

Abstract
Systemic lupus erythematosus (SLE) is frequently accompanied by neuropsychiatric (NP) and cognitive deficits of unknown etiology. By using autoimmune MRL-lpr mice as an animal model of NP-SLE, we examine the relationship between autoimmunity, hippocampal damage, and behavioral dysfunction. Fluoro Jade B (FJB) staining and anti-ubiquitin (anti-Ub) immunocytochemistry were used to assess neuronal damage in young (asymptomatic) and aged (diseased) mice, while spontaneous alternation behavior (SAB) was used to estimate the severity of hippocampal dysfunction. The causal relationship between autoimmunity and neuropathology was tested by prolonged administration of the immunosuppressive drug cyclophosphamide (CY). In comparison to congenic MRL +/+ controls, SAB acquisition rates and performance in the “reversal” trial were impaired in diseased MRL-lpr mice, suggesting limited use of the spatial learning strategy. FJB-positive neurons and anti-Ub particles were frequent in the CA3 region. Conversely, CY treatment attenuated the SAB deficit and overall FJB staining. Similarly to mouse brain, the hippocampus from a patient who died from NP-SLE showed reduced neuronal density in the CA3 region and dentate gyrus, as well as increased FJB positivity in these regions. Gliosis and neuronal loss were observed in the gray matter, and T lymphocytes and stromal calcifications were common in the choroid plexus. Taken together, these results suggest that systemic autoimmunity induces significant hippocampal damage, which may underlie affective and cognitive deficits in NP-SLE.
Keywords: autoimmunity, inflammation, lupus, hippocampus, Fluoro Jade B, ubiquitin, spontaneous alternation behavior, cyclophosphamide, MRL mice
INTRODUCTION
Cognitive and affective disorders are frequent manifestations of the systemic autoimmune/inflammatory disease lupus erythematosus (SLE). Largely due to multi-system involvement and confounding factors (e.g., uremia and treatment with corticosteroids), clinical and experimental studies have thus far not succeeded in elucidating the principal factors and mechanisms involved in the pathogenesis of neuropsychiatric (NP)-SLE (Bruyn, 1995). With an ultimate goal of understanding the central nervous system (CNS) targets and functional consequences of SLE-like disease, we have been examining the neuropathology and behavioral dysfunction in the MRL-MpJ-Tnfrsf6lpr (MRL-lpr) mouse substrain. This substrain is known to develop systemic autoimmune disease due to a lpr mutation on chromosome 19 and a dysfunctional Fas (CD95, APO-1) receptor in a negative selection of autoreactive T cells (Watanabe-Fukunaga et al., 1992a,b). Along with early onset of inflammation and autoimmunity, MRL-lpr mice display changes in emotional reactivity (Sakic et al., 1994b), deficits in spatial learning/memory tasks (Sakic et al., 1992, 1993; Hess et al., 1993), accumulation of serotonin in the hippocampus (Sakic et al., 2002), and atrophy of pyramidal neurons (Sakic et al., 1998). Consistent with a case report on isolated hippocampal damage (Schnider et al., 1995) and impaired cognitive function in SLE patients (Carbotte et al., 1995; Denburg et al., 1997; Hanly et al., 1999), these results suggested structural and functional damage of the hippocampus during systemic autoimmunity and inflammation.
Behavioral deficits in MRL-lpr mice are defined as the departure from the behavioral performance of congenic MRL-MpJ +/+ (MRL +/+) controls (Szechtman et al., 1997), which develop similar disease symptoms later in life (Theofilopoulos, 1992). Marked behavioral deficits in the MRL-lpr substrain have been observed in mice as early as 7 weeks of age (Sakic et al., 1994a) and coincide with the emergence of humoral autoimmunity, but antedate generalized lymphadenopathy, arthritis, glomerulo-nephritis, and skin lesions (Andrews et al., 1978). The initial support for the hypothesis that spatial learning and memory are impaired by lupus-like disease came from studies in which MRL-lpr mice perseverated in their response bias during extinction and reversal learning in the Morris water-maze task (Sakic et al., 1992, 1993). The aim of the present study is to evaluate the time course of spontaneous alternation behavior (SAB), a functional trait proposed to be highly sensitive to hippocampal damage (reviewed in Richman et al., 1986). More specifically, it is well documented that spatial working memory and the reliable alternation of rodents in a T-maze in two consecutive trials largely depend on an intact hippocampus (Lalonde, 2002). If development of autoimmunity induces hippocampal damage, one might expect that in comparison to congenic MRL +/+ controls, diseased MRL-lpr mice would show a reduced SAB rate at advanced stages of lupus-like disease, but not before the serological signs of autoimmunity are manifest.
Although neuronal degeneration in the CA2/CA3 regions of MRL-lpr brains was recently demonstrated with a novel Fluoro Jade B (FJB) cytochemical stain (Ballok et al., 2003), it has not been determined whether this damage is functionally important. In addition, it remains to be determined whether neuronal loss is associated with the progress of autoimmune disease or, alternatively, reflects a developmental deficiency related to impaired expression of the apoptotic Fas receptor in the MRL-lpr brain (Park et al., 1998). Although the latter possibility appears less viable in the light of evidence that cortical architectures in young, pre-diseased MRL-lpr and MRL +/+ mice are comparable (Sherman et al., 1990), and that size of hippocampal fields and neuronal density are not reduced in Fas-deficient lpr mice (Kovac et al., 2002), we assess age-related changes in densities of hippocampal neurons stained by the hematoxylin-eosin (H&E) method. The present analysis involves the comparison between young (asymptomatic) and old (diseased) MRL-lpr and MRL +/+ mice and employs ubiquitination as an additional marker of cell degeneration (Alves-Rodrigues et al., 1998). Specifically, ubiquitin (Ub) binds to damaged or misfolded proteins, targeting them for degradation by the Ub-proteasome pathway. If ubiquitinated proteins are not eliminated by this pathway, neurodegeneration may occur. In addition to cellular demise, ubiquitination of proteins may be involved in a DNA repair mechanism (Jentsch et al., 1987). Therefore, it was expected that an increased density of FJB-positive cells and alterations in the Ub-proteasome degradation system in the hippocampus would parallel both the emergence of autoimmunity and impaired SAB performance. To test for a cause-effect relationship between neuronal degeneration and autoimmunity, the immunosuppressive drug cyclophosphamide (CY) was used as previously reported (Farrell et al., 1997; Sakic et al., 1995, 2000a).
Despite the well-acknowledged construct validity and theoretical usefulness of animal models, it is equally important to demonstrate their face validity (Henn and McKinney, 1987). Our experimental study coincided with the death of an SLE patient who had CNS involvement. This provided us with a unique opportunity to compare neuropathological changes in animal and human forms of systemic autoimmune disease in which brain function is compromised.
MATERIALS AND METHODS
Experiment I: Age-Related Changes in SAB Response Rate and Neuromorphology
Animals
Three-week-old (±3 days) MRL-lpr and MRL +/+ male mice (n = 20 mice/substrain) were purchased from the Jackson Laboratory (Bar Harbor, ME); each group was left for 7 days to habituate to local laboratory conditions (light phase: 8 AM–8 PM, food, and water ad libitum; 5 mice/cage; level E, cages sanitized during regular housing). Five days prior to behavioral testing, mice were singly caged and habituated to the experimenter (Sakic et al., 1992). In addition, mouse cages were wheeled on three occasions from the colony room to the testing room, to reduce stress induced by transportation and a novel environment. Ten mice from each strain were tested and sacrificed at either 6 or 16 weeks of age (cohort 1). Because of dissimilar fixation protocols for the assessment of dying neurons and ubiquinated particles, a second batch of mice (cohort 2; 20 males/substrain) was obtained from our recently established MRL colony (housing level A, cages sterilized, replaced, and manipulated under laminar flow protection). At 3 weeks of age (±3 days), the mice were transferred to a housing room and left to habituate over 7 days, for assessment of brain morphology at 4 and 14 weeks of age. All experimental protocols were performed in accordance with the rules and regulations of the Canadian Council of Animal Care.
Spontaneous alternation behavior
There is substantial evidence that the hippocampus is one of the structures most intimately involved in SAB (Lalonde, 2002). Indirect but compelling evidence that supports this notion comes from developmental studies in which hippocampal maturation and level of SAB show parallel courses (Deacon et al., 2002). Considering that hippocampal damage reduces the rate of SAB in mice (Deacon et al., 2002), it was expected that young MRL-lpr mice would show a comparable SAB rate to congenic controls before the onset of autoimmunity and, conversely, that their SAB rate would decline with advancing manifestations of systemic autoimmune disease.
The T-maze (made of black Plexiglas) consisted of four perpendicular arms (H = 15 × L = 25 × W = 10 cm) and sliding separators that could modify the maze into an L, T, or + shape. The discrete-trial procedure (Richman et al., 1986) was employed, with a daily session consisting of trial 1 (5 s in a start position and entry into an unblocked arm), 60-s intertrial period (mouse restrained in the arm) and trial 2, in which both arms were open and a mouse was expected to choose an unvisited arm after leaving the start position. An arm was considered to have been chosen if all four limbs were within the selected arm. A guillotine door was lowered behind the mouse, immediately after the entry. Urinary trails were removed, and the maze was cleaned by a cloth moistened with Windex® after each mouse was tested. The sequence of blocked arms (left or right) in trial 1 was randomly generated by Microsoft Excel software. Following 5 days of SAB testing, mice were given a reversal trial on day 6, to examine whether the non-spatial learning strategy was employed (Bertholet and Crusio, 1991), such as taxis or praxis (Sutherland and McDonald, 1990; Whishaw, 1991). Trial 1 consisted of placing a mouse in the usual starting position. However, following a 60-s intertrial period, trial 2 was initiated from the arm located 180 degrees from the trial 1 arm. Assuming that a mouse had used a spatial strategy to memorize external cues, it was expected that after leaving the new start position, an unvisited arm would be chosen by a body turn identical to trial 1.
