

Abstract
Temporal lobe epilepsy may be associated with emotional difficulties such as depression or anxiety. Since the amygdala is involved in both epilepsy and emotion, common neural mechanisms in this temporal lobe structure may underlie the emotional disturbances observed in people with epilepsy. The neurotransmitter serotonin (5-hydroxytryptamine, or 5-HT) is implicated in many psychopathologies, and 5-HT also modulates amygdala excitability. Therefore, the present study used the fear potentiated startle (FPS) paradigm to investigate the effect of neuronal excitability on fear behavior in rats treated with p-chlorophenylalanine (PCPA) to chronically inhibit 5-HT synthesis. PCPA treatment selectively enhanced FPS in individually-housed rats. The exaggerated FPS response was reduced to control level by the anticonvulsant phenytoin (10mg/kg) and phenytoin (30 mg/kg) further decreased FPS behavior. These data suggest that a sub-seizure state of neuronal excitability mediated by low 5-HT in brain fear circuits may be associated with pathological fear behavior.
Keywords: Anticonvulsant, Amygdala, Hyperexcitability, Serotonin, Phenytoin, Fear-potentiated startle, Behavior, Rat
INTRODUCTION
The amygdala is a collection of nuclei in the temporal lobe that has important functional roles in both epilepsy and emotion. Imaging studies in humans have revealed the importance of the amygdala in processing fearful stimuli, showing that the amygdala is activated during the acquisition of conditioned fear [1] and recognition of emotional facial expressions [2]. Conversely, destruction of the amygdala selectively impairs emotional learning [3]. Amygdala dysfunction is implicated in several neurological and psychiatric disorders, especially mood and anxiety disorders [4]. Several lines of evidence suggest the neural mechanisms in the amygdala are part of a highly conserved network contributing to the evolutionarily advantageous ability to recognize and respond to potentially harmful stimuli in the environment, and functional deficits in this network can lead to altered emotional behavior.
The critical role of the amygdala in fear behavior has been analyzed using classical fear conditioning, in which a neutral conditioned stimulus (CS) is paired with a noxious unconditioned stimulus (US) such as a footshock. After conditioning, an animal will exhibit enhanced startle behavior in the presence of the CS. Fear potentiation of the acoustic startle reflex has been shown to involve well-defined neural circuits, with the amygdala playing a critical role in the learning of CS-US associations [5]. Hyperactivity in otherwise adaptive amygdala circuitry may produce abnormal emotional behavior and contribute to the neurobiology of mental illnesses such as mood and anxiety disorders .
In addition to its role in fear conditioning, the amygdala is often the focus of seizure activity in patients with temporal lobe epilepsy (TLE) [6]. These two seemingly disparate functional roles of the amygdala may be related. Converging evidence from human and animal studies suggests that there may be biological mechanisms in the amygdala that contribute to both epileptic activity and emotional disturbances [7, 8]. That is, the underlying cellular mechanisms in the amygdala that result in epilepsy may, at a sub-seizure level of hyperexcitability, result in abnormal emotional behavior. For example, TLE may be accompanied by emotional disturbances such as anxiety, depression, and aggression [6]. When anticonvulsant treatment is efficacious in controlling seizures in TLE, abnormal emotional behaviors are often normalized as well. And, in people without epilepsy, anticonvulsants are mood-stabilizing. Phenytoin, and other widely used anticonvulsants such as carbamazepine and valproate, selectively reduce the frequency of impulsive aggressive acts in both prison inmates [9] and non-incarcerated individuals [10]. Similarly, animal studies support common biological mechanisms in emotion and epilepsy. For example, amygdala kindling increases anxiety-like behavior in both rats and cats [11], and phenytoin reduces isolation-induced aggression in rats [12]. Altogether, these human and animal studies suggest that epilepsy-like hyperexcitability in the amygdala is involved in abnormal emotional behavior. Moreover, it has been suggested that the increased neuronal excitability in the amygdala and other limbic areas contribute to the emotional difficulties sometimes faced in people with epilepsy [7].
The neurotransmitter serotonin (5-HT) is a critical regulator of cell excitability in the amygdala and has been implicated in a variety of amygdala-dependent behaviors such as fear, aggression, and emotional learning. The lateral amygdala (LA) nucleus is the primary target of inputs to the amygdala, and the LA is thought to be the site of plasticity during fear learning [13–15]. 5-HT mediates a net inhibitory tone in the amygdala by enhancing GABA, and inhibiting glutamate neurotransmission [16, 17]. Serotonergic mechanisms also play an important role in emotional behavior in vivo. Chronic administration of selective serotonin reuptake inhibitors (SSRIs) reduces acquisition of auditory fear conditioning [18]. Studies in 5-HT receptor knock-out mice have shown that 5-HT1A receptor knock-outs have high anxiety [19], whereas 5-HT1B receptor knock-outs are less anxious and more aggressive [20]. Together, these studies suggest that serotonergic control over emotional behavior may involve regulation of cellular excitability in limbic brain areas such as the amygdala.
