

Abstract
Background
We previously reported successful therapeutic immunization in a chimpanzee having a relatively low viral load, which was immunized with recombinant plasmid hepatitis B surface antigen (HBsAg) DNA and boosted with recombinant HBsAg encoding canarypox virus. In the present study, we attempted to confirm these findings in an animal with a high virus load.
Methods and Results
We tested three immunization strategies successively over a 3-year period. In the first of these, we administered four monthly injections of DNA encoding HBsAg + PreS2 + hepatitis B core antigen (HBcAg) + DNA encoding interleukin (IL)-12, (given 3 days later), and boosted with canarypox expressing all of the above HBV genes 6 months after initial immunization. No reduction in viral load was observed. In the second trial, we administered lamivudine for 8 weeks, and then began monthly DNA-based immunization with plasmids expressing the above viral genes; however, viral loads rebounded 1 week after termination of lamivudine therapy. In a third trial, we continued lamivudine therapy for 30 weeks and immunized with vaccinia virus expressing the above viral genes 18 and 23 weeks after the start of lamivudine therapy. Again viral loads rebounded shortly after cessation of lamivudine treatment. Analysis of cell-mediated immune responses, and their avidity, revealed that DNA-based immunization produced the strongest enhancement of high avidity T-cell responses, while recombinant vaccinia immunization during lamivudine therapy enhanced low avidity responses only. The strongest low and high avidity responses were directed to the middle surface antigen.
Conclusions
Three strategies for therapeutic immunization failed to control HBV viremia in a chronically infected chimpanzee with a high viral load.
Keywords: chimpanzee studies, hepatitis B virus, immunotherapy, lamivudine
Introduction
We have previously reported that immunization of a chronically hepatitis B virus (HBV) infected chimpanzee with a prime boost regimen utilizing recombinant DNA expressing hepatitis B surface antigen (HBsAg), followed by boosting with a recombinant canarypox virus expressing the HBsAg + PreS1 + PreS2 antigens, resulted in a sustained 400-fold reduction of viral load [18]. In this study, viral load dropped shortly after the canarypox booster, in parallel to a marked increase in number of HBV specific interferon-γ (IFN-γ) secreting cells detected by ELISPOT assay. The animal used in this study had a relatively low viral load, averaging 105.4 HBV DNA copies/ml. The present studies were initiated in an attempt to confirm or extend these findings in an animal with a high viral load. Despite various potential improvements in the treatment protocol, downregulation of viral load was not achieved. Possible reasons for this failure are discussed.
Materials and Methods
Chimpanzee (Pan troglodytes)
Chimp X139 (Michelle) is a 20-year-old female who developed chronic HBV infection after inoculation with hepatitis B virus in 1979. She is housed at the Southwest Foundation for Biomedical Research (SFBR). During the past 4 years, except for times when lamivudine was administered, her viral load averaged 8.64 ± 0.47 log 10 DNA copies/ml. This animal received humane care exceeding the requirements of the NIH guide for the care and use of laboratory animals. She was housed together with HBV recovered animals.
Therapeutic immunization protocols
Phase 1
On December 28, 1999, chimp X139 was injected with 1 mg of a plasmid pCDNA-3 encoding HBsAg and PreS2 (kindly provided by Dr Robert Whalen) and 1 mg of plasmid pCDNA-3 encoding hepatitis B core antigen (HBcAg) intramuscularly (kindly provided by Dr Charles Tackney). The two plasmids were mixed and injected into thigh and deltoid muscles bilaterally and intradermally over the same sites. Three days later, 1 mg of a plasmid Rous sarcoma virus (pRSV) encoding human interleukin (IL)-12 was injected into the above sites i.m. The plasmids for this study were manufactured by Puresyn, Inc., Malvern, PA, USA. The above plasmid injections were repeated 4, 8, and 12 weeks later.
Twenty-four weeks after the first immunization, the animal was boosted by intravenous, and multiple site i.m., inoculations of 5 × 108 pfu (by each route) of recombinant canarypox virus (vcp 169) expressing HBsAg, PreS1, PreS2, and HBcAg, kindly provided by Virogenetics, Inc., Renselleer, NY, USA. (now Aventis Pasteur). Expression of the above antigens in transfected Escherichia coli (for plasmids) and in chick embryo fibroblasts (for canarypox) was confirmed by Western blotting (data not shown).
