compensatory proliferation and the ectopic expression of wg in these discs (Figures 3G and 3H; Figure S2). Furthermore, dp53 mutant larvae expressing Hid + P35 developed with normal timing (Figure S1A). Thus, dp53 activity is necessary for all of the growth and patterning alterations associated with undead cells, suggesting that it has a role in the mechanism of their deployment.
Dronc Is Required for dp53 to Regulate Growth
Our results suggest that the DNA-damage sensors ATM and Chk2 are not required for dp53 to function in wing discs containing undead cells. The AMP-activated protein kinase (AMPK) has been shown to activate dp53 upon metabolic stress [34], and therefore we examined levels of the active, phospho-AMPK in discs with undead cells. However, p-AMPK was present at levels similar to controls (Figures S3D and S3F).
We noticed that in spite of the complete lack of a response in dp53 mutants, the undead cells still contained high levels of cleaved caspase-3 (Figure 3I [n = 24]). Hid and Rpr initiate the death process by preventing the inhibitor of apoptosis, Diap-1, from blocking both initiator and effector caspase (e.g., caspase-3) activation [35]. Although the caspase inhibitor P35 prevents the activity of executioner caspases such as caspase-3, it does not prevent their processing by (for example) the P35-insensitive Dronc; thus, the presence of the cleaved form suggested that Dronc was still active in dp53 mutant discs [35]. Since Dronc had previously been implicated in a nonapoptotic role in undead cell-induced compensatory proliferation [5], we examined whether it was required for dp53 function. We generated undead cells by using EnGal4 to drive expression of Hid + P35 in animals carrying a null allele, dronc129 [36]. Loss of just one copy of dronc substantially reduced the growth alterations and allowed imaginal cells to differentiate and adults to form, suggesting that Dronc was haploinsufficient for the effects induced by undead cells (Figures S3A and S3B). Interestingly, high levels of cleaved caspase-3 were still induced in these dronc129 heterozygotes, which suggests that Dronc’s caspase-activating function may operate at a different activity threshold than its role in compensatory proliferation (Figure S3A) [5]. In homozygous dronc129 mutants with posterior undead cells, the extra growth and ectopic wg expression induced by undead cells were completely abolished (Figures 4A, 4B, 4D, and 4E). Complete loss of dronc also abolished caspase-3 cleavage in 100% of the wing discs, even though cells expressed high levels of Hid; this suggests that Dronc was responsible for caspase-3 cleavage under these conditions (Figure 4E [n = 21]). Moreover, dp53 mRNA was not increased in undead cells of dronc129 mutant wing discs (Figure 4J [n = 34]).
Figure 4. Dronc Activity Is Required for dp53 to Regulate Compensatory Proliferation.
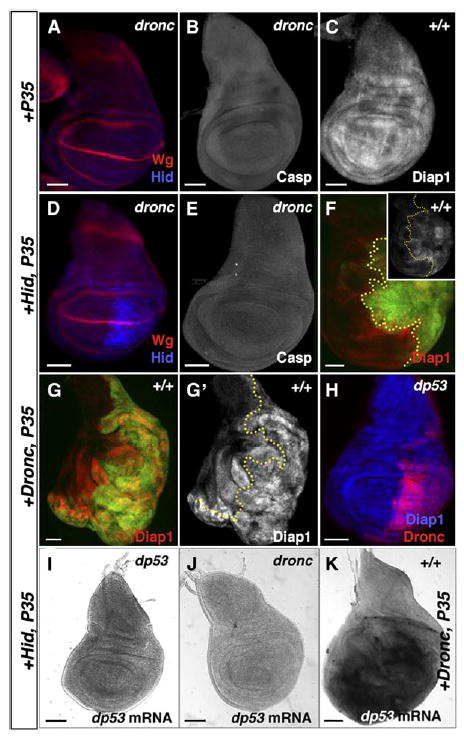
(A, B, D, E) Dronc is required for dp53 activation.
(A and B) Control dronc mutant wing discs stained for Wg (A) and cleaved caspase-3 (B).
(D and E) dronc mutant wing discs at LW stage.
(D) Wg expression is normal (red) despite the presence of undead cells, confirmed by the presence of Hid protein (blue).
(E) dronc mutant wing discs contain no cleaved caspase-3.
(C, F–H) Diap1 degradation is not necessary for undead-induced proliferation.
(C) Expression of Diap1 protein in a control wing disc.
(F) Expression of Hid + P35 induces loss of Diap1 protein (red) in many posterior cells (green). Inset shows single-channel image of Diap1 expression.