Based on the role of 5-HT in amygdala excitability, and the link between excitability and emotional behavior, it is hypothesized that low 5-HT results in an epilepsy-like state associated with disturbed amygdala-dependent emotional behavior. To test this, the effect of phenytoin on fear-potentiated startle (FPS) in animals treated with the 5-HT synthesis inhibitor p-chlorophenylalanine (PCPA) was examined. This study shows that FPS is increased in PCPA-treated rats. Consistent with previous findings, phenytoin dose-dependently reverses the PCPA-induced increase in FPS, suggesting that neuronal hyperexcitability may play an important role in the modulation of amygdala-dependent behaviors. These results are discussed in relation to emotional disturbances in people with epilepsy.
METHODS
Animals
A total of 77 male Sprague-Dawley rats (Harlan, Houston, TX), weighing 50–74 g at the time of arrival, were used. Rats were either group-housed or individually housed in 48 x 21 x 27 cm clear plastic cages. Animals were maintained on a 12-h light/dark cycle (lights on at 07:00) in a temperature-controlled (22°C) animal colony with food and water available ad libitum. Each rat was handled 5 min/day for four days prior to behavioral experimentation. Experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Baylor University.
Drugs
DL-p-chlorophenylalanine hydrochloride (PCPA, Sigma) was dissolved in saline and pH adjusted to 7.2 with KOH. Animals were injected intraperitoneally on days 1 and 10 after beginning the housing condition. Control rats received 0.9% saline (SAL) only. Phenytoin (PHN, 5,5-diphenylhydantoin sodium salt, Sigma) was dissolved in vehicle (VEH) composed of 40% dimethyl sulfoxide (Sigma), 40% propylene glycol (Fisher Scientific), and 20% dH2O at a pH of 7.2. Thirty minutes prior to fear potentiated startle testing, rats received intraperitoneal injections of VEH alone followed by phenytoin (10 and 30 mg/kg) on subsequent testing days.
Apparatus
Rats were trained and tested in an acoustic startle reflex system (Med Associates). Steel grid rod animal holders were attached to startle platforms and enclosed in unlit ventilated sound-attenuating chambers with inside dimensions 56 x 38 x 36 cm. Background noise was 45 dB, measured from the center of the animal holder using the Sound Pressure Level Measurement Package designed especially for use with this system.
Startle responses were evoked by 50 msec white noise bursts (5 msec rise-decay time) amplified by a two-channel power amplifier, and delivered through high frequency speakers located 5 cm behind each animal holder. Startle response amplitudes were quantified using an accelerometer located beneath each platform. The force of each rat’s startle response displaced the accelerometer below, causing a change in voltage output proportional to the velocity of platform movement. This output was amplified and digitized on a scale of 0–2047 units and analyzed by a PC. Startle response amplitude was defined as the maximum peak voltage that occurred during the first 200 msec after onset of the startle-eliciting stimulus.
The CS was a 3.7 sec light (80 Lux) produced by an 8 W fluorescent bulb (15 μsec onset) located 10 cm behind each animal holder. Luminosity was measured using a Vivitar 230 LX light meter. The US was a 0.5 sec, 0.6 mA shock (shock intensity measured using an oscilloscope and calculated as 0.707 x 0.5 base/peak voltage) generated by a stand-alone shocker/scrambler unit and delivered through the steel floorbars of the animal holder. The presentation and sequencing of all stimuli were under the control of a Dell PC using the Startle Reflex software package provided by Med Associates. Animal holders and platforms were cleaned between subjects with 1% acetic acid.
Behavioral Procedures
Behavioral procedures consisted of acclimation to the test chambers, habituation of the startle response, fear conditioning, and fear potentiated startle testing.
Acclimation
On each of two consecutive days, rats were placed into the test chambers for 10 min and then returned to their home cages.
Habituation
On each of the next two consecutive days, rats were placed into the test chambers and after 5 min presented with 30 startle-eliciting white noise bursts (10 each at 90, 95, and 105 dB, 50 msec duration). The three intensities were presented in a quasi-random order, with the restriction that each intensity occurred once within each successive block of three stimuli, at a 30 sec inter-stimulus interval (ISI).
Fear Conditioning
After 24 h, rats received 15 light-shock (CS-US) pairings. The first pairing occurred 5 min after rats were placed in the test chambers, and successive presentations occurred on average every 3 min (variable ISI 2–4 min). For each pairing the CS and US coterminated, with the 0.5 sec shock delivered 3.2 sec after onset of the 3.7 sec light CS.