Indices of autoimmunity
High levels of serum antinuclear antibodies (ANA) and splenomegaly are typical manifestations of systemic autoimmune lupus-like disease (Theofilopoulos, 1992) and were presently examined. Mice were anesthetized with Somnotol (i.p. 60 mg/kg body weight; MTC Pharmaceuticals, Cambridge, ON) and perfused with 40 ml of phosphate-buffered saline (PBS) after terminal bleeding from the vena cava. Blood samples were left to coagulate in 1.5-ml plastic vials and centrifuged for 10 min at 3,000 rpm. Serum was separated from the clot and stored at −20°C until further analysis. ANA concentration was measured using a sandwich Anti-Nuclear Antibody ELISA kit (Cat. no. 5200), according to the manufacturer’s instructions (Alpha Diagnostic International, San Antonio, TX) and the protocol previously described (Sakic et al., 2000a). In brief, serum samples were diluted 1:100 in the kit diluent and applied to both experimental and control wells to assess the specificity of binding. Optical density was determined using a microplate ELISA reader set to 450 nm. The wet spleen weight was determined on an analytical scale (Sartorius 2024 MP, VWR Scientific of Canada Ltd.) shortly after extraction.
Fluoro Jade B method
The Fluoro-Jade B stain has an affinity for the entire degenerating neuron, regardless of the type of cell death (Hopkins et al., 2000; Schmued and Hopkins, 2000). Despite incomplete knowledge of the staining mechanisms, the FJB method shows high reliability in the detection of dying neurons (Ye et al., 2001).
Extracted brains were fixed in 4% paraformaldehyde (PFA) for 24 h and were then immersed in 30% sucrose (in PBS) for 4 days before frozen sections were processed according to the previously published protocol (Ballok et al., 2003). In brief, slides were immersed in 100% ethanol for 3 min, followed by 1 min in 70% ethanol. The slides were rinsed in distilled water (dH2O) for 1 min before being transferred to a 0.06% potassium permanganate solution and gently shaken for 15 min. Slides were rinsed in dH2O for 1 min before immersion in a 0.001% FJB/0.1% acetic acid staining solution (prepared from a 0.01% stock solution; HistoChem, Jefferson, AR). In our experience, the stock solution produced optimal staining results after 2 months of storage at 4°C in darkness. After 30 min of gentle shaking in the staining solution, slides were rinsed for 1 min in each of three dH2O washes and left to dry for several hours in darkness. Subsequently, they were processed in three 2-min xylene washes before being coverslipped with DPX (Sigma, St. Louis, MO). The reactivity in the CA3 sector and nonhippocampal periventricular areas were examined using an epifluorescent microscope with 450 – 490-nm excitation light (Diastar Fluorescence Microscope, Reichert Scientific, Buffalo, NY) at ×200 magnification. To quantify the number of FJB-positive neurons, sections were photographed on 400 ASA 35-mm print film (Eastman Kodak, Rochester, NY), using a Nikon N90s camera. The pictures were scanned at 600-dpi resolution and digitized into TIFF files using Adobe Photoshop software (Adobe Systems, San Jose, CA) for assessment by NIH Image analysis software (Scion, Frederick, MD). To produce representative high-quality images, a Zeiss Laser Scanning Confocal Microscope (LSM 510, Carl Zeiss) argon laser (wavelength 488 nm) was employed for visualization of FJB. Confocal micrographs were obtained using a Fluar 20×/0.75 objective in combination with a 1,024 × 1,024-pixel resolution and were saved in the TIFF format.
The overall neuronal density was assessed by H&E staining of coronal sections (bregma approximately −1.0 mm) in the parietal cortex and CA2/CA3 area that were less densely packed with neurons and thus amenable to quantification. Sections were stained with an automated slide stainer (Leica Instruments, Germany), and 1-mm2 areas were examined with a stage micrometer by an unbiased observer. Sections of the highest quality were included in the analysis (n = 4 – 8 brains/group) and four counts (2 sections × 2 hemispheres) were performed at ×400 magnification.
Immunohistochemical localization of Ub
Mice were perfused with PBS, and the brains were extracted within 2 min, immersed into 10% neutral buffered formalin, and left in formalin for 4 days at room temperature (RT) until processing. Subsequent to fixation, brains were embedded in paraffin and cut in the coronal plane at 4 μm. Serial adjacent sections were stained with H&E and processed immunohistochemically for Ub, using an anti-Ub polyclonal antiserum (1:400; Dako, Burlington, Canada) and the labeled streptavidin-biotin-peroxidase technique (Vectastain; Shandon, Pittsburgh, PA). Sections were briefly counterstained with hematoxylin, dehydrated in a graded series of ethanol, mounted in xylene, and coverslipped. For each animal, the number of Ub-immunopositive dot-like structures was counted in five nonoverlapping fields within the strata oriens and pyramidale of the CA3 sector of the hippocampus. All counts were performed at the same coronal level (bregma −1.6 to −2.4 mm) under oil immersion (×1,000) by an experimenter blind to the experimental design.
Experiment II: Effects of Immunosuppression on SAB and Neuromorphology
Animals
To examine whether prolonged immunosuppressive treatment modifies SAB performance, 24 MRL-lpr and 20 MRL +/+ male mice (3 weeks old ±3 days) were purchased from the Jackson Laboratory. An additional batch of 12 MRL-lpr and 12 MRL +/+ mice was purchased to confirm behavioral observations and examine the effects of sustained immunosuppression on neuropathology seen in Experiment I. Each group was managed and maintained under conditions as outlined above. Two weeks later (i.e., 5 weeks of age), mice were housed singly to receive treatment.
Immunosuppressive treatment
The therapeutic effect of CY on the development of autoimmune symptoms was demonstrated previously (Shiraki et al., 1984; Grota et al., 1989, 1990; Sakic et al., 1995, 1996). In addition to the reduction of leukocyte numbers (Snippe et al., 1976), CY makes these cells unresponsive to stimuli, leading to generalized immunosuppression (ten Berge et al., 1982). CY was injected weekly (100 mg/kg i.p.; mouse LD50 = 405 mg/kg i.p.; Procytox, Horner, Montreal, Canada) to half of the mice in each group. The treatment started during the 5th week of life and ended during the14th week. The other half of the animals received nine injections of an equivalent volume (~0.2– 0.3 ml) of saline (SAL). Mice were assigned into one of four groups, according to substrain (MRL-lpr vs MRL +/+) and treatment (CY vs SAL). In both batches, two mice died prematurely in the CY group before treatment was completed. To avoid acute effects of CY and stress induced by injection, the SAB testing commenced 7 days after the last injection and mice were sacrificed at around 16 weeks of age. FJB and Ub staining was analyzed as described above, and all counts were performed by an observer blind to the experimental design. ANA levels, which correlate highly with the spleen weight (Sakic et al., 2000a), were assessed as in Experiment 1.
Experiment III: Human Specimens
Brain tissue was obtained from a 58-year-old woman who died from NP-SLE and had a history of psychosis and seizures. She presented with status epilepticus that was managed with phenytoin and diazepam. She remained comatose after her seizures. Despite aggressive immunosuppression, including high-dose methylpred-nisolone, plasmapheresis, and CY, her condition deteriorated; she developed disseminated intravascular coagulation and died. The brain was examined after 14-day fixation in 10% buffered formalin. Sections were taken from the meninges, blood vessels, pons, medulla oblongata, cerebellum, mamillary bodies, right and left hippocampus, basal ganglia, frontal watershed area, temporal, parietal and occipital lobes, periventricular area around lateral and third ventricles, and choroid plexus. Coronal sections of the brain were serially sectioned and representative paraffin wax embedded tissue sections were stained with Luxol fast blue/H&E. Other stains used were Bielschowsky silver and Congo red. Using standard immunohistochemical methods, sections of the periventricular area, hippocampus, and temporal gray matter were stained for Ub; sections of the choroid plexus were stained for leukocyte common antigen (LCA), T-cell markers (CD3, CD4, CD8), B-cell marker (CD20), and macrophage marker (CD68). All sections were examined under light microscopy. Additional sections were frozen, stained with FJB, and examined with confocal microscopy. Control sections were obtained from a female subject of similar age who died of causes unrelated to SLE and epilepsy.
Statistical Analysis
The data were analyzed by analysis of variance (ANOVA) with substrain (MRL-lpr vs MRL +/+), treatment (CY vs SAL), and age as between-group factors, and slide as the repeated measure. Student’s t-test was used in the post hoc analysis. Fisher’s exact test was used to assess the difference in group performance in the SAB tests. Significance level was set at P < 0.05, and all computations were performed using the SPSS 11.0 statistical package. Graphs show means ± SEM.
RESULTS
Experiment I
The SAB performance in young mice from the two MRL substrains was comparable at 6 weeks of age. However, 16-week-old MRL-lpr mice performed poorly in comparison to age-matched MRL+/+ controls (t18 = 2.228, P = 0.039, Table 1). This deficit was confirmed in the “reversal” trial, where the alternation rate of diseased MRL-lpr mice dropped to chance levels (Fisher’s exact test, one-tailed P = 0.016, Table 1). Neuropathological assessment revealed higher numbers of FJB-positive neurons (Fig. 1A) in the CA3 region (strain by age: F(1,36) = 39.944, P < 0.001, MRL-lpr vs age-matched MRL +/+, t18 = 6.583, P < 0.001, Fig. 2A). In a separate cohort of 14-week-old MRL-lpr mice, immunostaining for Ub showed intensely immunoreactive spherical dot-like structures (Fig. 1C), which were numerous in the neuropil of the stratum oriens and superficial stratum pyramidale of the CA3 region (strain F(1,18) = 8.093, P < 0.011, t18 = 3.000, P < 0.008; Fig. 2A). In addition to the hippocampus, the substantia nigra and brainstem tegmentum also showed more Ub particles than in MRL +/+ controls (strain F(1,18) = 8.998, P < 0.008, t18 = 2.845, P < 0.011; data not shown). Examination at higher magnification showed that the morphology of the structures was reminiscent of axon terminals. Indeed, occasional linear arrays of these structures indicated the presence of immunoreactive varicose axonal segments.