(G–G′) Expression of Dronc + P35 induces stronger overproliferation than Hid + P35, but Diap1 protein persists in most undead cells (G′).
The A/P boundary is marked with a dotted yellow line.
(H) In dp53 mutant discs, expression of Dronc + P35 has no effect on Diap1 expression (blue), and disc growth and morphology is normal. Dronc protein is shown in red.
(I–K) dronc is required for expression of dp53 in undead cells.
I and J) RNA in situ hybridization to detect dp53 mRNA in wing disc expressing Hid + P35 from dp53 mutant (I) and dronc mutant (J) larvae, showing that dp53 mRNA is not induced in either mutant.
(K) dp53 mRNA is strongly induced in wing discs expressing Dronc + P35. Bars indicate relative scale.
Dronc activity is held in check by Diap1, which is degraded upon binding by HRG (Hid-Rpr-Grim) proteins [35]. We tested whether Diap1 degradation was required for compensatory growth by coexpressing Dronc + P35 in the discs, bypassing Diap1. Diap1 protein was lost in most undead cells expressing Hid or Rpr + P35 (Figure 4F), but persisted in many Dronc + P35-expressing undead cells (Figure 4G). Despite the presence of Diap1 protein, expression of Dronc + P35 led to more severe growth effects than did expression of Hid or Rpr + P35 (Figure 4G [n = 36 discs]). These results confirm a previous report that loss of Diap1 is not required for compensatory proliferation induced by undead cells [5]. The expression of Dronc + P35 was sufficient to induce dp53 mRNA in wild-type wing discs (Figure 4K). However, Dronc expression was not able to induce overgrowth or loss of Diap1 in dp53 mutant wing discs (Figure 4H [n = 20 discs]). Taken together, our experiments indicate that Dronc is both necessary and sufficient to induce dp53 expression and activity in wing discs, leading to growth arrest and subsequently to compensatory proliferation.
dp53 Amplifies and Sustains Signaling from Undead Cells through a Positive Feedback Loop
Irradiation induces dp53-dependent expression of rpr and hid in embryos, and rpr is a direct target of dp53 [18, 19]. In our experiments, expression of Hid + P35 induced expression of rpr, and expression of Rpr + P35 induced expression of hid, suggesting a feedback relationship between the two genes (Figures 5A and 5B [n = 15 and 23 discs, respectively]). Expression from the endogenous rpr locus required dp53, as shown by the fact that dp53 mutant wing discs expressing Hid + P35 no longer expressed rpr (Figure 5C [n = 32]). Loss of rpr or hid had little effect on these phenotypes, consistent with the induction of both of these genes in response to undead cells (data not shown). However, feedback regulation between dp53 and hid and rpr required dronc, because dronc129 mutant discs that expressed exogenous Hid + P35 did not induce endogenous rpr expression (Figure 5D [n = 27]). Thus, a regulatory loop, consisting of the initiator caspase Dronc, dp53, and the endogenous hid and rpr loci, drives the disc response to undead cells. dp53 mutants containing undead cells were unhealthy, however, and only 30% of larvae survived to wandering stage, possibly due to the presence of TUNEL-positive cells despite inhibition of effector caspase activity by P35 (Figures S3C and S3E). It is possible that in the absence of a dp53-driven program of growth, caspase-3-independent cell death (presumably induced by Dronc activity), contributed to the demise of the animal. This interpretation is consistent with reports of the reduced survival of irradiated dp53 mutant animals [20, 23].
Figure 5. dp53, Dronc, and the Endogenous hid and rpr Loci Form a Regulatory Loop that Promotes Compensatory Proliferation.
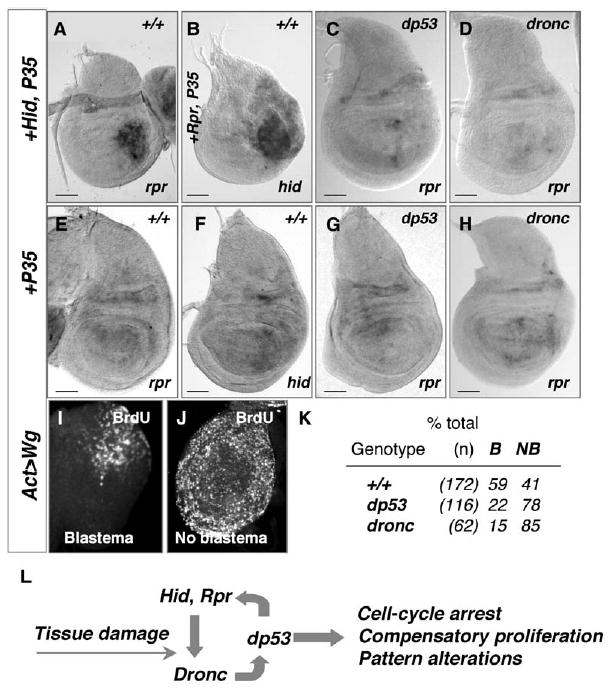
(A) 96 hr wing disc expressing Hid + P35 in posterior cells. rpr mRNA is highly induced in the UAS-Hid-expressing undead cells.