Phase 2
On May 16, 2001, chimp X139 was started on twice daily oral administration of 300 mg of lamivudine (Epivir; Glaxo, Philadephia, PA, USA) for 8 weeks. This resulted in a 3-log decline in viral load. Immediately after cessation of lamivudine, monthly injections of 4 mg of plasmid DNA (2 mg pNGL4a expressing HBsAg + PreS2 + 2 mg pNGVL3 expressing HbcAg) were administered into multiple sites (8/quadrant) intramuscularly and intradermally in deltoid and thigh muscles, bilaterally over each quadrant. Boosting with canarypox was planned for 6 months after the start of lamivudine therapy, but was not done as a result of the prompt return of high viral loads within 1 week after cessation of lamivudine therapy.
Phase 3
In the light of the rapid return to high viral loads shortly after cessation of lamivudine, we decided to extend the duration of lamivudine therapy, and to continue this after beginning therapeutic immunization. Lamivudine therapy was implemented on May 6, 2002 and continued for 18 weeks, resulting in a 4.3 log drop in viral load. As data indicated that recombinant vaccinia virus (WR) was more immunogenic than DNA/canarypox prime boost regimen (M. Shan et al, unpublished data), we decided to immunize with this vector. Vaccinia vp551 expressing HBsAg + PreS1 + PreS2, and vaccinia vp541 expressing HBcAg, were adjusted to 107 pfu/ml in phosphate buffered saline (PBS) 50% glycerol and held at −70°C. The above stock was administered by transdermal inoculation on the back between the shoulder blades with 15 sticks of a bifurcated needle routinely used in human vaccination. Recombinant vaccinia was administered as above 18 weeks after beginning lamivudine treatment. Lamivudine therapy continued for 8 weeks after the vaccinia virus immunization and then was inadvertently stopped for 9 days. A second vaccinia inoculation was carried out as above 10 weeks after the first, and lamivudine therapy was reinstituted for an additional 4 weeks.
Quantitation of viral loads
HBV DNA was quantitated using real time polymerase chain reaction (PCR) assay with molecular beacon technology as previously described [15], with modifications. Briefly, DNA was extracted from plasma using Qiagen DNA blood kits (Qiagen, Valencia, CA, USA). PCR mixtures were set up with a Beckman Biomek 2000 pipetting station (Beckman Coulter, Fullerton, CA, USA) in a laminar flow hood in a room dedicated to PCR set up. Primers used were 5′-AAA TTC GCA GTC CCC AACC3-′ and 5′-ATG AGG CAT AGC AGC AGG ATG-3′. The probe was TET-GGA CGG CTG GAT GTG TCT GCG GCG TTT TAT CCG TCG-DABCYL. The thermal cycling and data acquisition were done with a Perkin Elmer 7700 sequence detector (Perkin Elmer, Wellesley, MA, USA). This assay permits quantitation of both strong and weak specimens by comparison with a standard curve derived from serial dilutions of synthetic HBV plasmid DNA (pCDNA3-HBV). The method used was sensitive to approximately 100 DNA molecules/ml using a synthetic DNA standard, and gave linear results between 102.5 and 107 DNA molecules/ml. This methodology has two major advantages: the ability to quantitate strong and weak specimens from a single dilution, and that the amplified samples are not used for other assays (gel or ELISA), thus limiting sources of contamination. Quality control was performed routinely by including four HBV negative control sera and two positive sera during each PCR run as external negative and positive controls, respectively, to monitor extraction and amplification efficiencies. Assays in which positive control quantities are outside of the mean ± 2 SD for all assays run (QA curve) are discarded.
Assays for cell-mediated immunity
Isolation and propagation of peripheral blood lymphocytes
Peripheral blood mononuclear cells (PBMC) drawn at different intervals before and after immunotherapy were purified by Ficoll–Hypaque and then cryopreserved at 1°C/min in the presence of 20% autologous serum and 10% dimethyl sulfoxide (DMSO). Peripheral blood lymphocytes (PBLs) were rapidly thawed by swirling in a 37°C water bath, washed three times, and counted for functional analysis.