Fear Potentiated Startle Test
Fear potentiated startle was measured 24 h after fear conditioning. Rats were placed into the test chambers and after 5 min were presented with three consecutive blocks of stimuli. Block 1, called “leaders,” consisted of 15 startle-eliciting noise bursts (5 each at 90, 95, and 105 dB, 50 msec) presented in a quasi-random order. Thirty seconds after the final leader, the first of 60 test trials (Block 2) was presented. Half of the test trials presented the startle-eliciting noise burst 3.2 sec after onset of the light CS (light-noise trials) while the other half of the test trials presented a startle-eliciting noise burst without the CS (noise-alone trials). As with the leaders, equal numbers of 90, 95, and 105 dB 50 msec noise bursts were used, and the six resulting trial types were presented in a quasi-random order. Block 3, called “trailers,” began 30 sec after the final test trial and was identical to Block 1, consisting of 15 startle-eliciting noise bursts (90, 95, and 105 dB, 50 msec) presented in a quasi-random order. The ISI throughout fear potentiated startle testing was 30 sec.
Potentiation of the startle response was calculated as the difference between the light-noise trials and the average of the leaders and trailers, divided by the average of the leaders and trailers to be expressed as a percent. We used the average of the leaders and trailers as a baseline startle response for comparisons because we found this measurement to be more consistent within subjects than comparison to the noise-alone trials (data not shown).
The concentration-response relationship of phenytoin was examined within subjects. Animals were re-trained with CS-US pairing on intervening days to prevent extinction. Pilot data showed that rats exhibit consistent FPS on as many as six distinct testing days when retrained between FPS tests (data not shown). Data from VEH-treated and untreated controls (Figure 1) were pooled.
Figure 1.
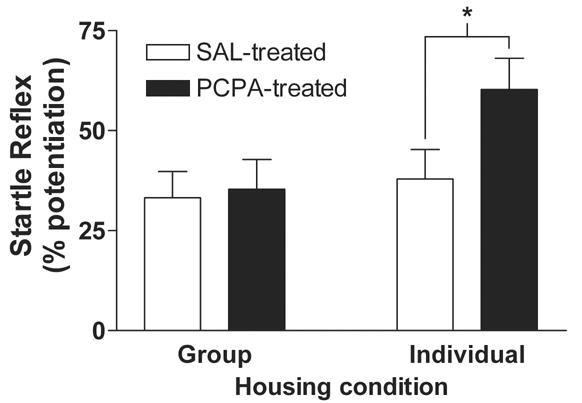
DL-p-chlorophenylalanine (PCPA) increases fear-potentiated startle (FPS) selectively in rats housed individually. Rats were assigned to one of four treatment groups in a 2 x 2 design with housing and drug treatment as between-groups factors: group/saline (n = 10), group/PCPA (n = 10), individual/saline (n = 12), or individual/PCPA (n = 13). Animals were either group or individually housed for 13 days, and received either PCPA (300 mg/kg i.p.; black bars) or isovolumetric 0.9% saline (SAL; white bars) on days 1 and 10. FPS was tested on day 13 and is expressed as percent (mean ± SE) potentiated startle amplitude (Methods). In the group housed condition, there is no significant difference in FPS between saline (33 ± 7%) and PCPA (35 ± 7) treated animals (p > 0.05). However, in the individually housed condition PCPA treated rats show 60 ± 8 % FPS compared to 38 ± 7 % in saline treated controls (p < 0.05). Thus, PCPA increased FPS selectively in the individually-housed rats. * indicates significant difference from individually-housed, SAL-treated control (P < 0.05, Bonferroni multiple comparisons post-hoc test).
Statistical Analyses
Two-way analysis of variance (ANOVA) was used to analyze percent potentiation and baseline startle responses. Housing and PCPA treatment were treated as between-groups factors. Repeated measures ANOVA was used to analyze effects of increasing dose of PHN. Following ANOVA, individual groups were compared using Bonferonni’s multiple comparison post-hoc test. All analyses were performed using Microsoft Excel and GraphPad Prism. The criterion of significance for all comparisons was p < 0.05.
RESULTS
The effect of chronic p-chlorophenylalanine (PCPA) treatment on fear-potentiated startle (FPS) was examined in both group-housed and individually-housed rats. PCPA is a competitive and irreversible inhibitor of tryptophan hydroxylase, the rate-limiting enzyme in 5-HT synthesis, used to deplete brain serotonin (5-HT) [21]. Fear potentiation of the acoustic startle reflex was measured as percent increase in startle amplitude during presentation of the CS (see Methods). During the housing condition, rats received either PCPA (300 mg/kg i.p) or isovolumetric injections of 0.9 % saline (SAL). In group-housed animals (Figure 1, Group), PCPA had no effect on FPS. FPS was 33 ± 7 % in SAL-treated (n = 10, open bar) and 35 ± 7 % in PCPA-treated (n = 10, filled bar) rats. However, in individually-housed rats (Figure 1, Individual), PCPA increased FPS. Startle reflex amplitude was 38 ± 7 % potentiated (n = 12, open bar) in SAL-treated, individually-housed rats, whereas FPS was enhanced to 60 ± 8 % (n = 13, filled bar) in PCPA-treated rats. Two-way analysis of variance (ANOVA) followed by post-hoc tests showed that PCPA treatment enhanced FPS in individually housed rats (p < 0.05).
