

Abstract
Post-natal human dental pulp stem cells (DPSCs) represent a unique precursor population in the dental pulp, which has multipotential and can regenerate a dentin/pulp-like structure. Because the dental pulp is frequently infected by oral bacteria due to dental decay, in this study, we examined whether lipopolysaccharide (LPS) and tumor necrosis factor (TNF) activated the immunologic transcription factor nuclear factor kappa B (NF-κB) in DPSCs. We found that both TNF and LPS activated the I-kappa B kinase complex (IKK) in DPSCs to induce the phosphorylation and degradation of IκBα, resulting in the nuclear translocation of NF-κB. Consistently, both TNF and LPS rapidly induced the expression of the NF-κB-dependent gene interleukin-8 (IL-8). However, unlike in monocytes, we found that LPS could not induce the phosphorylation of the NF-κB active subunit p65 in DPSCs. In summary, our studies suggest that DPSCs may be involved in immune responses during pulpal infection through activating NF-κB.
Keywords: NF-κB, dental pulp stem cells, tumor necrosis factor, LPS, inflammation
INTRODUCTION
DPSCs are specific mesenchymal stem cells that exist in human dental pulp tissues (Gronthos et al., 2000). DPSCs can be induced to differentiate into odontoblasts, adipocytes, and neural-like cells in vitro. In vivo transplantation has demonstrated that DPSCs were capable of forming dentin-pulp-like tissue. Moreover, DPSCs formed reparative dentin-like tissue on the surface of human dentin in vivo. They could be isolated from primary DPSC transplants and re-transplanted into immunocompromised mice to generate a dentin-pulp-like tissue, suggesting that DPSCs have self-renewal capability (Gronthos et al., 2000, 2002). Therefore, DPSCs may have excellent potential for dentin repair and tooth regeneration. However, before these cells can be used for clinical therapy, it is critical that we understand their biological properties in response to extrinsic and intrinsic stimuli.
The dental pulp tissues from which DPSCs are derived are frequently infected or inflamed due to bacterial infection from dental caries (Stashenko et al., 1998). LPS, a major component of the outer membrane of bacteria, has been identified in infected pulp tissues (Wang et al., 1997; Rupf et al., 2000; Darveau et al., 2002). Pro-inflammatory cytokines, such as TNF and IL-1, have been found to be highly expressed in inflamed pulp tissues (Stashenko et al., 1998; Coil et al., 2004). Importantly, elegant studies by Rutherford and Gu (2000) demonstrated that local dentin/pulp inflammation interfered with odontoblast differentiation and dentin repair. While monocytes or fibroblasts in dental pulp tissues have been found to express pro-inflammatory cytokines, it is not clear whether DPSCs are involved in host response and/or produce inflammatory mediators upon bacterial infection.
It is well-known that LPS stimulates monocytes, lymphocytes, and certain types of fibroblasts to produce pro-inflammatory cytokines, such as TNF and IL-1, in chronic oral inflammatory diseases such as pulpitis, and periapical and periodontal diseases (Nagaoka et al., 1996; Wang et al., 1997; Kawashima and Stashenko, 1999; Kent et al., 1999; Fouad and Acosta, 2001). Elevated levels of TNF and IL-1 can stimulate a variety of cell types to generate inflammatory mediators and chemokines to amplify inflammatory responses (Liu et al., 1996; Baqui et al., 1998). It is believed that the critical factor activated by LPS and TNF during infection is the transcription factor NF-κB (Baldwin, 2001). Genes known to be regulated by NF-κB include major histocompatibility molecules (MHC), IL-1β, IL2, IL-2 receptor, IL-6, IL-8, intercellular adhesion molecule-1, E-selectin, TNF, and interferon-γ. In the unstimulated condition, NF-κB is present in an inactive form that is retained in the cytoplasm by the inhibitory protein IκBα. The most common form of NF-κB is a heterodimer of p50 and p65/RelA proteins. Biochemical and genetic studies have found that the IKK complex plays a critical role in the activation of NF-κB. The IKK complex consists mainly of two catalytic subunits, IKKα and IKKβ, and a non-catalytic chaperone protein IKKγ (Baldwin, 2001). Recently, we have found that the zinc-finger structure of IKKγ also plays a critical role in IKK activation (Yang et al., 2004). Upon stimulation by LPS, TNF, or other stimuli, IKKα and IKKβ are activated following IKKγ ubiquitination by unknown mechanisms. The activated IKK complex phosphorylates the N-terminal region of IκBα at Serines 32 and 36. The phosphorylated IκBα is subsequently ubiquitinated and degraded by the 26S proteasome machinery. The degradation of IκBα releases NF-κB from cytoplasm to the nucleus, where NF-κB binds to specific elements in the promoter of NF-κB target genes and stimulates gene transcription (Wang et al., 1996, 1998; Baldwin, 2001). In this study, to understand the role of DPSCs in pulpal infection and host response, we examined whether LPS or TNF activated the NF-κB signaling pathway in DPSC cells.