Interferon-γ ELISPOT assay
Interferon-γ ELISPOT assay was done according to the manufacturer’s instructions (MABTECH cat no. M34201-A, Sweden) with modifications. Briefly, 96-well nitrocellulose bottomed Millititer plates (Millipore, Bedford, MA, USA) were coated with murine anti-human IFN-γ mAb at concentration of 15 μg/ml in PBS and incubated at 4°C. After 24 hours, the plates were washed and blocked with 10% human AB+ serum (1 h at 37°C). To estimate the number of HBV-specific IFN-γ secreting cells (ISC), ex-vivo unexpanded PBMCs, were added at different concentrations (104 – 105 cells/well) in 100 μl volume of complete medium (RPMI-1640 containing 10% AB+ serum). The stimulator cells, an autologous Epstein–Barr virus (EBV)-transformed cell line, were infected with either recombinant vaccinia- or parental vaccinia at multiplicity of infection (MOI) of 10 : 1. After 1 h the cells were treated with different doses of Cidofovir for 1 h to reduce background [20], then washed and resuspended in complete medium. The stimulator cells were added at a concentration of 105 cells/well in 100 μl volume of complete medium in duplicate. After 18 h incubation at 37°C, the plates were washed five times with washing buffer, PBS containing 0.05% (v/v) Tween 20 (Sigma, St Louis, MO, USA), using an ELISPOT plate washer (Millipore, Bedford, MA, USA). Biotinylated anti-human IFN-γ mAb (clone 4S.B3) at a concentration of 1 μg/ml in blocking buffer, PBS containing 0.05% (v/v) Tween-20 and 1% (w/v) bovine serum albumin (Sigma), was added, and the plates were incubated at room temperature for 2 h at 37°C. Plates were then again washed five times, and streptavidin-horse radish peroxidase (HRP) (1 : 1000) in blocking buffer was added and incubated at room temperature for 2 h, followed by washing five times with washing buffer and the addition of substrate 3-amino-9-ethyl carbazole (AEC) reagent in substrate buffer (Sigma). After developing the spots for 10–15 min, the plates were washed with distilled water and air-dried. The number of spots was enumerated using a computerized assisted ELISPOT image analyzer (Zeiss system, Axioplan 2 Imaging), and KS ELISPOT 4.2 program (Zeiss, Zeiss Inc., Thornwood, NY, USA) designed to detect spots using predetermined criteria based on size, shape and colorimetric density. Analysis of the data was calculated from the equation: Ag-specific ISC/106 cells = number of ISC in response to Ag/106 cells – number of ISC in the presence of media alone/106 cells. HBV-specific ISCs were plotted after subtracting the number of ISC induced by parental vaccinia.
To eliminate samples with poor viability, those having phytohemagglutinin (PHA) responses <2500 ISC/106 cells, equivalent to the mean – 2 SD of all the samples tested in 25 experiments, were excluded from analysis. The average number of ISC/106 cells, after PHA stimulation was 3783.3 ± 398.2.
Estimation of T-cell avidity
To evaluate the avidity of Ag-specific T cells after different therapeutic immunization approaches, IFN-γ ELISPOT assay was done as described above in presence of different stimulator cell concentration (105, 104 and 103 cells/well) to measure total, intermediate and high avidity Ag-specific responses, respectively (M.T.M. Shata et al., unpublished data). High avidity responses were calculated as the number of ISCs in the presence of low concentration of stimulator cells (103 cells/well). Total HCV-specific responses were calculated as the number of ISCs in the presence of high and intermediate stimulator cell concentrations (105 and 104 cells/well).
Evaluation of cytotoxic T cell and NK cell responses
Target cells
An immortalized B-lymphocyte cell line (B-LCL) from chimp X139 was generated by culturing chixmp X139 PBL in supernatant from the marmoset cell line B95-8 in 15% RPMI 1640 medium containing antibiotics and 2.5 mg/mL cyclosporin A.
Effector cells
Peripheral blood mononuclear cells were purified as described above and plated in 96-well plates at different concentrations starting at 2.5 × 106/ml in culture medium consisting of RPMI 1640, 10% heat-inactivated fetal bovine serum, antibiotics, and 2 mmol/l l-glutamine. B-LCL was used in the cytotoxic T lymphocyte (CTL) assay as target cells after infection with recombinant vaccinia virus vectors. All B-LCL was infected with either recombinant vaccinia expressing HBsAg (small: S), HBV (middle: HBsAg + PreS2), HBV (large: HBsAg + PreS1 + PreS2) proteins at a multiplicity of infection of 10 pfu/cell for 1 h at 37°C, and cultured 16 h prior to the assay. Vaccinia parent (without any insert) and recombinant vaccinia expressing β-galactosidase were included as controls. Vaccinia infected autologous B-LCL was labeled with 100 μCi of 51Cr for 1 h, washed three times and used as targets in a standard 6-h 51Cr release assay in triplicate at various effector target cell (E : T) ratios using 5000 target cells/well. Lysis was also assessed with K562 targets to assess the natural killer (NK) cell responses. The percent specific lysis was calculated as 100 × [(experimental release − spontaneous release)/(maximum release − spontaneous release)]. Maximum 51Cr release was determined from supernatants of target cells lysed by addition of 5% Triton X 100. Spontaneous release was measured in supernatants from target cells incubated without effector cells.