TABLE 1.
SAB Rates (%) of Young (6-wk) and Older (16-wk) MRL-lpr and Control MRL +/+ Mice Over 5-Day Response Acquisition and Single Reversal Trial†
| Group | Acquisition at 6 wk | Acquisition at 16 wk | Reversal at 16 wk |
|---|---|---|---|
| MRL-lpr (n = 10) | 76 ± 5 | 64 ± 6* | 50** (5/10) |
| MRL +/+ (n = 10) | 76 ± 5 | 80 ± 4 | 100 (10/10) |
SAB, spontaneous alternation behavior.
The alternation rate was lower in diseased 16-week-old MRL-lpr mice; a deficit in the spatial learning strategy was detected in the “reversal” task. This task differed from testing in the acquisition phase as Trial 2 was initiated 180 degrees away from the box arm where Trial 1 was given.
In comparison to MRL+/+, t18 = 2.228, P = 0.039.
In comparison with Fisher’s exact test P = 0.016.
FIGURE 1.
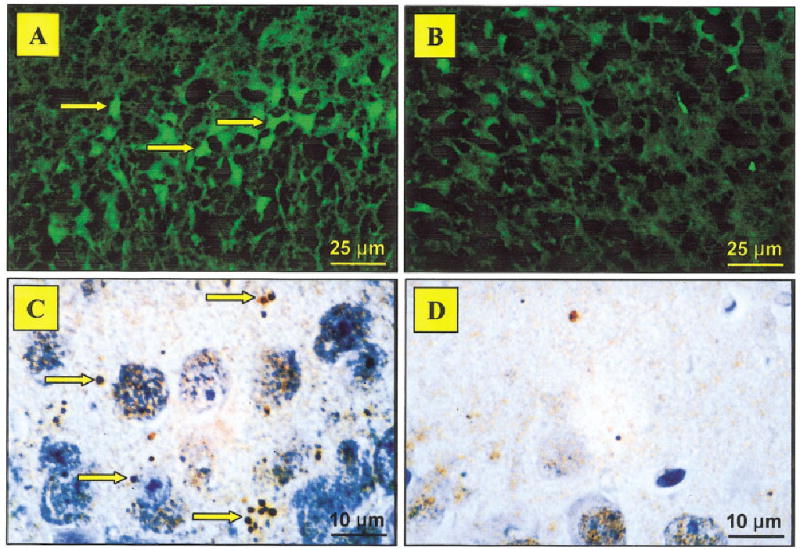
Representative micrographs showing Fluoro Jade B (FJB) and ubiquitin (Ub) labeling in the CA3 region. Numerous FJB-positive cells with neuronal morphology (shown by arrows) were common in brains from MRL-lpr mice (A) in comparison to asymptomatic MRL +/+ controls (B). Intensely immunoreactive Ub-positive spherical particles (shown by arrows) were detected in brains from diseased lupus-prone mice (C), likely reflecting degenerating axon terminals. Such particles were not abundant in the control brains (D). ×400 in A,B; ×1,000 in C,D. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com].
FIGURE 2.
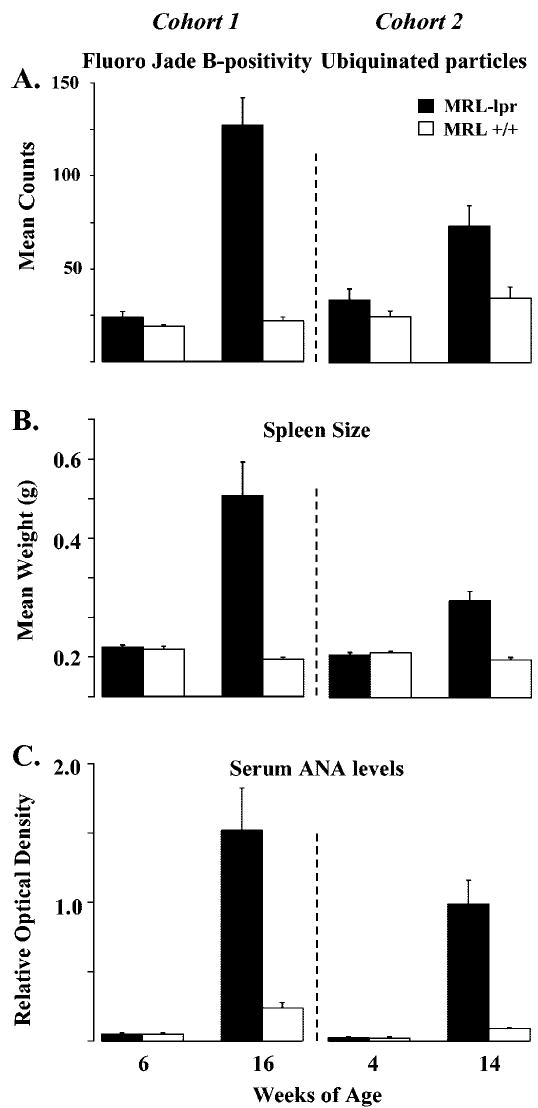
Total numbers of Fluoro Jade B (FJB)-positive and ubiquitin (Ub)-positive cells and the immune status in MRL mice at different ages. Cells/particles staining for FJB or Ub significantly increased following the onset of autoimmunity in older MRL-lpr mice (A). Spleen weight (B) and antinuclear antibody (ANA) levels (C) also increased significantly with age, confirming the autoimmune status in the MRL-lpr substrain.
Reduced density of cortical neurons in diseased MRL-lpr mice has been initially revealed with the cresyl violet method, but this observation was not quantified (Sakic et al., 1998). We presently confirmed this by assessing neuronal densities in the parietal cortex and CA2/CA3 region on sections stained with the standard H&E method. Comparisons across ages and to age-matched controls showed a paucity of cells in older MRL-lpr mice (Table 2). Despite a relatively small sample in some groups (n = 4 brains), the counts obtained suggest that significant group differences at an older age is a combination of an age-related increase in neuronal density in the MRL +/+ substrain and genuine loss of neurons during the development of systemic autoimmunity in the MRL-lpr substrain. This is supported by comparable immune statuses at younger ages, and the emergence of the SAB deficit when spleen weight and serum ANA titers increased in aged MRL-lpr mice (at 16 weeks of age, strain by age: for spleen, F(1,36) = 22.448, P < 0.001; for ANA, F(1,35) = 28.110, P < 0.001; at 14 weeks of age, strain by age: for ANA, F(1,34) = 15.006, P < 0.001; for spleen, F(1,36) = 36.568, P < 0.001; Fig. 2B,C).
TABLE 2.
Neuronal Density (Total Count ± SEM) as Assessed by Hematoxylin and Eosin Staining†
| Group | Parietal cortex | CA2/CA3 area |
|---|---|---|
| 6-wk MRL-lpr | 514 ± 32 (n = 4) | 210 ± 34 (n = 8) |
| 6-wk MRL +/+ | 573 ± 32 (n = 7) | 237 ± 33 (n = 7) |
| 16-wk MRL-lpr | 451 ± 43 (n = 8)* | 159 ± 28 (n = 8)** |
| 16-wk MRL +/+ | 691 ± 83 (n = 4) | 250 ± 29 (n = 7) |
The total number of H&E-stained neurons was obtained from four 1-mm2 areas (2 sections × 2 hemispheres) that were well preserved and amenable to counting (number of brains processed is shown in parentheses). Consistent with previous reports on reduced growth of brain mass and atrophy of pyramidal neurons, reduced neuronal density in diseased 16-week-old MRL-lpr mice supported the hypothesis that progress of systemic autoimmune disease impairs parenchymal growth in the parietal cortex and hippocampus, likely by inducing neuronal loss.
In comparison with age-matched MRL +/+ control
t10 = 2.868, P = 0.017, and
t13 = 2.238, P = 0.043.
Experiment II
The SAB deficit was less severe in CY-treated MRL-lpr mice compared to the SAL-treated MRL-lpr group (t22 = 2.691, P = 0.013; Table 2). CY-treated MRL-lpr mice also did not differ in the “reversal” test when compared to other groups (Table 3). Although the acquisition of SAB response in the CY-MRL +/+ group was comparable to that in the SAL-MRL +/+ group, their performance in the “reversal” task dropped to a chance level. This detrimental effect of CY on performance of control mice is not presently clear, but it is consistent with our previous observations (Sakic et al., 1995, 1996). Our recent study demonstrated that sustained CY treatment normalizes neuronal morphology, as evidenced by Golgi impregnation (Sakic et al., 2000a). In the present study, extensive FJB staining appeared to be attenuated after immunosuppressive treatment. However, likely due to the small sample size (n = 5–6 mice/group), a trend was seen in the CA3 region (strain by treatment: F(1,17) = 3.956, P = 0.063; Fig. 3A) and a statistically significant difference was detected in other periventricular gray matter regions (strain by treatment: F(3,17) = 5.549, P = 0.008; between SAL and CY MRL-lpr groups, t9 = 2.161, P = 0.05; Fig. 3B). This reduction in cytochemical staining was associated with the profound immunosuppressive effect of CY, as indicated by low ANA levels in the CY groups across two batches (batch: F(1,56) = 0.022, n.s.; strain by treatment: F(1, 56) = 47.376, P < 0.001; CY-lpr mice: 0.28 ± 0.06; SAL-lpr mice:1.58 ± 0.14; CY +/+ mice:0.13 ± 0.02; SAL +/+ mice: 0.24 ± 0.05). However, analysis of the CA3 region with the present sample size could not detect a beneficial effect of CY on the density of Ub-positive particles. Namely, sustained immunosuppression did not appear to change the difference between the MRL substrains (strain: t19 = 3.006, P = 0.007; treatment: t19 = 0.157, n.s.).