MATERIALS & METHODS
Cell Cultures
DPSCs were characterized as described previously (Gronthos et al., 2000). Tissues were collected at the Dental Clinic of the National Institute of Dental and Craniofacial Research under approved guidelines set by the National Institutes of Health Office of Human Subjects Research. Cells were grown in alpha-modified Eagle's medium (Invitrogen, Carlsbad, CA, USA) supplemented with 15% fetal bovine serum (FBS; Invitrogen). TNF was obtained from R&D Systems Inc. (Minneapolis, MN, USA). Porphyromonas gingivalis (P. gingivalis) LPS was kindly provided by Dr. Roland Arnold at the University of North Carolina at Chapel Hill. Porphyromonas endodontalis (P. endodontalis) LPS was prepared by phenol-chloroform extraction (Darveau et al., 2002). E. coli LPS and other chemicals were purchased from Sigma (St. Louis, MO, USA)
Western Blot Analysis
DPSCs were treated with TNF (10 ng/mL) or LPS (from 200 ng to 1 μg/mL) for different time periods. Cells underwent lysis in RIPA buffer (10 mM Tris-HCl, 1 mM EDTA, 1% sodium dodecyl sulfate [SDS], 1% Nonidet P-40, 1:100 proteinase inhibitor cocktail, 50 mM β-glycerophosphate, 50 mM sodium fluoride). Fifty-μg aliquots of protein extracts were subjected to 10% SDS-polyacrylamide gel electrophoresis and transferred to PVDF membrane by a semi-dry transfer apparatus (Bio-Rad, Hercules, CA, USA). Western blot analysis was performed as described previously (Yang et al., 2003, 2004). Primary antibodies were purchased from the following commercial sources: monoclonal antibodies against human IκBα (1:1000; Santa Cruz, CA, USA); monoclonal antibodies against phospho-specific IκBα and polyclonal antibodies against phospho-specific p65 (Cell Signaling, Beverly, MA, USA); and monoclonal antibodies against α tubulin (Sigma).
Northern Blot Analysis
DPSCs were treated with TNF or LPS, and total RNAs were extracted with Trizol reagents according to the manufacturer’s instructions (Invitrogen). Ten-μg aliquots of total RNAs were separated on a 1.4% agarose-formaldehyde gel, transferred onto a nylon membrane (Bio-Rad), and cross-linked with a UV cross-linker. IL-8 or IL-6 probes were generated with a random-primed labeling kit (Amersham Biosciences, Piscataway, NJ, USA) in the presence of [α-32P]dCTP (MP Biomedicals, Irvine, CA, USA), and purified via a micro-G-50 Sephadex column (Amersham Biosciences). The hybridization was performed as described previously (You et al., 2001).