Because 5-HT mediates a net inhibitory tone in the amygdala [16, 17] and because the amygdala is known to be important in fear learning [5], we hypothesized that the PCPA-induced increase in FPS is mediated by epilepsy-like mechanisms of neuronal excitability. To test this, we examined the effect of the anticonvulsant phenytoin (PHN) on FPS in PCPA-treated, individually-housed rats. FPS was measured 30 min after injection of PHN (0, 10, or 30 mg/kg; i.p.). Figure 2 shows the effect of PHN administration on startle reflex potentiation. In control, FPS was 26 ± 6 % (n = 20) in saline-treated animals, but was 47 ± 7 % (n = 22) in PCPA-treated rats. PHN (10 mg/kg) reduced FPS amplitude in PCPA-treated rats to 26 ± 10 %, but FPS in SAL-treated animals remained relatively unchanged (28 ± 13 %; n = 8). In PHN (30 mg/kg) treated rats, FPS was further reduced in PCPA-treated (6 ± 7 %; n = 8) but not in SAL-treated (19 ± 9 %; n = 8) rats. Two-way ANOVA showed a significant main effect of PHN (F2,68 = 3.19; p < 0.05) but no significant main effect of PCPA (F1,68 = 0.09; p > 0.05) and no significant interaction (F2,68 = 1.84; p > 0.05). Post-hoc comparisons show that FPS is enhanced in PCPA-treated rats compared to SAL-treated controls (p < 0.05) and that PHN (30 mg/kg) reduced FPS in PCPA-treated animals (p < 0.05). PHN had no effect on FPS in group-housed rats (data not shown).
Figure 2.
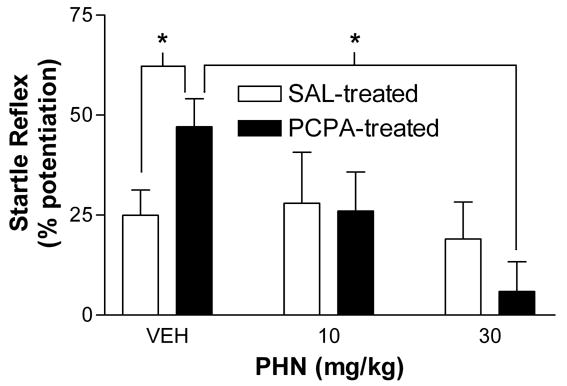
Phenytoin (PHN) reduces FPS selectively in PCPA-treated rats. Individually housed rats received i.p. injections of either saline (SAL) or PCPA (300 mg/kg) on days 1 and 10. FPS was measured 30 min. after injection of PHN (0, 10, or 30 mg/kg i.p.) within subjects on days 13, 15, and 17. On days 14 and 16, animals were retrained with CS-US pairing to minimize extinction. In individually-housed, PCPA-treated rats FPS was 47 ± 7% in control (n = 22), and reduced to 26 ± 10% (n = 8) and 6 ± 7% (n = 8) by 10 and 30 mg/kg PHN, respectively. In individually housed, SAL-treated rats FPS was 26 ± 6% in control (n = 20), and 28 ± 13% (n = 8) and 19 ± 9% (n = 8) after 10 and 30 mg/kg PHN, respectively. ANOVA shows that phenytoin significantly reduced FPS in PCPA-treated animals (p < 0.05), but had no effect in saline treated controls (p > 0.05). * indicates significant difference (p < 0.05, Bonferroni multiple comparison post-hoc test).
To determine whether phenytoin altered startle reflex nonspecifically, rather than fear potentiated startle responses, we examined baseline startle (amplitude during leaders and trailers, see Methods) at each dose of PHN (0, 10, and 30 mg/kg) for both saline and PCPA treated rats (Figure 3). PCPA treatment slightly reduced baseline startle reactivity, but this effect was not statistically significant. Two-way ANOVA revealed no significant effect of either PCPA treatment (p > 0.05) or PHN treatment (p > 0.05) on baseline startle behavior. However, the matching within subjects was significant (p < 0.05), indicating that baseline startle responses were consistent over several testing days in these animals.
Figure 3.