TABLE 3.
SAB Rates (%) of 14-Week-Old MRL-lpr and Control MRL +/+ Mice Following 8 Weekly i.p. Injections With Immunosuppressive Drug Cyclophosphamide (100 mg/kg body weight) or Saline (SAL)†
| Group | Acquisition | Reversal |
|---|---|---|
| MRL-lpr SAL (n = 12) | 60 ± 3* | 50 (6/12) |
| MRL +/+ SAL (n = 10) | 84 ± 7 | 90 (9/10) |
| MRL-lpr CY (n = 12) | 75 ± 4 | 67** (8/12) |
| MRL +/+ CY (n = 8) | 83 ± 8 | 50 (4/8) |
SAB, spontaneous alternation behavior; CY, cyclophosphamide; SAL, saline.
Sustained immunosuppressive treatment reduced the substrain difference in SAB acquisition rate. However, this treatment appeared to have detrimental effects in the MRL +/+ CY group in the “reversal” form of the test.
In comparison to MRL-lpr CY, t22 = 2.691, P = 0.013.
Not statistically different from other groups in multiple comparisons using the Fisher test.
FIGURE 3.
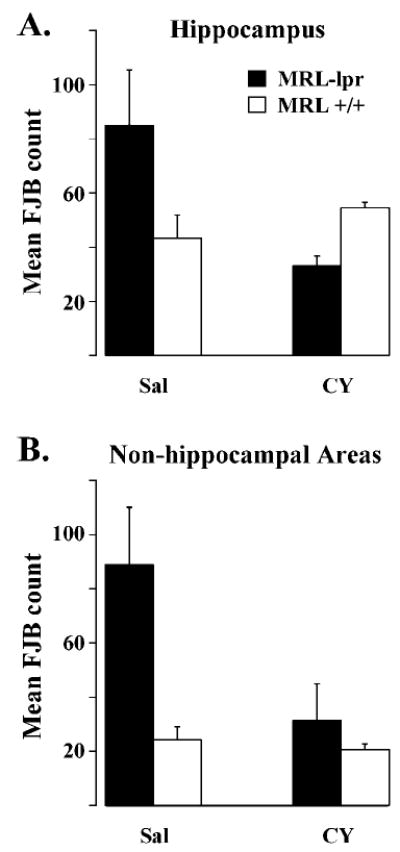
Effects of immunosuppressive treatment on brain pathology in MRL mice. Cyclophosphamide (CY) treatment appeared to attenuate the incidence of Fluoro Jade B (FJB)-positive cells in the hippocampus (A) and significantly reduced the staining in other brain regions of MRL-lpr mice (B).
Experiment III
The brain of the lupus patient weighed 1,145 g. There was no evidence of a subdural hematoma, and both hemispheres were symmetrical with no evidence of meningeal exudates or herniations. Blood vessels, dissected out from the circle of Willis, did not show any thromboembolic or atherosclerotic changes, but they did show myointimal proliferation with some hyalinization of smaller blood vessels (features suggestive of a history of benign hypertension). On serial sectioning, there was no evidence of infarction or of intracerebral or intraventricular bleeding. There was dilation of the lateral and third ventricles, left more than right, with thinning of the cortex around the ventricles. Some of the intraparenchymal blood vessels showed perivascular cuffing by chronic inflammatory cells and deposition of hemosiderin, features suggesting previously healed vasculitis. This is further corroborated by the fact that there were foci of demyelination around these vessels, indicating a vasculitic mechanism probably related to immune complex deposition. There was no microscopic evidence of abnormal neuronal migration, infarction, intraparenchymal, or intraventricular hemorrhage. Conversely, gross examination of the control brain did not show ventricular dilation of the lateral and third ventricles.
An interesting feature was the presence of increased densities of scattered inflammatory cells within the choroid plexus of the lupus patient (Fig. 4A). Mononuclear lymphocytes displayed positive immunostaining for LCA, CD3, CD4, and CD8, indicating that these were T lymphocytes. More CD8 than CD4 cells were observed, suggesting that most of the T lymphocytes were cytotoxic (Fig. 4C). These were not present in the control brain (Fig. 4D). Furthermore, periventricular tissue around the lateral and third ventricles showed loss of neurons, gliosis, satellitosis (microglial cells surrounding occasional viable neurons), and “brain sands”/corpora amylacea, not present in the control (Fig. 5A–D). The parietotemporal cortex also showed gliosis and ischemic changes in the neurons (likely secondary to status epilepticus). The neuronal loss in the CA3 region of the hippocampus was accompanied by surviving neurons, gliosis, and focal satellitosis (Fig. 6A). This was also seen above the dentate gyrus in the pyramidal layer, in addition to patchy areas of cell loss (Fig. 6C). In contrast, the hippocampus sections from the control brain did not show neuronal loss, gliosis, or satellitosis in the CA3 region. There were no patchy areas of cell loss in the pyramidal layer above the dentate gyrus. Neuronal demise in the hippocampus of the NP-SLE patient was confirmed by FJB staining. More specifically, FJB-positive neurons (Fig. 6E) and few scattered Ub-stained particles were observed in the CA3 and dentate gyrus. However, with respect to Ub staining, no significant difference between lupus and control brains could be detected.
FIGURE 4.
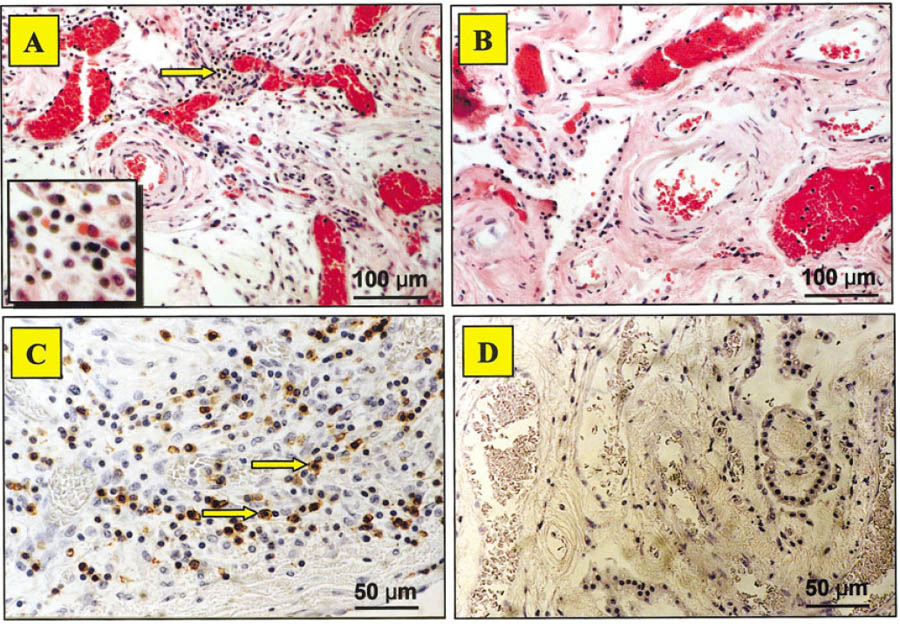
Chronic mononuclear inflammatory cells in the human brain. H&E staining showed a cluster of lymphocytes (arrow) in the choroid plexus of the lupus patient (A), but not in the control brain (B). Subsequent immunohistochemical staining for leukocyte common antigen (LCA), CD3, CD4, and CD8 antigens (shown by arrows) confirmed that the cells in the stroma were T lymphocytes (C), while the control brain was negative for the same markers (D). ×100 in A,B; ×200 in C,D; ×600 (inset in A).
FIGURE 5.
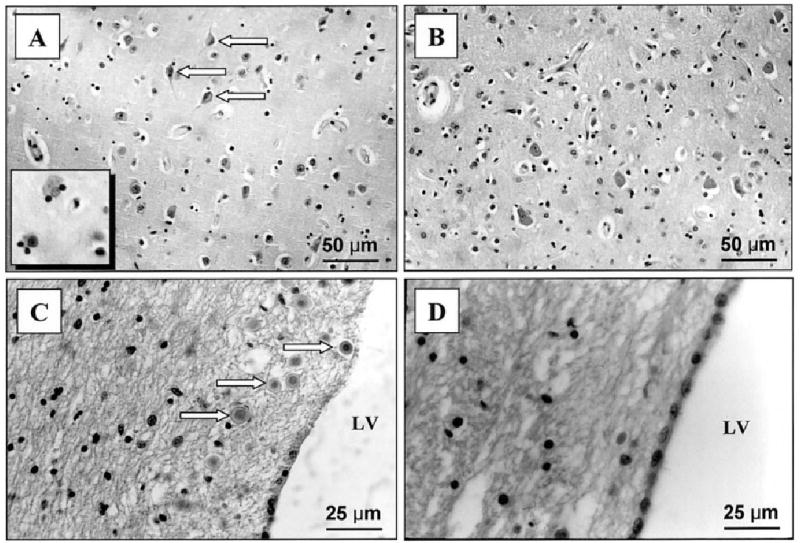
H&E staining of the parietotemporal cortex and periventricular regions of the human brain. “Dying neurons” (shown by arrows) and satellitosis (Inset) in the gray matter of the lupus patient (A). Normal neurons and resting glial cells in the control brain (B). Numerous particles identified as “brain sand” were common around the ventricles in the patient’s brain (C), and were rarely seen in the control (D). LV, lateral ventricle. ×200 in A,B); ×400 in C,D.