Results
Effect of immunotherapy on viral load
In phase 1 of this study, we attempted to reproduce the positive results of our first study [18]. Despite the addition of HBcAg to the immunogens, and addition of plasmids expressing human IL-12, no downregulation of viral load was observed (Fig. 1).
Fig. 1.
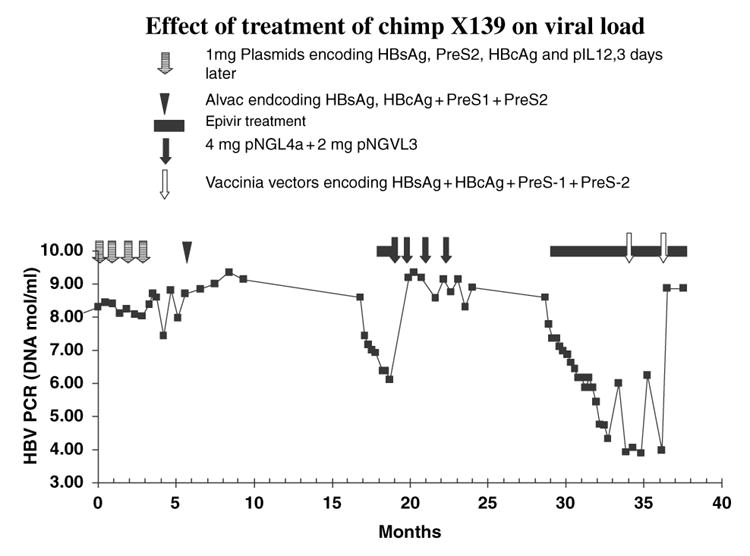
Effect of immunotherapy and lamivudine on viral load.
As lamivudine has been shown to effectively down-regulate HBV replication [20], and to restore T-cell responsiveness [7], we then embarked on a trial (phase 2) involving 8 weeks of lamivudine therapy prior to the onset of immunotherapy with HBV antigen expressing plasmids. The lamivudine treatment resulted in a 3 log reduction in HBV DNA levels. This trial unfortunately did not consider the rapid return in viremia levels after cessation of short-term lamivudine treatment [3], and viral load rebounded to original levels within 6 days of cessation of lamivudine treatment. DNA-based immunization did not significantly lower viral load.
We, therefore, embarked on a third trial (phase 3) in which immunotherapy was started after 18 weeks of lamivudine therapy resulting in a 4.3 log reduction in viral load. In this study, we used recombinant vaccinia viruses expressing HBsAg, PreS1, PreS2, and HBcAg as an immunogen. This was based on data (M. Shan et al., unpublished data) showing vaccinia to elicit far stronger cell mediated responses that those elicited by DNA-canarypox prime/boost regimens. Lamivudine treatment was continued for 8 weeks after vaccinia immunization, when it was inadvertently interrupted for 9 days. A second vaccinia boost was then given together with re-administration of lamivudine for an additional 4 weeks. The 9 day cessation of lamivudine treatment resulted in a 2 log increase in viral load; however, this returned to low levels, approximately five logs below baseline values, after re-initiation of treatment. However, within 1 week of cessation of lamivudine treatment viral load had returned to original levels, thus recombinant vaccinia virus proved to be ineffective for therapeutic immunization, even in the presence of lamivudine. It is possible that the effectiveness of the vaccinia vector was reduced by canary pox immunization in phrase 1, during which Elispot reactivity to the parental vaccinia virus rose to 3960 ISC/106 cells.
Analysis of cell mediated immune responses during attempted therapeutic immunization revealed that both cycles of DNA based immunization enhanced cell mediated immune responses revealed in the ELISPOT assays (Fig. 2). DNA based immunization preferentially boosted high avidity responses (Fig. 3), although the proportion of high avidity responses was low. The use of higher doses of DNA encoding HBsAg and PreS2 and HBcAg in phase 2 appeared to preferentially stimulate high avidity responses. It is of interest that high avidity responses were predominantly directed to the small surface antigen in most samples, while total T-cell responses were mostly directed toward HBcAg and the large surface antigen protein. The canarypox booster had no effect on the cell-mediated immune responses, and the vaccinia immunization appeared to decrease them. None of the immunization strategies significantly affected CTL or NK cell activity (Fig. 4).
Fig. 2.