(B) 96 hr wing disc expressing Rpr + P35 in posterior cells. The undead cells express high levels of hid mRNA, from the endogenous hid locus.
(C and D) dp53 and Dronc are required for expression from the rpr and hid loci in wing discs with undead cells.
(C) dp53 mutant wing disc expressing Hid + P35. Loss of dp53 prevents the expression of rpr mRNA.
(D) rpr expression is not induced by Hid + P35 in dronc129 mutant wing discs.
(E–H) rpr and hid mRNA expression in control discs expressing P35 alone.
(E and F) Wild-type wing imaginal discs.
(G) dp53 mutant wing disc.
(H) dronc129 mutant wing disc. Bars in (A)–(H) indicate relative scale.
(I–K) Regeneration induced by ectopic expression of Wg requires dp53 and dronc for blastema formation.
(I) BrdU labels only the dorsal proximal cells of the “weak point” in regenerating prothoracic leg discs.
(J) Prothoracic leg disc with no blastema.
(K) Percent of prothoracic leg discs that form a blastema after ectopic Wg expression in wild-type (+/+), dp53, or dronc mutant animals. See text for details. B, blastema; NB, no blastema.
(L) Model of dp53 activation and its regulation of the processes associated with compensatory proliferation. Tissue damage due to undead cells results in the activation of Dronc (possibly in a nonapoptotic role, see [5]), and results in a dramatic increase in expression of dp53. How dp53 expression is upregulated is unknown. The activity of dp53 causes G2 arrest, compensatory proliferation, and also ectopic expression of Wg and Dpp. The induction of hid and rpr expression by dp53 amplifies and sustains the process. All arrows indicate genetic relationships.
Blastema Formation during Leg Disc Regeneration Requires dronc and dp53
The parallels between the disc response to undead cells and the regeneration response of imaginal discs are striking, in that both induce growth arrest, changes in the expression of pattern regulating genes, and compensatory proliferation [12, 14, 17]. The reduced survival of irradiated dp53 mutants, combined with our observations that dp53 mutants containing undead cells were unable to induce compensatory proliferation, thus suggested the possibility that dp53 has a general role in allowing regeneration of damaged tissue. During imaginal disc regeneration, a proliferating blastema forms that supplies cells that will regenerate missing structures [10–12]. We tested the role of dp53 in blastema formation by inducing a regeneration response in leg discs via an established method whereby ectopic expression of Wg in the “weak point”—dorsal proximal cells—of the prothoracic leg disc induces a proliferating blastema and leads to a regenerative response in a process virtually identical to that after disc fragmentation [17, 37]. We monitored blastema formation by labeling wild-type, dp53 mutant, and dronc mutant prothoracic leg disc cells with BrdU (Figures 5I–5K). After expression of Wg for 40 hr, 59% of wild-type discs formed a blastema, consistent with previous reports [17]. In contrast, only 22% of dp53 mutant and 15% of dronc mutant leg discs formed a blastema (Figure 5K). These data suggest that in addition to their role in promoting compensatory proliferation in response to undead cells, dp53 and dronc contribute to blastema formation during a regeneration response. Collectively, these results support the idea that in the absence of dronc and dp53, extensive tissue damage leads to inefficient tissue repair.
Discussion
The repair of tissue after cellular damage can be critical to the survival of the animal. Previous studies demonstrated that undead cells stimulate the proliferation of neighboring cells, providing a model for how damaged and dying cells contribute to the replacement of lost tissue.