Electrophoretic Mobility Shift Assays (EMSAs)
DPSCs were treated with TNF (10 ng/mL) or P. gingivalis LPS (1 μg/mL), pelleted, and washed twice with ice-cold PBS. Nuclear protein isolation and EMSAs were performed as described previously (Yang et al., 2004). For EMSAs, 5-μg aliquots of nuclear extracts were incubated for 15 min at room temperature with 2 × 104 cpm of a 32P-labeled oligonucleotide probe containing a κB site (underlined) from the class I MHC promoter (5′-CAGGGCTGGGGATTCCCCATCTCCACAGTTTCACTTC-3′ in nuclear binding buffer plus 2 μg of poly (dI-dC) (Pharmacia Biotech, Piscataway, NJ, USA). For the super-shift assay to confirm NF-κB-binding specificity, we added 1 μL of rabbit polyclonal antibodies against p65 subunit (Rockland) of NF-κB to nuclear extracts for 15 min prior to the addition of poly(dI-dC)-poly(dI-dC) and a 32P-labeled oligonucleotide probe, and then separated them on 5% polyacrylamide gels (Wang et al., 1999a,b).
RESULTS
To investigate whether the NF-κB signaling pathway was intact in DPSCs, we first examined whether TNF induced the phosphorylation and degradation of IκBα by activating IKK. After TNF treatment, whole-cell extracts were prepared, and Western blot analysis was performed with monoclonal antibodies against phospho-specific IκBα (Yang et al., 2003, 2004). The phosphorylation of IκBα in DPSCs was rapidly induced upon TNF stimulation in 5 min (Fig. 1A). Subsequently, the degradation of IκBα by 26S proteasome occurred in DPSCs. Because IκBα was degraded, we were unable to detect the phosphorylated IκBα at 30-minute time points. However, due to the fact that the promoter of IκBα is also regulated by NF-κB, IκBα was rapidly re-synthesized after TNF treatment. Therefore, the phosphorylated IκBα was detected again at 60- and 120-minute time points (Fig. 1A). The induction of IκBα by NF-κB serves as a feedback mechanism for negative regulation of NF-κB activation.
Figure 1.
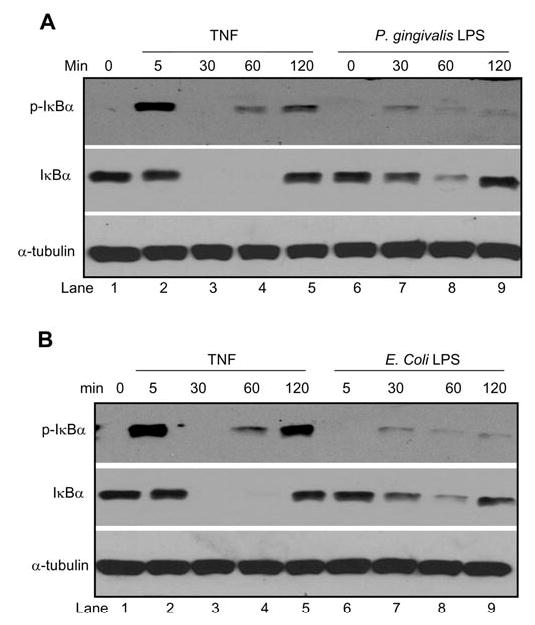
TNF or LPS activates IKK. (A) DPSCs were treated with TNF (10 ng/mL) and P. gingivalis LPS (1 μg/mL) for the indicated times. Fifty-μg aliquots of protein extracts were probed with polyclonal antibodies against IκBα (1:1000) or monoclonal antibodies against phospho-specific antibodies (1:1000). For loading control, the blots were stripped and re-examined with monoclonal antibodies against α-tubulin (1:15, 000). (B) E. coli LPS activates IKK. DPSCs were treated with E. coli LPS (200 ng/mL) or TNF for the indicated time points. Western blot analysis was performed as described in (A).