FIGURE 6.
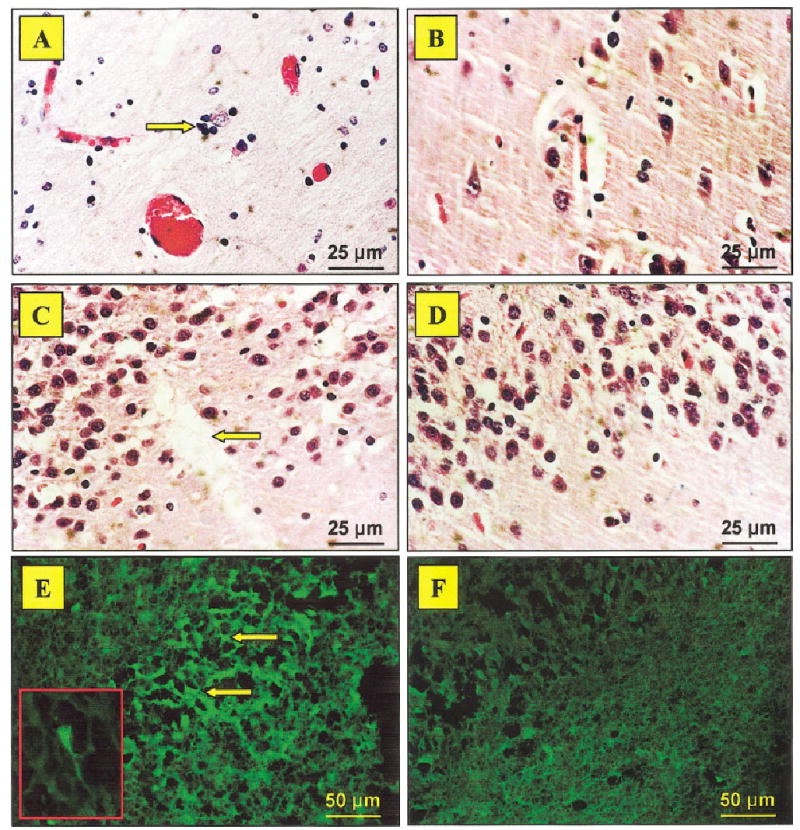
Neuronal loss and degeneration in the human hippocampus as revealed by H&E and FJB staining. The brain from the neuropsychiatric-systemic lupus erythematosus (NP-SLE) patient revealed a paucity of neurons and satellitosis (arrow) in the CA3 region (A), while the control brain showed normal neuronal density (B). A patchy area of cell loss was also seen in the dentate gyrus (arrow) of the lupus brain (C), but not in the control (D). Subsequently, bright green Fluoro Jade B (FJB)-positive neurons confirmed a degenerative process in the CA3 region (E), which was not observed in the control brain (F). ×400 in A–D; ×200 in E,F; ×600 (inset in E, under oil immersion). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com].
DISCUSSION
The emergence of FJB-positive neurons and Ub-positive structures in the hippocampus of mice with impaired SAB and the beneficial effects of immunosuppressive treatment point to the causal link between chronic autoimmunity/inflammation, structural damage, and aberrant behavior. These findings complement documented atrophy of basilar and apical dendritic branches in the CA1 region and parietal cortex (Sakic et al., 1998, 2000a) and neurotoxic properties of cerebrospinal fluid (CSF) from diseased MRL-lpr mice (Maric et al., 2001). In comparison to the murine form of the disease (Denenberg et al., 1992; Farrell et al., 1997; Sakic et al., 2000a,b), similar changes are seen in the brain from the NP-SLE patient. They include neuronal loss in the hippocampus and parietal regions, T-lymphocyte infiltration into the choroid plexus, and ventricular enlargement. Overall, the present results further support our hypothesis that systemic autoimmune disease induces brain damage and subsequently, behavioral dysfunction. In addition, the above evidence strengthens the face and construct validity of MRL-lpr mice as a model of NP-SLE.
The SAB paradigm is a procedure proposed to reflect exploratory behavior, which also depends on the formation of working memory (Richman et al., 1986). However, anxiety and stress may negatively affect SAB (Bats et al., 2001) to the extent that it does not reflect exploratory behavior (Gerlai, 2001). In our study, the animals were initially habituated to the experimenter and the testing environment to reduce these confounding effects. In addition to consistently poorer performance over successive trials, a profound deficiency in the “reversal” trial was observed in aged MRL-lpr mice. If the spatial strategy was employed, MRL-lpr mice would be expected to alternate to the novel arm by making the same body turn on the second trial. However, the MRL-lpr mice used the opposite body turn to enter the unvisited arm, thus ending up in the same arm as in trial 1. This observation suggests that during the acquisition phase (across-the-day SAB rate in MRL+/+ mice increased from 70% to 90%, in MRL-lpr mice from 40% to 80%) the diseased mice did not rely on an extramaze spatial map to the same extent as MRL +/+ controls. Detrimental effects of prolonged CY treatment on performance of control MRL +/+ mice in the “reversal” trial is consistent with its negative effects in paradigms that measure exploration and motivated behavior (Sakic et al., 1995, 1996). Although the principal mechanisms are unclear, one may hypothesize that by cross-linking strands of DNA/RNA and inhibiting protein synthesis, CY treatment impairs molecular mechanisms (e.g., phosphorylation) required for short-term memory formation and consolidation (Ng et al., 1991). Over the past 40 years, lesion studies demonstrating reduced SAB rate in rodents have implicated the role of the hippocampus in attention control and memory formation (Roberts et al., 1962). If so, increased degeneration in the CA3 region of MRL-lpr brains may account for deficits in formation of a spatial map. However, more recent evidence suggests that in addition to hippocampal damage, the SAB deficit may reflect severed connections with other brain regions vital for the formation of working memory (Lalonde, 2002).
The loss of hippocampal neurons may occur via several non-mutually exclusive mechanisms. First, a direct pathway may involve increased permeability of the blood-brain barrier and infiltration of circulating immune factors into the CNS. The choroid plexus appears to be the primary site of immune complex deposition (Lampert and Oldstone, 1973; Vogelweid et al., 1991), which facilitates increased entry of soluble immune factors, monocytes and lymphocytes into the parenchyma (Hess et al., 1993; Farrell et al., 1997) and hippocampal regions of lupus-prone mice (Kier, 1990). Subsequent leukocyte clustering may lead to the dissemination of autoreactive clones into the CSF (Sakic et al., 2000b). Whether systemically or intrathecally, these lymphocytes could produce neuroactive cytokines, chemokines, and/or brain-reactive antibodies (Hoffman and Madsen, 1990; Khin and Hoffman, 1993; Crimando and Hoffman, 1992, 1995; Hickey et al., 1997; Zameer and Hoffman, 2001), which compromise the survival of differentiated neurons (Maric et al., 2001). Indeed, when injected into the mouse hippocampus, the CSF from a demented NP-SLE patient induced apoptotic neuronal death via cross-reactive binding of anti-DNA antibodies to an N-methyl-D-aspartate (NMDA) receptor subtype (DeGiorgio et al., 2001). Given that neuronal survival is dependent on the activity of supporting cells, activation of microglia during the autoimmune disease (Hickey et al., 1997) may also lead to the generation of potentially neurotoxic factors (e.g., reactive oxygen species) which contribute to neuronal demise (Wood, 1998).
A second possibility is that glucococorticoids contribute to the loss of hippocampal neurons via cytokine-induced sustained activation of the hypothalamo-pituitary-adrenal (HPA) axis (Wick et al., 1993) and intracellular glucocorticoid receptors (McEwen et al., 1992). It is documented that glucocorticoids potentiate the release and postsynaptic actions of glutamate, which likely account for loss of neurons in the CA3 region (Magarinos et al., 1997). In vivo studies confirm that this region is vulnerable to endogenous glucocorticoids (Armanini et al., 1990) and that memory deficits and structural damage are prevented by an inhibitor of corticosterone synthesis (Roozendaal et al., 2001). Since MRL-lpr mice have chronically elevated basal levels of corticosterone (Hu et al., 1993; Lechner et al., 2000), this steroid may act in a similar fashion to induce neuronal loss in the CA3 region.
Pro-inflammatory cytokines are upregulated systemically (Tang et al., 1991; Tsai et al., 1995) and in the hippocampus of MRL-lpr mice (Tomita et al., 2001). Since they appear to alter central neurotransmission (Zalcman et al., 1994), one may hypothesize that an accumulation of serotonergic neurotoxins, such as 5,7-dihydroxytryptamine (Tabatabaie et al., 1993), could compromise the survival of hippocampal neurons. Indeed, we recently identified by HPLC excessive postmortem levels of serotonin (5-HT) in the hippocampus of MRL-lpr mice (Sakic et al., 2002). The observed increase in FJB and Ub staining in the CA3 region may reflect neuronal loss (due to an intracellular accumulation of toxic metabolites), which leads to impaired performance in tasks contingent upon the integrity of hippocampal circuits. However, more detailed assessment of the phenotype of dying neurons (e.g., serotonin transporter) combined with in situ analysis of central cytokines is required to confirm this relationship.