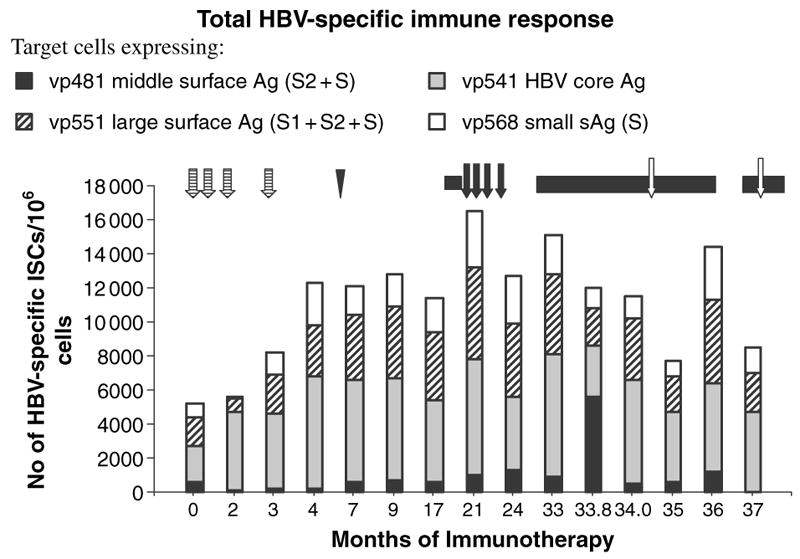
Effect of immunotherapy on T-cell responses to target cells expressing HBcAg and small, middle and large HBV surface antigen proteins. Symbols for treatment strategies are as shown in Fig. 1.
Fig. 3.
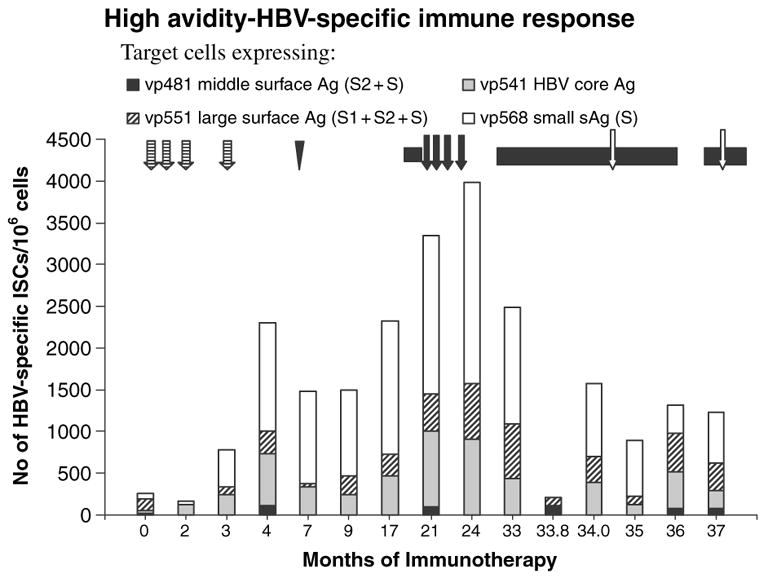
High avidity T-cell responses to target cells expressing HBcAg and small, middle, and large HBV surface antigen proteins. Symbols for treatment strategies are as shown in Fig. 1.
Fig. 4.
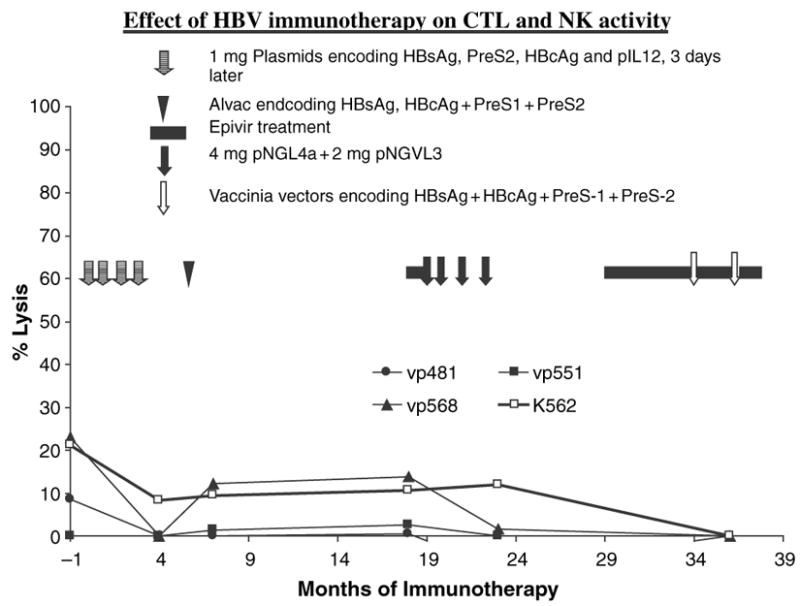
Effect of therapeutic immunization on cytotoxic T lymphocyte and NK activity. Target cells: Vγ 481 middle surface antigen; vp 541: 4BV core antigen; vp551: large surface antigen; K562 target for NK cells.