To determine whether DPSCs responded to bacterial infection, we treated DPSCs with LPS extracted from Porphyromonas gingivalis (P. gingivalis LPS), which has previously been found to activate NF-κB in monocytes and macrophages (Yang et al., 2003, 2004). Compared with TNF, P. gingivalis LPS induced modest phosphorylation of IκBα (Fig. 1A, top panel). However, it appeared that the level of IκBα phosphorylation was sufficient to trigger the degradation of IκBα in DPSCs (Fig. 1A, middle panel). To determine whether LPS from other bacteria also activated NF-κB in DPSCs, we utilized E. coli LPS. Like P. gingivalis LPS, E. coli LPS also induced the phosphorylation and degradation of IκBα in DPSCs (Fig. 1B). Taken together, our results suggest that both TNF and LPS induce IKK activity in DPSCs.
After IκBα degradation, it is believed that NF-κB is moved to the nucleus to bind to the promoter region of NF-κB target genes and to stimulate transcription. To determine whether TNF induced nuclear translocation of NF-κB in DPSCs, we isolated the nuclear proteins from DPSCs and subjected them to electrophoretic mobility shift assays (EMSAs), using 32P-labeled NF-κB oligonucleotide probes. TNF rapidly induced the nuclear translocation of NF-κB in DPSCs (Fig. 2A). To confirm the specificity of NF-κB-binding activity, we pre-incubated the nuclear proteins from TNF-treated DPSCs with anti-p65 antibodies for 10 min and then incubated them with NF-κB oligonucleotide probes. The pre-treatment of anti-p65 antibodies supershifted NF-κB-binding activity in DPSCs (Fig. 2A, compare lane 3 with lane 6), confirming the specificity of the NF-κB-binding activity. Similarly, the nuclear translocation of NF-κB in DPSCs was also induced by P. gingivalis LPS (Fig. 2B) and E. coli LPS (data not shown).
Figure 2.
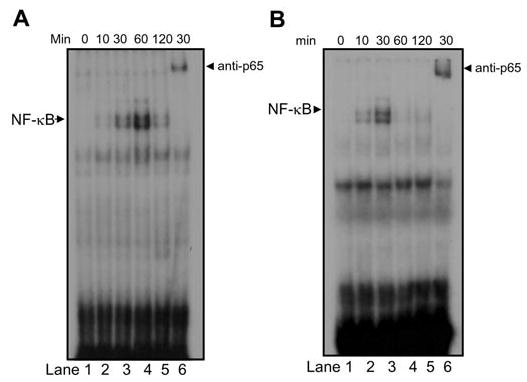
TNF or LPS induces the nuclear translocation of NF-κB. (A) DPSCs were treated with TNF (10 ng/mL) for the indicated time. Five-μg aliquots of nuclear proteins were incubated with 32P-labeled NF-κB oligonucleotide probes. For the supershift assay, nuclear proteins were pre-incubated with polyclonal antibodies against p65 for 10 min and then probed with32P-labeled NF-κB oligonucleotide probes. (B) DPSCs were treated with P. gingivalis LPS (1 μg/mL) for the indicated times. EMSA was performed as described in (A).
Recently, we also found that the transactivation domain of NF-κB subunit p65 was phosphorylated by the IKK complex in monocytes or macrophages upon stimulation of TNF or P. gingivalis LPS. The phosphorylation of p65 was required for optimal activation of NF-κB-dependent gene transcription in monocytes or macrophages (Yang et al., 2003). Thus, we also examined whether TNF or LPS induced phosphorylation of p65 in DPSCs. On the contrary, we found that TNF weakly induced the phosphorylation of p65 on serine 536, as detected by phospho-specific p65 antibodies (Fig. 3A). Moreover, we did not find that P. gingivalis LPS induced the phosphorylation of p65 on serine 536 in DPSCs. Our previous studies found that the phosphorylation of p65 had effects on NF-κB transcription in monocytes or macrophages. Since TNF or LPS weakly induced, or could not induce, p65 phosphorylation, we were interested in determining whether TNF or LPS functionally stimulated NF-κB-dependent transcription. However, because DPSCs were resistant to transfection, we were unable to utilize NF-κB-dependent luciferase reporter assays for our studies. Since it is well-known that the expression of IL-8 is dependent on NF-κB activation, we performed Northern blot analysis to determine whether TNF or LPS induced IL-8 expression through activating NF-κB. Both TNF and P. gingivalis LPS rapidly induced the expression of IL-8 expression, as detected by Northern blot analysis (Figs. 3B, 3C), suggesting that they activated NF-κB transcription in DPSCs. To confirm our results, we examined whether the lack of p65 phosphorylation affected the expression of IL-6, another NF-κB-dependent pro-inflammatory cytokine. We found IL-6 was also rapidly induced by both TNF and E. coli LPS in DPSCs (Figs. 3D, 3E).