Highly Ub-immunoreactive structures were observed in brains from autoimmune MRL-lpr mice. They likely represent degenerating axon terminals, and their location suggests that they belong to mossy fibers. The damage observed in the hippocampal synapses in human neuropathological diseases (DeKosky and Scheff, 1990; Samuel et al., 1994) and aging (Gray et al., 2003) is proposed to have significant cognitive and behavioral consequences. Interestingly, antibodies reacting with Ub and ubiquitinated histones are present in SLE, with almost 80% of patients having antibodies against Ub-protein conjugates (Muller and Schwartz, 1995). However, the present treatment with CY neither reduced expression of Ub in brains from MRL-lpr mice nor did the analysis of the brain from a NP-SLE patient show increased density of Ub-positive particles. With respect to the animal model, it needs to be determined whether overexpression of Ub is associated with anti-brain reactivity, such as autoantibodies to Ub particles in kidneys (Elouaai et al., 1994), is a consequence of nonimmune pathogenic mechanisms (e.g., hormonal), or is merely an epiphenomenon of a systemic autoimmune disease.
The CNS and choroid plexus pathology in the MRL-lpr strain has been reported extensively (Alexander et al., 1983; Vogelweid et al., 1991; Hess et al., 1993; Farrell et al., 1997; Sakic et al., 2000b). Similarly, clinical reports point to the possibility that the choroid plexus is a site for immune complex and leukocyte deposition (Atkins et al., 1972; Gershwin et al., 1975; Peress et al., 1981; Duprez et al., 2001). In the present study, we provide a direct comparison of animal and human brain pathology by using the same protocols and reagents for FJB staining. In addition, the cortical thinning, dilation of ventricles, and infiltration of T lymphocytes into the stroma of the choroid plexus are comparable to ventricular enlargement (Denenberg et al., 1992) and the infiltration of cells immunoreactive for CD3, CD4, and CD8 antigens in the choroid plexus of MRL-lpr mice (Sakic et al., 2000b; Ballok et al., 2003). The neuropsychiatric manifestations reported in human SLE have also been shown to accompany cerebral atrophy (Chinn et al., 1997) and progressive neuronal loss (Sibbitt and Sibbitt, 1993; Brooks et al., 1997). In line with this evidence, the parietotemporal cortex of our patient showed gliosis and ischemic changes in neurons. Consistent with the notion of cortical damage, reduced neuronal density was presently observed by H&E in the parietal cortex of diseased MRL-lpr mice.
Schnider et al. (1995) described an SLE patient who presented with severe amnesia due to isolated hippocampal damage. Similar to this case report, the present analysis revealed neuronal loss in the CA3 region and the dentate gyrus, with surviving neurons in the pyramidal layer showing focal satellitosis and gliosis. Although CY treatment may have beneficial effects in preventing NP-SLE manifestations (Boumpas et al., 1991; Ramos et al., 1996), it neither prevented a fatal outcome in our patient nor showed effects comparable to those seen in lupus-prone mice. One explanation is that the dose of CY used in mice (100 mg/kg) is essentially myeloablative in humans and substantially higher then the standard treatment for human autoimmune diseases (1–2 mg/kg i.v. was given daily to our patient). Second, in experimental studies, effective immunosuppressive treatments are started well before overt signs of the disease (O’Sullivan et al., 1995; Sakic et al., 1995; Walker, 2001). This is similar to the clinical protocols in which CY treatment (often associated with side effects) needs to be employed early in order to prevent or minimize irreversible organ damage (Ioannou and Isenberg, 2002). However, the SLE patient in our study received the CY treatment when she became comatose (i.e., 4 days after she presented with status epilepticus), which was probably not sufficient to reduce existing brain damage.
In summary, the present results support the hypothesis that systemic autoimmune/inflammatory disease impairs hippocampal function and compromises neuronal survival in lupus-prone mice. Damage in the CA3 region is detectable after the spontaneous onset of the disease, affects performance in the SAB and “reversal” test, and can be attenuated by immunosuppressive treatment. Further studies are required to elucidate neuropathogenic mechanisms and neuronal phenotypes susceptible to the disease process.
Acknowledgments
The authors are grateful to the Cicci family, to the Lupus Society of Hamilton for supporting research and allowing the human studies to take place, and to Dr. J.A. Denburg for help in procuring patient material postmortem. This work was supported by funds from the National Institutes of Health (grant 1R21 AR49163-01) and the Canadian Institutes of Health Research (to B.S.). D. Ballok is a student fellow of the Father Sean O’Sullivan Research Centre (FSORC). B. Sakic is an Intermediate Fellow of the Ontario Mental Health Foundation (OMHF) and recipient of the FSORC career development award.
Footnotes
Grant sponsor: Canadian Institutes of Health Research (CIHR); Grant number: MOP 38065; Grant sponsor: National Institutes of Health (NIH); Grant number: 1R21 AR49163-01.
References
- Alexander EL, Murphy ED, Roths JB, Alexander GE. Congenic autoimmune murine models of central nervous system disease in connective tissue disorders. Ann Neurol. 1983;14:242–248. doi: 10.1002/ana.410140211. [DOI] [PubMed] [Google Scholar]
- Alves-Rodrigues A, Gregori L, Figueiredo-Pereira ME. Ubiquitin, cellular inclusions and their role in neurodegeneration. Trends Neurosci. 1998;21:516–520. doi: 10.1016/s0166-2236(98)01276-4. [DOI] [PubMed] [Google Scholar]
- Andrews BS, Eisenberg RA, Theofilopoulos AN, Izui S, Wilson CB, Mc-Conahey PJ, Murphy ED, Roths JB, Dixon FJ. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med. 1978;148:1198–1215. doi: 10.1084/jem.148.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armanini MP, Hutchins C, Stein BA, Sapolsky RM. Glucocorticoid endangerment of hippocampal neurons is NMDA-receptor dependent. Brain Res. 1990;532:7–12. doi: 10.1016/0006-8993(90)91734-x. [DOI] [PubMed] [Google Scholar]
- Atkins CJ, Kondon JJ, Quismorio FP, Friou GJ. The choroid plexus in systemic lupus erythematosus. Ann Intern Med. 1972;76:65–72. doi: 10.7326/0003-4819-76-1-65. [DOI] [PubMed] [Google Scholar]
- Ballok DA, Millward JM, Sakic B. Neurodegeneration in autoimmune MRL-lpr mice as revealed by Fluoro Jade B staining. Brain Res. 2003;964:200–210. doi: 10.1016/s0006-8993(02)03980-x. [DOI] [PubMed] [Google Scholar]
- Bats S, Thoumas JL, Lordi B, Tonon MC, Lalonde R, Caston J. The effects of a mild stressor on spontaneous alternation in mice. Behav Brain Res. 2001;118:11–15. doi: 10.1016/s0166-4328(00)00285-0. [DOI] [PubMed] [Google Scholar]
- Bertholet JY, Crusio WE. Spatial and non-spatial spontaneous alternation and hippocampal mossy fibre distribution in nine inbred mouse strains. Behav Brain Res. 1991;43:197–202. doi: 10.1016/s0166-4328(05)80071-3. [DOI] [PubMed] [Google Scholar]
- Boumpas DT, Yamada H, Patronas NJ, Scott D, Klippel JH, Balow JE. Pulse cyclophosphamide for severe neuropsychiatric lupus. Q J Med. 1991;81:975–984. doi: 10.1093/qjmed/81.3.975. [DOI] [PubMed] [Google Scholar]
- Brooks WM, Sabet A, Sibbitt WL, Jr, Barker PB, van Zijl PC, Duyn JH, Moonen CT. Neurochemistry of brain lesions determined by spectroscopic imaging in systemic lupus erythematosus. J Rheumatol. 1997;24:2323–2329. [PubMed] [Google Scholar]
- Bruyn GAW. Controversies in lupus: nervous system involvement. Ann Rheum Dis. 1995;54:159–167. doi: 10.1136/ard.54.3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbotte RM, Denburg SD, Denburg JA. Cognitive dysfunction in systemic lupus erythematosus is independent of active disease. J Rheumatol. 1995;22:863–867. [PubMed] [Google Scholar]
- Chinn RJS, Wilkinson ID, Hallcraggs MA, Paley MNJ, Shorthall E, Carter S, Kendall BE, Isenberg DA, Newman SP, Harrison MJG. Magnetic resonance imaging of the brain and cerebral proton spectroscopy in patients with systemic lupus erythematosus. Arthritis Rheum. 1997;40:36–46. doi: 10.1002/art.1780400107. [DOI] [PubMed] [Google Scholar]
- Crimando J, Hoffman SA. Detection of brain-reactive autoantibodies in the sera of autoimmune mice using ELISA. J Immunol Methods. 1992;149:87–95. doi: 10.1016/s0022-1759(12)80052-4. [DOI] [PubMed] [Google Scholar]