With this model, we find that the wing imaginal disc responds to this damage as a whole by deploying a multi-step process that ends with compensatory growth. We find that dp53, the sole p53 family member in Drosophila, functions in a dronc-dependent manner at each step of the tissue-replacement process. Furthermore, our experiments suggest that dp53 and the initiator caspase dronc may be generally required for tissue recovery in imaginal discs, because we found that blastema formation was significantly impaired during regeneration induced in either dp53 or dronc mutant leg discs.
dp53 Is Induced Independently of DNA Damage, but Requires Dronc Activity
Our data suggest that dp53 is induced and becomes functional in undead cells by a mechanism that does not require DNA-damage sensing or activation of the stress kinase AMPK. Rather, we find that Dronc, an initiator caspase homologous to caspase-9, is necessary and sufficient to induce all aspects of the growth regulation by dp53. We do not know how Dronc activity results in dp53 expression and activity in these cells, but many caspase substrates are not directly involved in apoptosis. As an example, one of the first substrates identified was the cytokine IL-1β, which regulates many aspects of the inflammatory response [38]. We found that induction of dp53 mRNA in undead cells is prevented in dronc mutant discs, and thus it is possible that a regulator of dp53 is cleaved by Dronc, leading to its expression and ultimately to its ability to regulate the compensatory growth response in the imaginal discs. Regardless of the molecular mechanism, our data argue for direct communication between Dronc and dp53 in response to tissue damage.
dp53: A Damage Monitor that Induces Compensatory Growth
Collectively, our experiments imply that dp53 serves as a master coordinator of tissue repair in imaginal discs, regulating both cell-autonomous and non-cell-autonomous cell-cycle arrests, the expression of the pattern-regulating genes wg and dpp, and compensatory cell proliferation and growth. Based on our results, we suggest that cellular damage activates Dronc, which in a nonapoptotic role causes the induction of dp53 mRNA and leads to dp53 activity. We propose that dp53 then acts as an overall damage monitor, in a role that includes its conserved functions in apoptosis (here, induction of hid and rpr expression) and growth arrest (by repression of stg/cdc25), but also allows for induction of signals that promote compensatory growth of the disc. Our results suggest that dp53 monitors tissue damage through a feed-forward loop with Dronc and the pro-apoptotic genes hid and rpr, which both amplifies and sustains the growth-regulating signal (Figure 5L).
An intriguing puzzle left unanswered by our results is why the growth response to undead cells occurs only several days after they are generated: both HhGal4 and EnGal4 drive expression of Hid or Rpr from early embryonic stages, yet even with careful observation we detected no growth phenotype until the middle part of the third instar. Caspases are active in cells expressing Hid or Rpr + P35 at early time points, indicating that these cells are not immune to the apoptotic response early in development (data now shown). The genes involved in the apoptotic response are subject to many levels of control, including that by micro-RNAs (miRNAs). Hid protein expression, for example, is suppressed by Bantam, a miRNA highly expressed early in imaginal disc development, but declining as development progresses [39]. It is likely that rpr is also regulated by miRNA gene silencing [40]. Hence, the delay of the growth response in discs with undead cells may reflect a requirement for threshold levels of these factors to fully activate the feedback loop. At the very least it emphasizes that the regulation of growth and cell death during wing disc development is complex and has multiple inputs, many of which we have little knowledge.
Activity thresholds appear to play an important role in the processes induced by undead cells. We found that Dronc, for instance, is haploinsufficient for its effect in compensatory proliferation. It is possible that the apoptotic functions of Dronc require a relatively low activity level, but that high Dronc activity allows activation of the dp53-dependent tissue-damage response. Regulation of Dronc by critical activity thresholds could provide the animal some regenerative capacity and increase its chances for survival when conditions are appropriate for tissue repair.
dp53 Regulates string/cdc25 to Activate a G2 Checkpoint
As expected given its role in coordinating many cellular behaviors, p53 modulates the activity or expression of myriad effectors [1]. Regulatory effectors of Drosophila p53 are only beginning to be identified, and our data add stg/cdc25 to the list. In our experiments, one of the first detectable disc responses to undead cells is G2 arrest, mediated by loss of stg mRNA. Cdc25 is also regulated by vertebrate p53 but is inhibited post-transcriptionally by p53-dependent 14-3-3 activity [1]. Experiments with irradiated dp53 mutant animals have not revealed a cell-cycle arrest role previously [19, 20, 23]. However, recent work indicates that dp53 also regulates a G1 checkpoint under conditions of metabolic stress [34]; thus, like vertebrate p53, dp53 can activate both a G1 and a G2 checkpoint in response to tissue stress. Other effectors and targets involved in the compensatory proliferation process remain unknown, although expression profiling experiments from irradiated dp53 mutants identified several potential targets, several of which do not have obvious roles in cell death or DNA repair [19].