Discussion
The relatively poor response to IFN and the requirement for long-term chemotherapy in chronic HBV infection, as well as the strong cellular immune response in subjects who recover from HBV infection [4], has led investigators to attempt to control these infections by therapeutic immunization. Various animal models of hepadnavirus infection have been used for such studies. Mancini et al. immunized HBsAg transgenic mice with HBaAg + PreS2 encoding DNA and observed development of anti-HBs, clearance of HBsAg in serum, and decrease of HBV messenger RNA [16]. Unfortunately these findings have been difficult to reproduce [21]. Other investigators used monthly administration of HBV vaccine in complete Freund’s adjuvant in HBV transgenic mice and observed a disappearance of HBs antigenemia in 25 of 32 immunized mice, after eight to 12 injections [1]. Chronically infected woodchucks immunized with woodchuck HBsAg together with a T-helper peptide developed anti-HBs, and in some cases a reduction in viremia; however, in two cases developing the highest anti-HBs levels, severe liver damage was observed [13]. This represents an obvious danger for clinical trials of immunotherapy. Mature dendritic cells, activated by pulsing with HBsAg peptides or particles, induced HBsAg specific CTL when injected into HBsAg transgenic mice. These cells produced severe hepatitis when passively transferred to isogenic transgenic mice [11]. However, these dendritic cells did not downregulate virus replication or antigenemia, possibly because the numbers used, or the numbers of HBsAg specific T-cells induced in vivo, were not sufficient. Long lasting downregulation (>400-fold) of viremia was induced in a chronically HBV infected chimpanzee by HBsAg DNA priming and recombinant canarypox boosting [18]. Significantly, downregulation in this study was temporarily associated with an increased number of HBV specific IFN-γ secreting lymphocytes, which occurred shortly after boosting, as well as increase in NK activity, but not with the number of CTL. This is in accord with numerous reports from Chisari’s laboratory indicating control of HBV replication in vivo by cytokine dependant mechanisms [11]. The above study was done with a chimpanzee with a relatively low viral load (105.4 DNA mol/ml of plasma), and did not succeed in an animal with a high (108.6±0.47) viral load, as shown in the present study. We reasoned that this may have been due to the 10,000-fold higher titer of viremia in the chimpanzee used for the present study. Such high viral loads could have resulted in peripheral T-cell tolerance or anergy, although this was not reflected in the ELISPOT results which showed moderately high IFN-γ secretion after exposure of effector cells to target cells infected with recombinant vaccinia virus expressing core and large and small surface antigens even prior to immunotherapy. These responses increased moderately after DNA based immunization, but, in contrast to our previous results [18], were not increased by the canarypox booster (Fig. 2).
Although high viral load may have been the determinant of this failure, we cannot rule out an effect of differences between the animals tested. A similar failure has been reported in woodchucks treated with the antiviral drug l-FMAU and then with HBV vaccine [17].
So far, relatively few therapeutic immunization studies for HBV have been done in man. Dienstag et al. gave monthly HBV vaccine to 16 chronic carriers and observed no beneficial or harmful effects [5, 6]. Other investigators observed termination of chronic infection in some patients who received HBV vaccine [19]. Intriguingly, Lau et al. have reported that four of six chronic carriers receiving allogeneic bone marrow transplants from anti-HBs and anti-HBc positive donors, but none of seven who received anti-HBs (+), anti-HBc (−) marrow, terminated their infections [14]. Heathcote et al. immunized chronically infected subjects with a dominant core antigen lipopeptide, together with a T-helper epitope [12]. This vaccine induced CTL responses, but did not affect viral replication.
Antigen-specific CD8+ T-cells play an important role in anti-viral and anti-tumor immunity. Strong evidence has suggested that high avidity, and not low avidity, CD8+ T-cells mediate protective immunity in both viral infection [2, 8–10] and tumors [23, 24]. It is thus likely that the low frequency of high avidity T-cells observed in this study may in part account for the inability of the immunization regimens tested to control HBV replication. High viral loads may preferentially cause elimination of the high avidity effector cells. It is also possible that at the lowest number of targets used, the responses reflect sparse spatial distribution of stimulators and not competition of T cells of different affinities. Evaluation of this mechanism is presently underway in our laboratory by using high and low avidity murine T-cell clones isolated in other system [2]. However, sparse target cells do not explain the differences in responses using the lowest number of target cells under different immunization protocols as shown by our data. Our data are in agreement with a recent report in the mouse model which highlights the potential positive effects of therapeutic vaccination on viral control during chronic infection, but also provides evidence that a high viral load at the time of immunization is likely to limit the effectiveness of therapeutic immunization [22].