Figure 3.
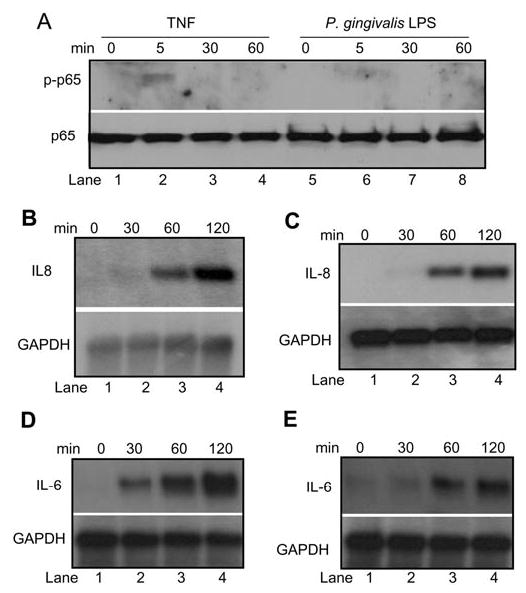
LPS induces IL-8 expression in DPSCs. (A) DPSCs were treated with TNF or P. gingivalis LPS for the indicated times. Fifty-μg aliquots of protein extracts were probed with polyclonal antibodies against phospho-specific p65. For loading control, the membrane was stripped and re-probed with polyclonal antibodies against p65. (B) DPSCs were treated with TNF for the indicated times. Ten-μg aliquots of total RNAs were hybridized with 32P-labeled IL-8 cDNA probes. For loading control, the blot was stripped and re-probed with 32P-labeled glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA probes. (C) DPSCs were treated with P. gingivalis LPS for the indicated times. Northern blot analysis was performed as described in (B). (D) DPSCs were treated with TNF for the indicated times. Total RNAs were probed with 32P-labeled IL-6 cDNA probes. (E) DPSCs were treated with E. coli LPS for the indicated times. Total RNAs were probed with 32P-labeded IL-6 cDNA probes.
Since P. gingivalis or E. coli LPS is not associated with endodontic infection, we further examined whether LPS isolated from the common endodontic pathogen P. endodontalis could activate NF-κB in DPSCs. P. endodontalis LPS also stimulated the IKK complex to phosphorylate IκBα. The level of IκBα was decreased following stimulation, suggesting that IκBα was partially degraded (Fig. 4A). Consistently, we found that P. endodontalis induced the nuclear translocation of NF-κB. However, P. endodontalis LPS did not induce the phosphorylation of p65 (Fig. 4B).
Figure 4.