dp53 Coordinates Signaling Processes Leading to Damage-Induced Growth
How does dp53 control the signaling that leads to compensatory proliferation? The events that we observe—G2 arrests in two different cell populations, ectopic expression of wg, and compensatory growth—are all regulated by dp53. It is possible that dp53 directly and coordinately controls each of these processes by regulating the expression of specific effectors. However, because the response is both cell autonomous and non-cell autonomous, we favor the idea that these processes are interdependent, but sequentially activated. We envision that as a result of Dronc activation in undead cells, dp53 induces loss of stg, leading to G2 arrest, and hid and rpr expression, initiating the feedback loop. We postulate that cells then synthesize factors that stimulate their survival and proliferation. The non-cell-autonomous arrest in the anterior compartment may be a secondary effect of undead cells in the posterior. We have observed high levels of TUNEL activity in the anterior cells of these discs (data not shown), which could feasibly activate dp53 in those cells. However, we did not detect dp53 mRNA in anterior cells. One possibility is that the DNA fragmentation resulting from dying anterior cells could activate ATM and Chk2 in those cells. Consistent with this, although loss of either of these kinases did not affect undead cell induction of Wg expression or compensatory growth, the cell-cycle arrest in anterior cells was reduced in a fraction of atm and chk2 mutants (Figure S2 and data not shown).
What is the growth-stimulating signal induced by undead cells? While its identity is still unclear, both Wg and Dpp have been implicated in this role [7, 13]. This makes sense, because Wg and Dpp are the major pattern organizers of all imaginal discs and are also involved in regulating their growth, and furthermore they are known to be induced in disc regeneration [14, 15, 41]. However, although wg and dpp are ectopically expressed in undead cells, we find that targets of both are sharply downregulated, specifically in the undead cells (Figure S4). Our data also show that undead cells are able to proliferate and contribute to the compensatory growth. Thus, although the nonautonomous stimulation of growth (anterior cells near the A/P boundary) could be due to increased Dpp signaling, we suspect that the autonomous growth stimulation is due to other, unidentified factors.
Evolutionary Considerations
We have identified a growth-regulatory role for dp53 that seems counter to its role as a tumor suppressor in vertebrates. However, we speculate that the ability of dp53 to sense and respond to tissue damage and promote compensatory proliferation and regeneration in Drosophila reflects an ancestral function, aspects of which have been appropriated for developmental processes and distributed among p53, p63, and p73 during vertebrate evolution. Although p63 and p73 initially were proposed to have evolved as duplications of p53, reanalysis of the phylogenetic relationship between the three family members has suggested that p63 may be the ancestral gene [26, 42, 43]. p63 and p73 are structurally similar to p53 but contain an additional SAM domain. dp53 is the sole member of the family encoded in the Drosophila genome, and although dp53 does not contain a SAM domain, based on the sequence of the DNA binding domain, the most highly conserved region of p53, it is more related to vertebrate p63 than to p53 [26]. After irradiation, cell-cycle arrest is not p53 dependent in either Drosophila or the nematode C. elegans, and therefore it has been proposed that the ancestral p53 function is apoptosis, rather than a “repair, then death” response when damage cannot be repaired [20]. Our experiments argue that as in vertebrates, dp53 plays a role in cell-cycle arrest after tissue damage. The additional functions of dp53 in promoting cell proliferation may have been conserved in p63, which regulates progenitor cell renewal in the epidermis. Other processes that require cell renewal may also be regulated by dp53. For example, dp53 mutants are reported to have fertility defects [20, 44], so it is tempting to speculate that stem cell renewal in the gonad requires this previously unappreciated role of dp53.
Conclusions
How cell death and cell proliferation are coordinated during development and after stress is a fundamental question that is key to our understanding of growth regulation. We have found that dp53 is required for the compensatory growth induced after tissue damage, which could reflect an ancestral function of p53 in tissue repair and cell renewal.
Experimental Procedures
Fly Strains
The following strains were used: UAS-P35 [45]; UAS-Hid [5]; UAS-Rpr (gift of J. Abrams), Act>Draf>lacZ [27]; dp53ns [20]; dp535a-1-4 [21]; atm6, atm3 [33]; chk2p6 (also called mnkp6) [19]; EnGal4, UAS-GFP [28]; HhGal4, UAS-GFP; dronc129/TM6B [36]; Df(3L)XR38/TM6B; hidA22/TM6B; Df(3L)H99/TM6B; hidP05014/TM6B; hidX14/TM6B; UAS-Dronc80 [5]; UAS-Wg; Act>y+>Gal4, UAS-GFP; Dpp-lacZ; Brk-Z, VgBE-Z; VgQE-lacZ. Unless otherwise indicated, fly strains are described on http://flybase.bio.indiana.edu.
Fly Husbandry
Eggs from appropriate crosses were collected on yeasted grape plates for short periods (2–3 hr). After hatching, larvae were transferred to standard molasses food vials (≤50/vial) supplemented with fresh yeast and raised at 25°C for defined periods of time, as described [28, 29].