Numerous questions need to be answered if a rationally based therapeutic immunization strategy is to be achieved. We need to know more the mechanism for immune tolerance, or anergy, in chronic HBV infections. We also need to know how best to induce strong, highly avid, cell mediated immune responses in the face of pre-existing anergy, and which viral proteins and epitopes should be targeted. The present study underscores the need to answer these questions.
Acknowledgments
Virogenetics, Inc. (presently Aventis Pasteur) supplied the recombinant poxviruses. Drs Charles Tackney and Robert Whalen generously provided us with plasmids encoding HBcAg and HBsAg + PreS1. We are grateful to Mussa Konneh, John Zeonyuway, George Saycoyah, Joseph Thomas, and their colleagues for expert animal and laboratory support for these studies.
The Starr Foundation, NIH grant no. RO1 AI47349, and institutional funds from the New York Blood Center supported this study.
This study was partially supported by NHLBI contract NO1HB27091 and the authors would like to thank Dr Luiz Barbosa for his enthusiastic support.
Footnotes
Present address
Mohamed Tarek M. Shata, Viral Immunology Laboratory, Digestive Diseases Division, Department of Internal Medicine, Hepatology Research Group, University of Cincinnati, College of Medicine, Cincinnati, OH, USA.
References
- 1.Akbar SM, Kajino K, Tanimoto K, et al. Placebo-controlled trial of vaccination with hepatitis B virus surface antigen in hepatitis B virus transgenic mice. J Hepatol. 1997;26:131–137. doi: 10.1016/s0168-8278(97)80019-0. [DOI] [PubMed] [Google Scholar]
- 2.Alexander-Miller MA, Leggatt GR, Sarin A, Berzofsky J. A role of antigen, CD8, and cytotoxic T lymphocyte (CTL) avidity in high dose antigen induction of apoptosis of effector CTL. J Exp Med. 1996;184:485–492. doi: 10.1084/jem.184.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boni C, Bertoletti A, Penna A, et al. Lamivudine treatment can restore T cell responsiveness in chronic hepatitis B. J Clin Invest. 1998;102:968–975. doi: 10.1172/JCI3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chisari FV, Ferrari C. Hepatitis B virus immunopathogenesis. Annu Rev Immunol. 1995;13:29–60. doi: 10.1146/annurev.iy.13.040195.000333. [DOI] [PubMed] [Google Scholar]
- 5.Dienstag JL, Stevens CE, Bhan AK, Szmuness W. Hepatitis B vaccine administered to chronic carriers of hepatitis b surface antigen. Ann Intern Med. 1982;96:575–579. doi: 10.7326/0003-4819-96-5-575. [DOI] [PubMed] [Google Scholar]
- 6.Dienstag JL, Perrillo RP, Schiff ER, Bartolomew M, Vicar C, Rubin M. A preliminary trial of lamivudine for chronic hepatitis B infection. N Engl J Med. 1995;333:1657–1661. doi: 10.1056/NEJM199512213332501. [DOI] [PubMed] [Google Scholar]
- 7.Dienstag JL, Schiff ER, Wright TL, Perrillo RP, et al. Lamivudine as initial treatment for chronic hepatitis B in the United States. N Engl J Med. 2003;341:1256–1263. doi: 10.1056/NEJM199910213411702. [DOI] [PubMed] [Google Scholar]
- 8.Gallimore A, Hombach J, Dumrese T, Rammensee HG, Zinkernagel RM, Hengartner H. A protective cytotoxic T cell response to a subdominant epitope is influenced by the stability of the MHC class I/peptide complex and the overall spectrum of viral peptides generated within infected cells. Eur J Immunol. 1998;28:3301–3311. doi: 10.1002/(SICI)1521-4141(199810)28:10<3301::AID-IMMU3301>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 9.Gallimore A, Hengartner H, Zinkernagel R. Hierarchies of antigen-specific cytotoxic T-cell responses. Immunol Rev. 1998;164:29–36. doi: 10.1111/j.1600-065x.1998.tb01205.x. [DOI] [PubMed] [Google Scholar]