- Crimando J, Hoffman SA. Characterization of murine brain-reactive monoclonal IgG autoantibodies. Brain Behav Immun. 1995;9:165–181. doi: 10.1006/brbi.1995.1016. [DOI] [PubMed] [Google Scholar]
- Deacon RM, Bannerman DM, Kirby BP, Croucher A, Rawlins JN. Effects of cytotoxic hippocampal lesions in mice on a cognitive test battery. Behav Brain Res. 2002;133:57– 68. doi: 10.1016/s0166-4328(01)00451-x. [DOI] [PubMed] [Google Scholar]
- DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med. 2001;7:1189–1193. doi: 10.1038/nm1101-1189. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- Denenberg VH, Sherman GF, Rosen GD, Morrison L, Behan PO, Galaburda AM. A behavior profile of the MRL/Mp lpr/lpr mouse and its association with hydrocephalus. Brain Behav Immun. 1992;6:40–49. doi: 10.1016/0889-1591(92)90058-v. [DOI] [PubMed] [Google Scholar]
- Denburg SD, Carbotte RM, Denburg JA. Psychological aspects of systemic lupus erythematosus: cognitive function, mood, and self-report. J Rheumatol. 1997;24:998–1003. [PubMed] [Google Scholar]
- Duprez T, Nzeusseu A, Peeters A, Houssiau FA. Selective involvement of the choroid plexus on cerebral magnetic resonance images: a new radiological sign in patients with systemic lupus erythematosus with neurological symptoms. J Rheumatol. 2001;28:387–391. [PubMed] [Google Scholar]
- Elouaai F, Lule J, Benoist H, Appolinaire-Pilipenko S, Atanassov C, Muller S, Fournie GJ. Autoimmunity to histones, ubiquitin, and ubiquitinated histone H2A in NZB × NZW and MRL-lpr/lpr mice. Anti-histone antibodies are concentrated in glomerular eluates of lupus mice. Nephrol Dial Transplant. 1994;9:362–366. [PubMed] [Google Scholar]
- Farrell M, Sakic B, Szechtman H, Denburg JA. Effect of cyclophosphamide on leucocytic infiltration in the brain of MRL/lpr mice. Lupus. 1997;6:268–274. doi: 10.1177/096120339700600310. [DOI] [PubMed] [Google Scholar]
- Gerlai R. Behavioral tests of hippocampal function: simple paradigms, complex problems. Behav Brain Res. 2001;125:269–277. doi: 10.1016/s0166-4328(01)00296-0. [DOI] [PubMed] [Google Scholar]
- Gershwin ME, Hyman LR, Steinberg AD. The choroid plexus in CNS involvement of systemic lupus erythematosus. J Pediatr. 1975;87:588–590. doi: 10.1016/s0022-3476(75)80831-6. [DOI] [PubMed] [Google Scholar]
- Gray DA, Tsirigotis M, Woulfe J. Ubiquitin, proteasomes, and the aging brain. SAGE KE. 2003;34:1– 6. doi: 10.1126/sageke.2003.34.re6. [DOI] [PubMed] [Google Scholar]
- Grota LJ, Schachtman TR, Moynihan JA, Cohen N, Ader R. Voluntary consumption of cyclophosphamide by Mrl mice. Brain Behav Immun. 1989;3:263–273. doi: 10.1016/0889-1591(89)90041-x. [DOI] [PubMed] [Google Scholar]
- Grota LJ, Ader R, Moynihan JA, Cohen N. Voluntary consumption of cyclophosphamide by nondeprived Mrl-lpr/lpr and Mrl +/+ mice. Pharm Biochem Behav. 1990;37:527–530. doi: 10.1016/0091-3057(90)90023-b. [DOI] [PubMed] [Google Scholar]
- Hanly JG, Hong C, Smith S, Fisk JD. A prospective analysis of cognitive function and anticardiolipin antibodies in systemic lupus erythematosus. Arthritis Rheum. 1999;42:728–734. doi: 10.1002/1529-0131(199904)42:4<728::AID-ANR16>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Henn FA, McKinney WT. Animal models in psychiatry. In: Meltzer HY, editor. Psychopharmacology: the third generation of progress. New York: Raven Press; 1987. pp. 687–695. [Google Scholar]
- Hess DC, Taormina M, Thompson J, Sethi KD, Diamond B, Rao R, Feldman DS. Cognitive and neurologic deficits in the MRL/lpr mouse: a clinicopathologic study. J Rheumatol. 1993;20:610–617. [PubMed] [Google Scholar]
- Hickey WF, Lassmann S, Cross AH. Lymphocyte entry and the initiation of inflammation in the central nervous system. In: Keane RW, Hickey WF, editors. Immunology of the nervous system. New York: Oxford University Press; 1997. pp. 200–225. [Google Scholar]
- Hoffman SA, Madsen CS. Brain specific autoantibodies in murine models of systemic lupus erythematosus. J Neuroimmunol. 1990;30:229–237. doi: 10.1016/0165-5728(90)90107-x. [DOI] [PubMed] [Google Scholar]
- Hopkins KJ, Wang G, Schmued LC. Temporal progression of kainic acid induced neuronal and myelin degeneration in the rat fore-brain. Brain Res. 2000;864:69–80. doi: 10.1016/s0006-8993(00)02137-5. [DOI] [PubMed] [Google Scholar]
- Hu Y, Dietrich H, Herold M, Heinrich PC, Wick G. Disturbed immuno-endocrine communication via the hypothalamo-pituitary-adrenal axis in autoimmune disease. Int Arch Allergy Immunol. 1993;102:232–241. doi: 10.1159/000236531. [DOI] [PubMed] [Google Scholar]
- Ioannou Y, Isenberg DA. Current concepts for the management of systemic lupus erythematosus in adults: a therapeutic challenge. Postgrad Med J. 2002;78:599–606. doi: 10.1136/pmj.78.924.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch S, McGrath JP, Varshavsky A. The yeast DNA repair gene RAD6 encodes a ubiquitin-conjugating enzyme. Nature. 1987;329:131–134. doi: 10.1038/329131a0. [DOI] [PubMed] [Google Scholar]
- Khin NA, Hoffman SA. Brain reactive monoclonal auto-antibodies: production and characterization. J Neuroimmunol. 1993;44:137–148. doi: 10.1016/0165-5728(93)90035-w. [DOI] [PubMed] [Google Scholar]
- Kier AB. Clinical neurology and brain histopathology in NZB/NZW F1 lupus mice. J Comp Pathol. 1990;102:165–177. doi: 10.1016/s0021-9975(08)80122-3. [DOI] [PubMed] [Google Scholar]
- Kovac AD, Grammig J, Mahlo J, Steiner B, Roth K, Nitsch R, Bechmann I. Comparison of neuronal density and subfield sizes in the hippocampus of CD95L-deficient (gld), CD95-deficient (lpr) and nondeficient mice. Eur J Neurosci. 2002;16:159–163. doi: 10.1046/j.1460-9568.2002.02060.x. [DOI] [PubMed] [Google Scholar]
- Lalonde R. The neurobiological basis of spontaneous alternation. Neurosci Biobehav Rev. 2002;26:91–104. doi: 10.1016/s0149-7634(01)00041-0. [DOI] [PubMed] [Google Scholar]
- Lampert PW, Oldstone MB. Host immunoglobulin G and complement deposits in the choroid plexus during spontaneous immune complex disease. Science. 1973;180:408–410. doi: 10.1126/science.180.4084.408. [DOI] [PubMed] [Google Scholar]
- Lechner O, Dietrich H, Oliveira dos SA, Wiegers GJ, Schwarz S, Harbutz M, Herold M, Wick G. Altered circadian rhythms of the stress hormone and melatonin response in lupus-prone MRL/MP-fas(Ipr) mice. J Autoimmun. 2000;14:325–333. doi: 10.1006/jaut.2000.0375. [DOI] [PubMed] [Google Scholar]
- Magarinos AM, Verdugo JM, McEwen BS. Chronic stress alters synaptic terminal structure in hippocampus. Proc Natl Acad Sci USA. 1997;94:14002–14008. doi: 10.1073/pnas.94.25.14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maric D, Millward JM, Ballok DA, Szechtman H, Barker JL, Denburg JA, Sakic B. Neurotoxic properties of cerebrospinal fluid from behaviorally impaired autoimmune mice. Brain Res. 2001;920:183–193. doi: 10.1016/s0006-8993(01)03060-8. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Gould EA, Sakai RR. The vulnerability of the hippocampus to protective and destructive effects of glucocorticoids in relation to stress. Br J Psychiatry. 1992;(Suppl):18–23. [PubMed] [Google Scholar]
- Muller S, Schwartz LM. Ubiquitin in homeostasis, development and disease. BioEssays. 1995;17:677–684. doi: 10.1002/bies.950170804. [DOI] [PubMed] [Google Scholar]
- Ng KT, Gibbs ME, Crowe SF, Sedman GL, Hua F, Zhao W, O’Dowd B, Rickard N, Gibbs CL, Sykova E. Molecular mechanisms of memory formation. Mol Neurobiol. 1991;5:333–350. doi: 10.1007/BF02935556. [DOI] [PubMed] [Google Scholar]
- O’Sullivan FX, Vogelweid CM, Beschwilliford CL, Walker SE. Differential effects of CD4+ T cell depletion on inflammatory central nervous system disease, arthritis and sialadenitis in MRL/lpr mice. J Autoimmun. 1995;8:163–175. doi: 10.1006/jaut.1995.0013. [DOI] [PubMed] [Google Scholar]
- Park C, Sakamaki K, Tachibana O, Yamashima T, Yamashita J, Yonehara S. Expression of Fas antigen in the normal mouse brain. Biochem Biophys Res Commun. 1998;252:623–628. doi: 10.1006/bbrc.1998.9572. [DOI] [PubMed] [Google Scholar]
- Peress NS, Roxburgh VA, Gelfand MC. Binding sites for immune components in human choroid plexus. Arthritis Rheum. 1981;24:520–526. doi: 10.1002/art.1780240312. [DOI] [PubMed] [Google Scholar]
- Ramos PC, Mendez MJ, Ames PRJ, Khamashta MA, Hughes GRV. Pulse cyclophosphamide in the treatment of neuropsychiatric systemic lupus erythematosus. Clin Exp Rheumatol. 1996;14:295–299. [PubMed] [Google Scholar]