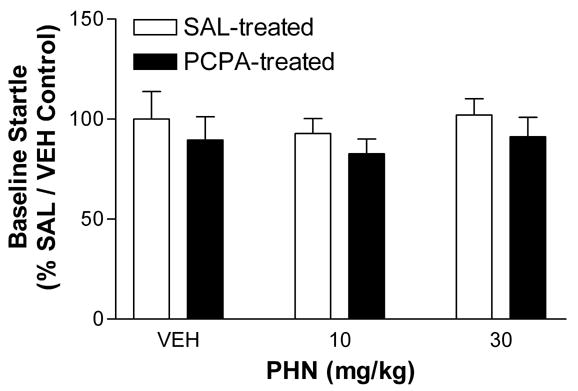
Phenytoin does not alter baseline startle responses in individually housed rats. Rats were individually housed for 13 days prior to testing, and received either PCPA (300 mg/kg i.p.) injections on days 1 and 10 or isovolumetric injections of 0.9 % saline. Baseline startle was measured 30 min after injection of PHN (0, 10, or 30 mg/kg i.p.) on days 13, 15, and 17 within subjects, and is expressed as percent (mean ± SE) of control (group housed, saline, 0 mg/kg PHN) startle amplitude during leaders and trailers (Methods). The baseline startle amplitude of saline treated animals was 970 ± 130, 900 ± 70, and 990 ± 80 at 0, 10, and 30 mg/kg PHN, respectively. In PCPA treated rats, baseline startle amplitude was 870 ± 110, 800 ± 70, and 890 ± 100 at 0, 10, and 30 mg/kg PHN, respectively. Two-way ANOVA shows no significant main effect of PHN (p > 0.05) or PCPA (p > 0.05) and no significant interaction (p > 0.05). However, the matching within subjects was significant (p < 0.05), indicating that phenytoin did not alter baseline startle reactivity in these animals.
DISCUSSION
This study investigated the effect of PCPA on FPS in group and individually-housed rats, and the effect of the anticonvulsant phenytoin in PCPA-treated, individually-housed rats. The results show that FPS is significantly enhanced in PCPA-treated, individually housed animals and that phenytoin dose-dependently reduces FPS in PCPA-treated rats but has no effect in saline treated controls. Because both PCPA [21] and isolation-induced stress [22] lower brain levels of serotonin, together these data suggest that serotonergic modulation of neuronal excitability in brain structures comprising the fear circuitry is important in fear learning in vivo.
The neural systems mediating fear conditioning include limbic structures (amygdala, hypothalamus), as well as higher cortical areas and brainstem nuclei [5]. Much of the detailed analysis of fear circuitry has focused on the amygdala. Of the many nuclei located in the amygdala, the lateral (LA) and central (CeA) nuclei are implicated in having critical roles in acquisition and expression of fear learning. The LA serves as a sensory input area, receiving both cortical and thalamic sensory input, and synaptic plasticity in the LA underlies US-CS association [13–15]. The CeA is a final common output pathway to other structures involved in fear behavior. Information about the emotional salience of environmental stimuli (CS-US associations from the LA) is sent via the CeA to brain areas involved in fear responses. Output from the CeA to the periaqueductal gray (PAG) and nucleus reticularis pontis caudalis (RPC) mediate fear behaviors such as freezing and potentiated acoustic startle reflex, respectively. Additional CeA output to the lateral hypothalamus, dorsal vagal motor nucleus, parabrachial nucleus and bed nucleus of the stria terminalis mediate the autonomic and endocrine responses to environmental danger. Because of the nature of systemic drug administration used in the present study, these results shown here could be mediated by increased excitability in any or all of these structures. Given the importance the amygdala in fear learning and the RPC in acoustic startle reflex, it is proposed that these structures have critical roles in the PCPA-induced exaggerated fear behavior shown in this study. Although it remains to be shown that the amygdala is critically involved in the PCPA-induced exaggerated FPS response, we have previously observed epilepsy-like excitability in LA neurons in vitro from rats treated chronically with PCPA in vivo [40].
Previous studies have addressed the role of serotonin in fear learning. Increased serotonergic activity generally reduces fear behavior in animal models. Systemic administration of the 5-HT1A agonists buspirone, flesinoxan, or 8-hydroxy-2-(di-n-propylamino)tetralin (8-OH-DPAT) prior to testing dose-dependently reduces the expression of FPS in rodents [23–25]. Intra-amygdala infusion of the selective 5-HT1A receptor agonist flesinoxan blocks FPS in rats [26]. Similarly, intra-amygdala injection of the 5-HT1A agonist 8-OH-DPAT impairs inhibitory avoidance of the open arms of the elevated T-maze, a task representing conditioned fear [27]. Also, bilateral microinjection of the selective serotonin reuptake inhibitor, citalopram, into the amygdala before testing significantly reduces freezing behavior induced by conditioned fear stress [28]. In contrast, decreasing serotonergic activity often enhances fear behavior. Mice lacking the 5-HT3A receptor show enhanced fear conditioning as indexed by fear-induced freezing behavior [29]. Similarly, 5-HT1A receptor knockout (5-HT1AR KO) mice have been shown to be more fearful than wild type mice in tests of behavioral anxiety [19, 30, 31]. Also, 5-HT1B KO mice have been shown to be more reactive to nonentrained stimuli and more aggressive in the resident-intruder paradigm [20, 32]. The results of the present study show that PCPA-treated, individually-housed rats exhibit an exaggerated FPS response. These data are consistent with studies showing an inverse relationship between 5-HTR activation and fear learning. Altogether with our results, these studies further show that enhancing 5-HT decreases fear and reducing 5-HT increases fear behavior.