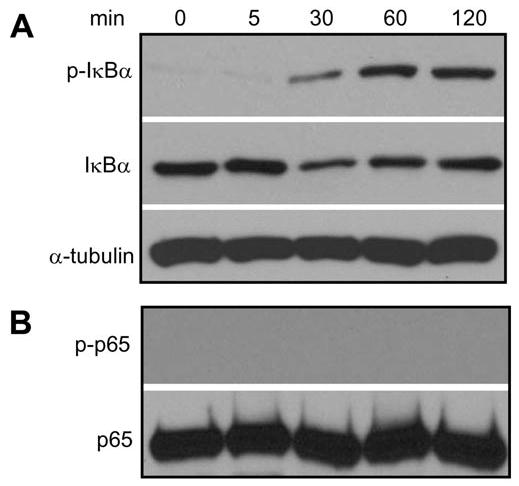
P. endodontalis LPS activates IKK. (A) Cells were treated with P. endodontalis LPS (1 μg/mL) for the indicated times. Western blot analysis was performed as described in Fig. 1. (B) Western blot was performed as described in Fig. 3A.
DISCUSSION
The present study demonstrated that both LPS and TNF activated the NF-κB signaling pathway in DPSCs. NF-κB has been considered as a master transcription factor that regulates a variety of inflammatory mediators, including TNF, IL-1, IL-6, and IL-8 (Baldwin, 2001). Previously, it has been reported that LPS or TNF can induce the production of pro-inflammatory cytokines in dental pulp fibroblasts, cementum-like fibroblasts, and oral epithelial cells (Nakane et al., 1995; Salvi et al., 1997; Chiang et al., 1999; Martin et al., 2001; Ogawa et al., 2002; Nociti et al., 2004). However, the underlying molecular mechanisms which control the expression of these inflammatory mediators in these cells have not been explored. Our results are the first demonstration that NF-κB is functionally activated by both TNF and LPS in DPSCs. Also, according to our results, the activation of NF-κB by TNF or LPS might be responsible for the production of pro-inflammatory cytokines in dental pulp fibroblasts and other cells. NF-κB may be an important target for inhibiting oral and dental inflammation.
While the NF-κB subunit p50 has been found to bind to DNA, the NF-κB subunit p65 has transactivation domains which are critical for NF-κB transcription (Baldwin, 2001). Recently, we have found that the IKK complex activated by TNF and P. gingivalis LPS also phosphorylated the p65 transactivation domain on serine 536 in human monocytes or mouse macrophages. The phosphorylation of p65 potentiated NF-κB transactivation (Yang et al., 2003). In contrast, we found that LPS could not induce phosphorylation of p65, while TNF only weakly stimulated p65 phosphorylation in DPSCs. Currently, we cannot provide an explanation for this difference. It is possible that the conformation of the IKK complex in DPSCs may be different from that in monocytes or macrophages. Although the activated IKK complex could sufficiently phosphorylate IκBα, it might be unable to phosphorylate p65 in DPSCs. Alternatively, it is also possible that the transactivation domain of p65 might be masked by other molecules in DPSCs which interfere with the phosphorylation of p65 by the IKK complex. Nevertheless, we found that both LPS and TNF could rapidly induce the expression of the NF-κB-dependent gene IL-8, suggesting that the phosphorylation of p65 may not be required for NF-κB transcription in DPSCs. Finally, it should be mentioned that the phosphorylation of p65 may be required for the induction of a specific group of NF-κB target genes. It is possible that these genes may be induced by TNF or LPS in monocytes or macrophages, but not in DPSCs. The identification of these genes may further help us to understand the biological properties of DPSCs.
Interestingly, we also observed that TNF and LPS exhibited different patterns of NF-κB activation. NF-κB-binding activities were induced by LPS in 30 min and disappeared by 60 min. In contrast, NF-κB-binding activities were further increased after 60 min following TNF stimulation. This difference may be due to the fact that TNF induced the phosphorylation and degradation of IκBα more potently than did LPS in DPSCs. However, since TNF and LPS utilized different receptor pathways to activate IKK, NF-κB-binding activities may be regulated at the second level by different components associated with the TNF or LPS intracellular signaling pathways. In the future, it will be interesting to compare the gene expression profiles induced by TNF and LPS in DPSCs.
Acknowledgments
The study was supported by USPHS Research Grants R01-DE015973 and R01-DE016513 from the National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD 20892, USA.