- 10.Gallimore A, Dumrese T, Hengartner H, Zinkernagel RM, Rammensee HG. Protective immunity does not correlate with the hierarchy of virus-specific cytotoxic T cell responses to naturally processed peptides. J Exp Med. 1998;187:647–1657. doi: 10.1084/jem.187.10.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guidotti LG, Chisari FV. To kill or to cure: options in host defense against viral infection. Curr Opin Immunol. 1996;8:478–483. doi: 10.1016/s0952-7915(96)80034-3. [DOI] [PubMed] [Google Scholar]
- 12.Heathcote J, McHutchison J, Lee S, et al. A pilot study of the CY-1899 T-cell vaccine in subjects chronically infected with hepatitis B virus. The CY-1899 T Cell Vaccine Study Group. Hepatology. 1999;30:531–536. doi: 10.1002/hep.510300208. [DOI] [PubMed] [Google Scholar]
- 13.Hervas-Stubbs SJ, Lasarte J, Sarobe P, et al. Therapeutic vaccination of woodchucks against chronic woodchuck hepatitis virus infection. J Hepatol. 1997;27:726–737. doi: 10.1016/s0168-8278(97)80090-6. [DOI] [PubMed] [Google Scholar]
- 14.Lau GK, Liang R, Lee CK, et al. Clearance of persistent hepatitis B virus infection in Chinese bone marrow transplant recipients whose donors were anti-hepatitis B core- and anti-hepatitis B surface antibody-positive. J Infect Dis. 1998;178:1585–1591. doi: 10.1086/314497. [DOI] [PubMed] [Google Scholar]
- 15.Lee DH, Prince AM. Automation of nucleic acid extraction for NAT screening of individual blood units. Transfusion. 2001;41:483–487. doi: 10.1046/j.1537-2995.2001.41040483.x. [DOI] [PubMed] [Google Scholar]
- 16.Mancini M, Hadchouel M, Davis HL, Whalen RG, Tiollais P, Michel ML. DNA-mediated immunization in a transgenic mouse model of the hepatitis B surface antigen chronic carrier state. Proc Natl Acad Sci USA. 1996;93:12496–12501. doi: 10.1073/pnas.93.22.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menne SC, Roneker A, Korba BE, Gerin JL, Tennant BC, Cote PJ. Immunization with surface antigen vaccine alone and after treatment with 1-(2-fluoro-5-methyl-beta-L-arabinofuranosyl)-uracil (L-FMAU) breaks humoral and cell-mediated immune tolerance in chronic woodchuck hepatitis virus infection. J Virol. 2002;76:5305–5314. doi: 10.1128/JVI.76.11.5305-5314.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pancholi P, Lee DH, Liu Q, et al. DNA prime/canarypox boost-based immunotherapy of chronic hepatitis B virus infection in a chimpanzee. Hepatology. 2001;33:448–454. doi: 10.1053/jhep.2001.21594. [DOI] [PubMed] [Google Scholar]
- 19.Pol S, Driss F, Michel ML, Nalpas B, Berthelot P, Brechot C. Specific vaccine therapy in chronic hepatitis B infection. Lancet. 1994;344:342. doi: 10.1016/s0140-6736(94)91384-6. [DOI] [PubMed] [Google Scholar]
- 20.Shata MT, Shan MM, Tricoche N, Talal A, Perkus M, Prince AM. Optimization of recombinant vaccinia-based ELISPOT assay. J Immunol Methods. 2003;283:281–289. doi: 10.1016/j.jim.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 21.Shimizu Y, Guidotti LG, Fowler P, Chisari FV. Dendritic cell immunization breaks cytotoxic T lymphocyte tolerance in hepatitis B virus transgenic mice. J Immunol. 1998;61:4520–4529. [PubMed] [Google Scholar]
- 22.Wherry EJ, Blattman JN, Ahmed R. Low CD8 T-cell proliferative potential and high viral load limit the effectiveness of therapeutic vaccination. J Virol. 2005;79:8960–8968. doi: 10.1128/JVI.79.14.8960-8968.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang S, Linette GP, Longerich S, Haluska FG. Anti-melanoma activity of CTL generated from peripheral blood mononuclear cells after stimulation with autologous dendritic cells pulsed with melanoma gp100 peptide G209–2M is correlated to TCR avidity. J Immunol. 2002;169:531–539. doi: 10.4049/jimmunol.169.1.531. [DOI] [PubMed] [Google Scholar]
- 24.Zeh HJ, III, Perry-Lalley D, Dudley ME, Rosenberg SA, Yang JC. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J Immunol. 1999;162:989–994. [PubMed] [Google Scholar]