Phenytoin reduced FPS selectively in PCPA-treated, individually-housed rats as shown in Figure 2. Phenytoin is a widely used anticonvulsant which is effective in treating both human seizure disorders [33] and animal models of epilepsy [34]. While phenytoin is known to have several biological activities, its ability to limit the spread of seizure activity is thought to result primarily from its inhibitory effects on voltage-gated sodium channels. We and others [7, 8] have suggested that increasing excitability in limbic brain areas may sensitize these regions, resulting in dysfunctional emotional behavior. In support of this hypothesis, many antiepileptic drugs (AEDs) have been used to treat an array of conditions in people without epilepsy. Pain disorders, impulsive aggression, personality disorders, mood and anxiety disorders and schizophrenia have been treated with AEDs [9, 10, 33]. Recent studies show that phenytoin reduces symptoms of hyperarousal in patients with a DSM-IV diagnosis of PTSD [35], consistent with the reduction in startle potentiation shown here. Together, these studies show that many human pathologies involving emotional dysfunction are sensitive to treatment with anticonvulsants. The present finding that phenytoin reduces the exaggerated FPS response seen in PCPA-treated animals further suggests that epilepsy-like hyperexcitability in brain fear circuitry, such as the amygdala, is associated with emotional dysfunction.
In rats, the kindling model of epilepsy has been used to examine the relationship between neuronal hyperexcitability and abnormal emotional behavior. Rosen and colleagues [36] found that FPS was exaggerated in amygdala-kindled, but not in hippocampus-kindled, rats compared to sham-kindled controls. Partial amygdala kindling also renders aggressive cats less aggressive but more defensively fearful [37]. Full kindling is anxiogenic as measured with the elevated plus maze [38, 39]. The results of these experiments suggest that the kindling process alters the brain’s response to fear provoking stimuli. Phenytoin is an effective anticonvulsant in the kindling model [34]. In the present study, we show that phenytoin (30mg/kg) also reduces FPS in PCPA-treated, individually-housed rats. Together, these findings suggest that the neural mechanisms contributing to epilepsy may also contribute to abnormal emotional behaviors.
The present study supports the hypothesis that neuronal hyperexcitability in limbic areas may contribute to human emotional disorders [7]. PCPA treatment combined with the stress of individual housing significantly lowers brain serotonin levels [21, 22]. We have previously shown that phenytoin reduces aggressive behavior in animals with low 5-HT, and that individually housed, PCPA treated rats may be a useful model of human disorders characterized by impulsive aggressive acts [12]. Furthermore, systemic PCPA treatment is associated with epilepsy-like hyperexcitability in LA neurons recorded in vitro [40]. Here, we show that emotional learning, as indexed within the well-established fear potentiated startle paradigm, is also responsive to phenytoin treatment. The fear-related behaviors controlled by the amygdala require particularly fast neural processing to ensure the survival of an organism. The same adaptive neurochemical and neurobiological mechanisms that allow this rapid processing to occur may also predispose fear circuits to neuronal hyperexcitability. Also, 5-HT mediates an inhibitory tone in the amygdala, the putative location of synaptic plasticity during classical fear conditioning. The present data suggest that low 5-HT in brain structures such as the amygdala that are important in fear behavior results in a sub-seizure state of neuronal hyperexcitability that underlies an exaggerated FPS response. Also, individually-housed, PCPA-treated rats show pharmacological sensitivities similar to those of patients with mood and anxiety disorders. Thus, these animals may provide a useful model to investigate the neurobiological mechanisms involved in human conditions characterized by pathological fear. Understanding the mechanisms of cell excitability in the amygdala, and how they impact emotional behavior may improve treatment options for patients suffering from fear, anxiety, and other disorders involving amygdala dysfunction. Further studies will be necessary to define the critical role of the amygdala in PCPA-induced exaggerated fear behavior, and to elucidate the pharmacological and physiological mechanisms underlying low 5-HT-mediated hyperexcitability and emotional disturbance.
Acknowledgments
This project was supported by the National Institutes of Health (MH64639 to N.B.K) and the Baylor University Research Committee (to N.B.K.) C.R.H. is a 2006 Barry M. Goldwater scholar. The authors express their appreciation to Dr. Matthew Stanford and Trent Terrell for reading an early version of this paper and contributing helpful comments.