References
- Baldwin AS. Series introduction: the transcription factor NF-kappaB and human disease. J Clin Invest. 2001;107:3–6. doi: 10.1172/JCI11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baqui AA, Meiller TF, Chon JJ, Turng BF, Falkler WA., Jr Granulocyte-macrophage colony-stimulating factor amplification of interleukin-1beta and tumor necrosis factor alpha production in THP-1 human monocytic cells stimulated with lipopolysaccharide of oral microorganisms. Clin Diag Lab Immun. 1998;5:341–347. doi: 10.1128/cdli.5.3.341-347.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CY, Kyritsis G, Graves DT, Amar S. Interleukin-1 and tumor necrosis factor activities partially account for calvarial bone resorption induced by local injection of lipopolysaccharide. Infect Immun. 1999;67:4231–4236. doi: 10.1128/iai.67.8.4231-4236.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coil J, Tam E, Waterfield JD. Proinflammatory cytokine profiles in pulp fibroblasts stimulated with lipopolysaccharide and methyl mercaptan. J Endodont. 2004;30:88–91. doi: 10.1097/00004770-200402000-00006. [DOI] [PubMed] [Google Scholar]
- Darveau RP, Arbabi S, Garcia I, Bainbridge B, Maier RV. Porphyromonas gingivalis lipopolysaccharide is both agonist and antagonist for p38 mitogen-activated protein kinase activation. Infect Immun. 2002;70:1867–1873. doi: 10.1128/IAI.70.4.1867-1873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouad AF, Acosta AW. Periapical lesion progression and cytokine expression in an LPS hyporesponsive model. Int Endodont J. 2001;34:506–513. doi: 10.1046/j.1365-2591.2001.00423.x. [DOI] [PubMed] [Google Scholar]
- Gronthos S, Mankani M, Brahim J, Robey PG, Shi S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci USA. 2000;97:13625–13630. doi: 10.1073/pnas.240309797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronthos S, Brahim J, Li W, Fisher LW, Cherman N, Boyde A, et al. Stem cell properties of human dental pulp stem cells. J Dent Res. 2002;81:531–535. doi: 10.1177/154405910208100806. [DOI] [PubMed] [Google Scholar]
- Kawashima N, Stashenko P. Expression of bone-resorptive and regulatory cytokines in murine periapical inflammation. Arch Oral Biol. 1999;44:55–66. doi: 10.1016/s0003-9969(98)00094-6. [DOI] [PubMed] [Google Scholar]
- Kent LW, Rahemtulla F, Michalek SM. Interleukin (IL)-1 and Porphyromonas gingivalis lipopolysaccharide stimulation of IL-6 production by fibroblasts derived from healthy or periodontally diseased human gingival tissue. J Periodontol. 1999;70:274–282. doi: 10.1902/jop.1999.70.3.274. [DOI] [PubMed] [Google Scholar]
- Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector function: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- Martin M, Katz J, Vogel SN, Michalek SM. Differential induction of endotoxin tolerance by lipopolysaccharides derived from Porphyromonas gingivalis and Escherichia coli. J Immunol. 2001;167:5278–5285. doi: 10.4049/jimmunol.167.9.5278. [DOI] [PubMed] [Google Scholar]
- Nagaoka S, Tokuda M, Sakuta T, Taketoshi Y, Tamura M, Takada H, et al. Interleukin-8 gene expression by human dental pulp fibroblast in cultures stimulated with Prevotella intermedia lipopolysaccharide. J Endod. 1996;22:9–12. 19. doi: 10.1016/S0099-2399(96)80228-7. [DOI] [PubMed] [Google Scholar]
- Nakane A, Yoshida T, Nakata K, Horiba N, Nakamura H. Effects of lipopolysaccharides on human dental pulp cells. J Endod. 1995;21:128–130. doi: 10.1016/s0099-2399(06)80437-1. [DOI] [PubMed] [Google Scholar]