Cell-Proliferation Rate Measurements
The Flp-out Lac-Z cassette [27] was used to generate neutral clones. Larval heat shocks to activate Flp recombinase expression were performed at 37°C for 20 min at 70 hr AEL and allowed to grow until 96 hr AEL. The location of each clone in the wing disc was recorded as medial or lateral (with respect to the A/P boundary) in the anterior or posterior. Cell doubling times were determined by counting the number of cells in each clone with the formula log 2 (hr)/log N, where N = median cell number/clone and hr = age of the clone [28].
Flow Cytometry
Wing discs were dissected at the time points indicated and dissociated into single-cell suspensions in Trypsin-EDTA supplemented with Hoechst 33342 [28]. Cell cycles were analyzed as described previously [28], with a Becton Dickinson LSR II with FACS Diva software.
Leg Disc Regeneration
Regeneration was induced in leg imaginal discs as described [17]. In brief, larvae of the genotype ywhsflp; Act>y+>Gal4, UAS-GFP/UAS-Wingless; dp53ns (or dronc129) were subjected to a 2 hr heat shock at 37°C at 72 hr AEL. This induced Wingless in >95% of cells and led to regeneration in the first leg discs. After 40 hr, blastema formation was monitored by BrdU labeling of discs for 10 min, and the percent of discs with a regenerating blastema was scored as described [17, 37].
Immunocytochemistry
Fixation and immunocytochemistry of imaginal discs were carried out as described [31]. RNA in situ hybridizations were carried out with digoxigenin-labeled RNA probes [31]. TUNEL assays were carried out with Apoptag Red (Intergen); a detailed protocol is available upon request. BrdU assays were carried out as described [29] with modifications (protocol available upon request). Images were acquired with Apotome software and a Zeiss Axioplan 2 microscope with an Orca-100 CCD camera (Hammatsu) and processed with Photoshop (Adobe) software. The following antibodies and dilutions were used: rabbit anti-p-Smad, 1:4000 (gift of E. Laufer); rabbit anti-Vg, 1:100 (gift of S. Carroll); mouse anti-Cut, 1:50 (DSHB); mouse anti-Wg, 1:30 (DSHB); rabbit anti-β-gal, 1:2000 (Cappel); mouse anti-Diap1, 1:200, and rabbit anti-Hid, 1:1000 (gifts of B. Hay); rabbit anti-Diap, 1:100 (gift of H. Steller); rabbit anti-cleaved Caspase 3, 1:100 (Cell Signaling); mouse anti-Digoxigenin, 1:2000 (Roche); mouse anti-BrdU, 1:100 (Roche); rabbit anti-p-AMPK, 1:250 (Cell Signaling); guinea pig anti-Dronc, (1:1000) [36].
Supplementary Material
Acknowledgments
We thank N. Senoo-Matsuda for help with some experiments; M. Brodsky, B. Hay, A. Bergmann, E. Laufer, J. Abrams, P. Meier, D. Rio, H. Steller, and T.T. Su for generously sharing fly strains and antibodies; T.T. Su for an enlightening discussion; and A. Tomlinson and members of the Johnston lab for critical reading of the manuscript. Supported by grants from the NIH (HD42770 to L.A.J; T32-CA009503 to B.S.W) and the Rita Allen Foundation (L.A.J.).
Footnotes
Supplemental Data
Four Supplemental Figures can be found with this article online at http://www.current-biology.com/cgi/content/full/16/16/1606/DC1/.