References
- 1.LaBar KS, Gatenby JC, Gore JC, LeDoux JE, Phelps EA. Human amygdala activation during conditioned fear acquisition and extinction: a mixed-trial fMRI study. Neuron. 1998;20(5):937–45. doi: 10.1016/s0896-6273(00)80475-4. [DOI] [PubMed] [Google Scholar]
- 2.Adolphs R, Tranel D, Damasio AR. The human amygdala in social judgment. Nature. 1998;393(6684):470–4. doi: 10.1038/30982. [DOI] [PubMed] [Google Scholar]
- 3.Bechara A, Tranel D, Damasio H, Adolphs R, Rockland C, Damasio AR. Double dissociation of conditioning and declarative knowledge relative to the amygdala and hippocampus in humans. Science. 1995;269(5227):1115–8. doi: 10.1126/science.7652558. [DOI] [PubMed] [Google Scholar]
- 4.Davidson RJ, Abercrombie H, Nitschke JB, Putnam K. Regional brain function, emotion and disorders of emotion. Curr Opin Neurobiol. 1999;9(2):228–34. doi: 10.1016/s0959-4388(99)80032-4. [DOI] [PubMed] [Google Scholar]
- 5.LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–84. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- 6.Gloor P. Role of the amygdala in temporal lobe epilepsy. In: Aggleton J, editor. The amygdala: Neurobiological aspects of emotion, memory and mental dysfunction. New York: Wiley-Liss; 1992. pp. 505–538. [Google Scholar]
- 7.Keele NB. The role of serotonin in impulsive and aggressive behaviors associated with epilepsy-like neuronal hyperexcitability in the amygdala. Epilepsy Behav. 2005;7(3):325–35. doi: 10.1016/j.yebeh.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 8.Rosen JB, Schulkin J. From normal fear to pathological anxiety. Psychol Rev. 1998;105(2):325–50. doi: 10.1037/0033-295x.105.2.325. [DOI] [PubMed] [Google Scholar]
- 9.Barratt ES, Stanford MS, Felthous AR, Kent TA. The effects of phenytoin on impulsive and premeditated aggression: a controlled study. J Clin Psychopharmacol. 1997;17(5):341–9. doi: 10.1097/00004714-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Stanford MS, Helfritz LE, Conklin SM, Villemarette-Pittman NR, Greve KW, Adams D, et al. A comparison of anticonvulsants in the treatment of impulsive aggression. Exp Clin Psychopharmacol. 2005;13(1):72–7. doi: 10.1037/1064-1297.13.1.72. [DOI] [PubMed] [Google Scholar]
- 11.Adamec R, Young B. Neuroplasticity in specific limbic system circuits may mediate specific kindling induced changes in animal affect-implications for understanding anxiety associated with epilepsy. Neurosci Biobehav Rev. 2000;24(7):705–23. doi: 10.1016/s0149-7634(00)00032-4. [DOI] [PubMed] [Google Scholar]
- 12.Keele NB. Phenytoin inhibits isolation-induced aggression specifically in rats with low serotonin. Neuroreport. 2001;12(6):1107–12. doi: 10.1097/00001756-200105080-00012. [DOI] [PubMed] [Google Scholar]
- 13.Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308(5718):83–8. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- 14.Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390(6660):604–7. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- 15.McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390(6660):607–11. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- 16.Rainnie DG. Serotonergic modulation of neurotransmission in the rat basolateral amygdala. J Neurophysiol. 1999;82(1):69–85. doi: 10.1152/jn.1999.82.1.69. [DOI] [PubMed] [Google Scholar]
- 17.Stutzmann GE, LeDoux JE. GABAergic antagonists block the inhibitory effects of serotonin in the lateral amygdala: a mechanism for modulation of sensory inputs related to fear conditioning. J Neurosci. 1999;19(11):RC8. doi: 10.1523/JNEUROSCI.19-11-j0005.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burghardt NS, Sullivan GM, McEwen BS, Gorman JM, LeDoux JE. The selective serotonin reuptake inhibitor citalopram increases fear after acute treatment but reduces fear with chronic treatment: a comparison with tianeptine. Biol Psychiatry. 2004;55(12):1171–8. doi: 10.1016/j.biopsych.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 19.Ramboz S, Oosting R, Amara DA, Kung HF, Blier P, Mendelsohn M, et al. Serotonin receptor 1A knockout: an animal model of anxiety-related disorder. Proc Natl Acad Sci U S A. 1998;95(24):14476–81. doi: 10.1073/pnas.95.24.14476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saudou F, Amara DA, Dierich A, LeMeur M, Ramboz S, Segu L, et al. Enhanced aggressive behavior in mice lacking 5-HT1B receptor. Science. 1994;265(5180):1875–8. doi: 10.1126/science.8091214. [DOI] [PubMed] [Google Scholar]