- Nociti FH, Jr, Foster BL, Barros SP, Darveau RP, Somerman MJ. Cementoblast gene expression is regulated by Porphyromonas gingivalis lipopolysaccharide partially via toll-like receptor-4/MD-2. J Dent Res. 2004;83:602–607. doi: 10.1177/154405910408300804. [DOI] [PubMed] [Google Scholar]
- Ogawa T, Asai Y, Hashimoto M, Takeuchi O, Kurita T, Yoshikai Y, et al. Cell activation by Porphyromonas gingivalis lipid A molecule through Toll-like receptor 4- and myeloid differentiation factor 88-dependent signaling pathway. Infect Immun. 2002;14:1325–1332. doi: 10.1093/intimm/dxf097. [DOI] [PubMed] [Google Scholar]
- Rupf S, Kannengiesser S, Merte K, Pfister W, Sigusch B, Eschrich K. Comparison of profiles of key periodontal pathogens in periodontium and endodontium. Endod Dent Traumatol. 2000;16:269–275. doi: 10.1034/j.1600-9657.2000.016006269.x. [DOI] [PubMed] [Google Scholar]
- Rutherford RB, Gu K. Treatment of inflamed ferret dental pulps with recombinant bone morphogenetic protein-7. Eur J Oral Sci. 2000;108:202–206. doi: 10.1034/j.1600-0722.2000.108003202.x. [DOI] [PubMed] [Google Scholar]
- Salvi GE, Collins JG, Yalda B, Arnold RR, Lang NP, Offenbacher S. Monocytic TNF alpha secretion patterns in IDDM patients with periodontal diseases. J Clin Periodontol. 1997;24:8–16. doi: 10.1111/j.1600-051x.1997.tb01178.x. [DOI] [PubMed] [Google Scholar]
- Stashenko P, Teles R, D'Souza R. Periapical inflammatory responses and their modulation. Crit Rev Oral Biol Med. 1998;9:498–521. doi: 10.1177/10454411980090040701. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo M, Baldwin AS. TNF- and cancer therapy-induced apoptosis potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- Wang CY, Tani-Ishii N, Stashenko P. Bone-resorptive cytokine gene expression in periapical lesions in the rat. Oral Microbiol Immunol. 1997;12:65–71. doi: 10.1111/j.1399-302x.1997.tb00619.x. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Korneluk RC, Goeddel DV, Baldwin AS. NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Wang CY, Cusack J, Liu R, Baldwin AS. Control of inducible chemoresistance: enhanced anti-tumor therapy via increased apoptosis through inhibition of NF-κB. Nature Med. 1999a;5:412–417. doi: 10.1038/7410. [DOI] [PubMed] [Google Scholar]
- Wang CY, Guttridge DC, Mayo MW, Baldwin AS. NF-κB induces expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy-induced apoptosis. Mol Cell Biol. 1999b;19:5923–5929. doi: 10.1128/mcb.19.9.5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Tang E, Guan K, Wang CY. IKKβ plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J Immunol. 2003;170:5630–5635. doi: 10.4049/jimmunol.170.11.5630. [DOI] [PubMed] [Google Scholar]
- Yang F, Yamashita J, Tang E, Wang H, Guan G, Wang CY. The zinc finger mutation C417R of I-κB kinase g impairs lipopolysaccharide- and TNF-mediated NF-κB activation through inhibiting phosphorylation of the I-κB kinase B activation loop. J Immunol. 2004;172:2446–2452. doi: 10.4049/jimmunol.172.4.2446. [DOI] [PubMed] [Google Scholar]
- You Z, Ouyang H, Lopatin D, Polverini PJ, Wang CY. Nuclear factor-kappa B-inducible death effector domain-containing protein suppresses TNF-mediated apoptosis by inhibiting caspase-8 activity. J Biol Chem. 2001;276:26398–26404. doi: 10.1074/jbc.M102464200. [DOI] [PubMed] [Google Scholar]