References
- 1.Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13:1027–1036. doi: 10.1038/sj.cdd.4401910. [DOI] [PubMed] [Google Scholar]
- 2.McGowan CH, Russell P. The DNA damage response: sensing and signaling. Curr Opin Cell Biol. 2004;16:629–633. doi: 10.1016/j.ceb.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 4.Wyllie AH, Kerr JF, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 5.Huh JR, Guo M, Hay BA. Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr Biol. 2004;14:1262–1266. doi: 10.1016/j.cub.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 6.Perez-Garijo A, Martin FA, Morata G. Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development. 2004;131:5591–5598. doi: 10.1242/dev.01432. [DOI] [PubMed] [Google Scholar]
- 7.Ryoo HD, Gorenc T, Steller H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell. 2004;7:491–501. doi: 10.1016/j.devcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 8.Kim R, Emi M, Tanabe K. Cancer cell immune escape and tumor progression by exploitation of anti-inflammatory and pro-inflammatory responses. Cancer Biol Ther. 2005;4:924–933. doi: 10.4161/cbt.4.9.2101. [DOI] [PubMed] [Google Scholar]
- 9.Janes KA, Gaudet S, Albeck JG, Nielsen UB, Lauffenburger DA, Sorger PK. The response of human epithelial cells to TNF involves an inducible autocrine cascade. Cell. 2006;124:1225–1239. doi: 10.1016/j.cell.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 10.Bryant PJ, Fraser SE. Wound healing, cell communication, and DNA synthesis during imaginal disc regeneration in Drosophila. Dev Biol. 1988;127:197–208. doi: 10.1016/0012-1606(88)90201-1. [DOI] [PubMed] [Google Scholar]
- 11.Schubiger G. Regeneration, duplication and transdetermination in fragments of the leg disc of Drosophila melanogaster. Dev Biol. 1971;26:277–295. doi: 10.1016/0012-1606(71)90127-8. [DOI] [PubMed] [Google Scholar]
- 12.Haynie JL, Bryant PJ. Intercalary regeneration in imaginal wing disk of Drosophila melanogaster. Nature. 1976;259:659–662. doi: 10.1038/259659b0. [DOI] [PubMed] [Google Scholar]
- 13.Perez-Garijo A, Martin FA, Struhl G, Morata G. Dpp signaling and the induction of neoplastic tumors by caspase-inhibited apoptotic cells in Drosophila. Proc Natl Acad Sci USA. 2005;102:17664–17669. doi: 10.1073/pnas.0508966102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brook WJ, Ostafichuk LM, Piorecky J, Wilkinson MD, Hodgetts DJ, Russell MA. Gene expression during imaginal disc regeneration detected using enhancer-sensitive P-elements. Development. 1993;117:1287–1297. doi: 10.1242/dev.117.4.1287. [DOI] [PubMed] [Google Scholar]
- 15.Russell MA, Ostafichuk L, Scanga S. Lethal P-lacZ insertion lines expressed during pattern respecification in the imaginal discs of Drosophila. Genome. 1998;41:7–13. doi: 10.1139/g97-099. [DOI] [PubMed] [Google Scholar]
- 16.Couso JP, Bate M, Martinez-Arias A. A wingless-dependent polar coordinate system in Drosophila imaginal discs. Science. 1993;259:484–489. doi: 10.1126/science.8424170. [DOI] [PubMed] [Google Scholar]
- 17.Sustar A, Schubiger G. A transient cell cycle shift in Drosophila imaginal disc cells precedes multipotency. Cell. 2005;120:383–393. doi: 10.1016/j.cell.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 18.Brodsky MH, Nordstrom W, Tsang G, Kwan E, Rubin GM, Abrams JM. Drosophila p53 binds a damage response element at the reaper locus. Cell. 2000;101:103–113. doi: 10.1016/S0092-8674(00)80627-3. [DOI] [PubMed] [Google Scholar]
- 19.Brodsky MH, Weinert BT, Tsang G, Rong YS, McGinnis NM, Golic KG, Rio DC, Rubin GM. Drosophila melanogaster MNK/Chk2 and p53 regulate multiple DNA repair and apoptotic pathways following DNA damage. Mol Cell Biol. 2004;24:1219–1231. doi: 10.1128/MCB.24.3.1219-1231.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sogame N, Kim M, Abrams JM. Drosophila p53 preserves genomic stability by regulating cell death. Proc Natl Acad Sci USA. 2003;100:4696–4701. doi: 10.1073/pnas.0736384100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rong YS, Titen SW, Xie HB, Golic MM, Bastiani M, Bandyopadhyay P, Olivera BM, Brodsky M, Rubin GM, Golic KG. Targeted mutagenesis by homologous recombination in D. melanogaster. Genes Dev. 2002;16:1568–1581. doi: 10.1101/gad.986602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ollmann M, Young LM, Di Como CJ, Karim F, Belvin M, Robertson S, Whittaker K, Demsky M, Fisher WW, Buchman A, et al. Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell. 2000;101:91–101. doi: 10.1016/S0092-8674(00)80626-1. [DOI] [PubMed] [Google Scholar]