- Richman CL, Dember WN, Kim P. Spontaneous alternation behavior in animals: a review. Curr Psychol Res Rev. 1986;5:358–391. [Google Scholar]
- Roberts WW, Dember WN, Brodwick M. Alternation and exploration in rats with hippocampal lesions. J Comp Physiol Psychol. 1962;55:695–700. doi: 10.1037/h0045168. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Phillips RG, Power AE, Brooke SM, Sapolsky RM, Mc-Gaugh JL. Memory retrieval impairment induced by hippocampal CA3 lesions is blocked by adrenocortical suppression. Nat Neurosci. 2001;4:1169–1171. doi: 10.1038/nn766. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Keffer M, Talangbayan H, Stead R, Denburg JA. A behavioral profile of autoimmune lupus-prone MRL mice. Brain Behav Immun. 1992;6:265–285. doi: 10.1016/0889-1591(92)90048-s. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Denburg SD, Carbotte RM, Denburg JA. Spatial learning during the course of autoimmune disease in MRL mice. Behav Brain Res. 1993;54:57–66. doi: 10.1016/0166-4328(93)90048-u. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Talangbayan H, Denburg SD, Carbotte RM, Denburg JA. Behaviour and immune status of MRL mice in the postweaning period. Brain Behav Immun. 1994a;8:1–13. doi: 10.1006/brbi.1994.1001. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Talangbayan H, Denburg SD, Carbotte RM, Denburg JA. Disturbed emotionality in autoimmune MRL-lpr mice. Physiol Behav. 1994b;56:609–617. doi: 10.1016/0031-9384(94)90309-3. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Denburg SD, Denburg JA. Immunosuppressive treatment prevents behavioral deficit in autoimmune MRL-lpr mice. Physiol Behav. 1995;58:797–802. doi: 10.1016/0031-9384(95)00135-6. [DOI] [PubMed] [Google Scholar]
- Sakic B, Denburg JA, Denburg SD, Szechtman H. Blunted sensitivity to sucrose in autoimmune MRL-lpr mice: a curve-shift study. Brain Res Bull. 1996;41:305–311. doi: 10.1016/s0361-9230(96)00190-6. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Denburg JA, Gorny G, Kolb B, Whishaw IQ. Progressive atrophy of pyramidal neuron dendrites in autoimmune MRL-lpr mice. J Neuroimmunol. 1998;87:162–170. doi: 10.1016/s0165-5728(98)00085-x. [DOI] [PubMed] [Google Scholar]
- Sakic B, Kolb B, Whishaw IQ, Gorny G, Szechtman H, Denburg JA. Immunosuppression prevents neuronal atrophy in lupus-prone mice: evidence for brain damage induced by autoimmune disease? J Neuroimmunol. 2000a;111:93–101. doi: 10.1016/s0165-5728(00)00364-7. [DOI] [PubMed] [Google Scholar]
- Sakic B, Maric I, Koeberle PD, Millward JM, Szechtman H, Maric D, Denburg JA. Increased TUNEL-staining in brains of autoimmune Fas-deficient mice. J Neuroimmunol. 2000b;104:147–154. doi: 10.1016/s0165-5728(99)00277-5. [DOI] [PubMed] [Google Scholar]
- Sakic B, Lacosta S, Denburg J, Szechtman H. Altered neurotransmission in brains of autoimmune mice: pharmacological and neurochemical evidence. J Neuroimmunol. 2002;129:84–96. doi: 10.1016/s0165-5728(02)00171-6. [DOI] [PubMed] [Google Scholar]
- Samuel W, Masliah E, Hill LR, Butters N, Terry R. Hippocampal connectivity and Alzheimer’s dementia: effects of synapse loss and tangle frequency in a two-component model. Neurology. 1994;44:2081–2088. doi: 10.1212/wnl.44.11.2081. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Schnider A, Bassetti C, Gutbrod K, Ozdoba C. Very severe amnesia with acute onset after isolated hippocampal damage due to systemic lupus erythematosus. J Neurol Neurosurg Psychiatry. 1995;59:644–646. doi: 10.1136/jnnp.59.6.644-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman GF, Morrison L, Rosen GD, Behan PO, Galaburda AM. Brain abnormalities in immune defective mice. Brain Res. 1990;532:25–33. doi: 10.1016/0006-8993(90)91737-2. [DOI] [PubMed] [Google Scholar]
- Shiraki M, Fujiwara M, Tomura S. Long term administration of cyclophosphamide in MRL/1 mice. I. The effects on the development of immunological abnormalities and lupus nephritis. Clin Exp Immunol. 1984;55:333–339. [PMC free article] [PubMed] [Google Scholar]
- Sibbitt WL, Jr, Sibbitt RR. Magnetic resonance spectroscopy and positron emission tomography scanning in neuropsychiatric systemic lupus erythematosus. Rheum Dis Clin North Am. 1993;19:851–868. [PubMed] [Google Scholar]
- Snippe H, Davidse RP, Belder M, Willers JM. Effects of cyclophosphamide treatment on the in vitro activity of mouse lymphoid cells after nonspecific and specific stimulation. Int Arch Allergy Appl Immunol. 1976;50:536–547. doi: 10.1159/000231558. [DOI] [PubMed] [Google Scholar]
- Sutherland RJ, McDonald RJ. Hippocampus, amygdala, and memory deficits in rats. Behav Brain Res. 1990;37:57–79. doi: 10.1016/0166-4328(90)90072-m. [DOI] [PubMed] [Google Scholar]
- Szechtman H, Sakic B, Denburg JA. Behaviour of MRL mice: an animal model of disturbed behaviour in systemic autoimmune disease. Lupus. 1997;6:223–229. doi: 10.1177/096120339700600302. [DOI] [PubMed] [Google Scholar]
- Tabatabaie T, Goyal RN, Blank CL, Dryhurst G. Further insights into the molecular mechanisms of action of the serotonergic neurotoxin 5,7-dihydroxytryptamine. J Med Chem. 1993;36:229–236. doi: 10.1021/jm00054a006. [DOI] [PubMed] [Google Scholar]
- Tang B, Matsuda T, Akira S, Nagata N, Ikehara S, Hirano T, Kishimoto T. Age-associated increase in interleukin 6 in MRL/lpr mice. Int Immunol. 1991;3:273–278. doi: 10.1093/intimm/3.3.273. [DOI] [PubMed] [Google Scholar]
- ten Berge RJ, van Walbeek HK, Schellekens PT. Evaluation of the immunosuppressive effects of cyclophosphamide in patients with multiple sclerosis. Clin Exp Immunol. 1982;50:495–502. [PMC free article] [PubMed] [Google Scholar]
- Theofilopoulos AN. Murine models of lupus. In: Lahita RG, editor. Systemic lupus erythematosus. New York: Churchill Livingstone; 1992. pp. 121–194. [Google Scholar]
- Tomita M, Holman BJ, Santoro TJ. Aberrant cytokine gene expression in the hippocampus in murine systemic lupus erythematosus. Neurosci Lett. 2001;302:129–132. doi: 10.1016/s0304-3940(01)01679-2. [DOI] [PubMed] [Google Scholar]
- Tsai CY, Wu TH, Huang SF, Sun KH, Hsieh SC, Han SH, Yu HS, Yu CL. Abnormal splenic and thymic IL-4 and TNF-alpha expression in MRL-lpr/lpr mice. Scand J Immunol. 1995;41:157–163. doi: 10.1111/j.1365-3083.1995.tb03548.x. [DOI] [PubMed] [Google Scholar]
- Vogelweid CM, Johnson GC, Besch-Williford CL, Basler J, Walker SE. Inflammatory central nervous system disease in lupus-prone MRL/lpr mice: comparative histologic and immunohistochemical findings. J Neuroimmunol. 1991;35:89–99. doi: 10.1016/0165-5728(91)90164-3. [DOI] [PubMed] [Google Scholar]
- Walker SE. Bromocriptine treatment of systemic lupus erythematosus. Lupus. 2001;10:762–768. doi: 10.1191/096120301717165010. [DOI] [PubMed] [Google Scholar]
- Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992a;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- Watanabe-Fukunaga R, Brannan CI, Itoh N, Yonehara S, Copeland NG, Jenkins NA, Nagata S. The cDNA structure, expression, and chromosomal assignment of the mouse Fas antigen. J Immunol. 1992b;148:1274–279. [PubMed] [Google Scholar]
- Whishaw IQ. Latent learning in a swimming pool place task by rats: evidence for the use of associative and not cognitive mapping processes. Q J Exp Psychol B. 1991;43:83–103. [PubMed] [Google Scholar]
- Wick G, Hu Y, Schwarz S, Kroemer G. Immunoendocrine communication via the hypothalamo-pituitary-adrenal axis in autoimmune diseases. Endocr Rev. 1993;14:539–563. doi: 10.1210/edrv-14-5-539. [DOI] [PubMed] [Google Scholar]
- Wood PL. Roles of CNS macrophages in neurodegeneration. In: Wood PL, editor. Neuroinflammation: mechanisms and management. Totowa, NJ: Humana Press; 1998. pp. 1–59. [Google Scholar]
- Ye X, Carp RI, Schmued LC, Scallet AC. Fluoro-Jade and silver methods: application to the neuropathology of scrapie, a transmissible spongiform encephalopathy. Brain Res Brain Res Protoc. 2001;8:104–112. doi: 10.1016/s1385-299x(01)00086-1. [DOI] [PubMed] [Google Scholar]
- Zalcman S, Greenjohnson JM, Murray L, Nance DM, Dyck D, Anisman H, Greenberg AH. Cytokine-specific central monoamine alterations induced by interleukin-1, -2 and -6. Brain Res. 1994;643:40–49. doi: 10.1016/0006-8993(94)90006-x. [DOI] [PubMed] [Google Scholar]
- Zameer A, Hoffman SA. Immunoglobulin binding to brain in autoimmune mice. J Neuroimmunol. 2001;120:10–18. doi: 10.1016/s0165-5728(01)00412-x. [DOI] [PubMed] [Google Scholar]