- 23.Jaklevic BR, Su TT. Relative contribution of DNA repair, cell cycle checkpoints, and cell death to survival after DNA damage in Drosophila larvae. Curr Biol. 2004;14:23–32. doi: 10.1016/j.cub.2003.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koster MI, Kim S, Mills AA, DeMayo FJ, Roop DR. p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev. 2004;18:126–131. doi: 10.1101/gad.1165104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKeon F. p63 and the epithelial stem cell: more than status quo? Genes Dev. 2004;18:465–469. doi: 10.1101/gad.1190504. [DOI] [PubMed] [Google Scholar]
- 26.Yang A, Kaghad M, Caput D, McKeon F. On the shoulders of giants: p63, p73 and the rise of p53. Trends Genet. 2002;18:90–95. doi: 10.1016/s0168-9525(02)02595-7. [DOI] [PubMed] [Google Scholar]
- 27.Struhl G, Basler K. Organizing activity of Wingless protein in Drosophila. Cell. 1993;72:527–540. doi: 10.1016/0092-8674(93)90072-x. [DOI] [PubMed] [Google Scholar]
- 28.Neufeld T, de la Cruz AF, Johnston LA, Edgar BA. Coordination of growth and cell division in the Drosophila wing. Cell. 1998;93:1183–1193. doi: 10.1016/s0092-8674(00)81462-2. [DOI] [PubMed] [Google Scholar]
- 29.Johnston LA, Sanders AL. Wingless promotes cell survival but constrains growth during Drosophila wing development. Nat Cell Biol. 2003;5:827–833. doi: 10.1038/ncb1041. [DOI] [PubMed] [Google Scholar]
- 30.Edgar BA, O’Farrell PH. Genetic control of cell division patterns in the Drosophila embryo. Cell. 1989;57:177–187. doi: 10.1016/0092-8674(89)90183-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnston LA, Edgar BA. Wingless and Notch regulate cell-cycle arrest in the developing Drosophila wing. Nature. 1998;394:82–84. doi: 10.1038/27925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.James AA, Bryant PJ. Mutations causing pattern deficiencies and duplications in the imaginal wing disk of Drosophila melanogaster. Dev Biol. 1981;85:39–54. doi: 10.1016/0012-1606(81)90234-7. [DOI] [PubMed] [Google Scholar]
- 33.Silva E, Tiong S, Pedersen M, Homola E, Royou A, Fasulo B, Siriaco G, Campbell SD. ATM is required for telomere maintenance and chromosome stability during Drosophila development. Curr Biol. 2004;14:1341–1347. doi: 10.1016/j.cub.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 34.Mandal S, Guptan P, Owusu-Ansah E, Banerjee U. Mitochondrial regulation of cell cycle progression during development as revealed by the tenured mutation in Drosophila. Dev Cell. 2005;9:843–854. doi: 10.1016/j.devcel.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 35.Salvesen GS, Abrams JM. Caspase activation—stepping on the gas or releasing the brakes? Lessons from humans and flies. Oncogene. 2004;23:2774–2784. doi: 10.1038/sj.onc.1207522. [DOI] [PubMed] [Google Scholar]
- 36.Xu D, Li Y, Arcaro M, Lackey M, Bergmann A. The CARD-carrying caspase Dronc is essential for most, but not all, developmental cell death in Drosophila. Development. 2005;132:2125–2134. doi: 10.1242/dev.01790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnston LA, Schubiger G. Ectopic expression of wingless in imaginal discs interferes with decapentaplegic expression and alters cell determination. Development. 1996;122:3519–3529. doi: 10.1242/dev.122.11.3519. [DOI] [PubMed] [Google Scholar]
- 38.Green DR, Melino G. ICE heats up. Cell Death Differ. 2001;8:549–550. doi: 10.1038/sj.cdd.4400887. [DOI] [PubMed] [Google Scholar]
- 39.Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113:25–36. doi: 10.1016/s0092-8674(03)00231-9. [DOI] [PubMed] [Google Scholar]
- 40.Stark A, Brennecke J, Russell RB, Cohen SM. Identification of Drosophila microRNA targets. PLoS Biol. 2003;1:e60. doi: 10.1371/journal.pbio.0000060. 10.1371/journal. pbio.0000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bryant PJ. The polar coordinate model goes molecular. Science. 1993;259:471–472. doi: 10.1126/science.8424169. [DOI] [PubMed] [Google Scholar]
- 42.Mills AA. p53: link to the past, bridge to the future. Genes Dev. 2005;19:2091–2099. doi: 10.1101/gad.1362905. [DOI] [PubMed] [Google Scholar]
- 43.Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, Saville MK, Lane DP. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005;19:2122–2137. doi: 10.1101/gad.1339905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JH, Lee E, Park J, Kim E, Kim J, Chung J. In vivo p53 function is indispensable for DNA damage-induced apoptotic signaling in Drosophila. FEBS Lett. 2003;550:5–10. doi: 10.1016/s0014-5793(03)00771-3. [DOI] [PubMed] [Google Scholar]
- 45.Hay BA, Wassarman DA, Rubin GM. Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell. 1995;83:1253–1262. doi: 10.1016/0092-8674(95)90150-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.