- 21.Valzelli L, Bernasconi S, Dalessandro M. Time-courses of p-CPA-induced depletion of brain serotonin and muricidal aggression in the rat. Pharmacol Res Commun. 1983;15(4):387–95. doi: 10.1016/s0031-6989(83)80048-4. [DOI] [PubMed] [Google Scholar]
- 22.Heidbreder CA, Weiss IC, Domeney AM, Pryce C, Homberg J, Hedou G, et al. Behavioral, neurochemical and endocrinological characterization of the early social isolation syndrome. Neuroscience. 2000;100(4):749–68. doi: 10.1016/s0306-4522(00)00336-5. [DOI] [PubMed] [Google Scholar]
- 23.Risbrough VB, Brodkin JD, Geyer MA. GABA-A and 5-HT1A receptor agonists block expression of fear-potentiated startle in mice. Neuropsychopharmacology. 2003;28(4):654–63. doi: 10.1038/sj.npp.1300079. [DOI] [PubMed] [Google Scholar]
- 24.Joordens RJ, Hijzen TH, Peeters BW, Olivier B. Fear-potentiated startle response is remarkably similar in two laboratories. Psychopharmacology (Berl) 1996;126(2):104–9. doi: 10.1007/BF02246344. [DOI] [PubMed] [Google Scholar]
- 25.Joordens RJ, Hijzen TH, Olivier B. The effects of 5-HT1A receptor agonists, 5-HT1A receptor antagonists and their interaction on the fear-potentiated startle response. Psychopharmacology (Berl) 1998;139(4):383–90. doi: 10.1007/s002130050729. [DOI] [PubMed] [Google Scholar]
- 26.Groenink L, Joordens RJ, Hijzen TH, Dirks A, Olivier B. Infusion of flesinoxan into the amygdala blocks the fear-potentiated startle. Neuroreport. 2000;11(10):2285–8. doi: 10.1097/00001756-200007140-00043. [DOI] [PubMed] [Google Scholar]
- 27.Zangrossi H, Jr, Viana MB, Graeff FG. Anxiolytic effect of intra-amygdala injection of midazolam and 8-hydroxy-2-(di-n-propylamino)tetralin in the elevated T-maze. Eur J Pharmacol. 1999;369(3):267–70. doi: 10.1016/s0014-2999(99)00075-8. [DOI] [PubMed] [Google Scholar]
- 28.Inoue T, Li XB, Abekawa T, Kitaichi Y, Izumi T, Nakagawa S, et al. Selective serotonin reuptake inhibitor reduces conditioned fear through its effect in the amygdala. Eur J Pharmacol. 2004;497(3):311–6. doi: 10.1016/j.ejphar.2004.06.061. [DOI] [PubMed] [Google Scholar]
- 29.Bhatnagar S, Sun LM, Raber J, Maren S, Julius D, Dallman MF. Changes in anxiety-related behaviors and hypothalamic-pituitary-adrenal activity in mice lacking the 5-HT-3A receptor. Physiol Behav. 2004;81(4):545–55. doi: 10.1016/j.physbeh.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 30.Hen R. Mean genes. Neuron. 1996;16(1):17–21. doi: 10.1016/s0896-6273(00)80019-7. [DOI] [PubMed] [Google Scholar]
- 31.Gross C, Santarelli L, Brunner D, Zhuang X, Hen R. Altered fear circuits in 5-HT(1A) receptor KO mice. Biol Psychiatry. 2000;48(12):1157–63. doi: 10.1016/s0006-3223(00)01041-6. [DOI] [PubMed] [Google Scholar]
- 32.Bouwknecht JA, Hijzen TH, van der Gugten J, Maes RA, Hen R, Olivier B. Absence of 5-HT(1B) receptors is associated with impaired impulse control in male 5-HT(1B) knockout mice. Biol Psychiatry. 2001;49(7):557–68. doi: 10.1016/s0006-3223(00)01018-0. [DOI] [PubMed] [Google Scholar]
- 33.Rogawski MA, Loscher W. The neurobiology of antiepileptic drugs for the treatment of nonepileptic conditions. Nat Med. 2004;10(7):685–92. doi: 10.1038/nm1074. [DOI] [PubMed] [Google Scholar]
- 34.McNamara JO, Rigsbee LC, Butler LS, Shin C. Intravenous phenytoin is an effective anticonvulsant in the kindling model. Ann Neurol. 1989;26(5):675–8. doi: 10.1002/ana.410260514. [DOI] [PubMed] [Google Scholar]
- 35.Douglas Bremner J, Mletzko T, Welter S, Siddiq S, Reed L, Williams C, et al. Treatment of posttraumatic stress disorder with phenytoin: an open-label pilot study. J Clin Psychiatry. 2004;65(11):1559–64. doi: 10.4088/jcp.v65n1120. [DOI] [PubMed] [Google Scholar]
- 36.Rosen JB, Hamerman E, Sitcoske M, Glowa JR, Schulkin J. Hyperexcitability: exaggerated fear-potentiated startle produced by partial amygdala kindling. Behav Neurosci. 1996;110(1):43–50. doi: 10.1037//0735-7044.110.1.43. [DOI] [PubMed] [Google Scholar]
- 37.Adamec RE, Stark-Adamec C. Partial kindling and emotional bias in the cat: lasting aftereffects of partial kindling of the ventral hippocampus. I. Behavioral changes. Behav Neural Biol. 1983;38(2):205–22. doi: 10.1016/s0163-1047(83)90212-1. [DOI] [PubMed] [Google Scholar]
- 38.Adamec RE, Morgan HD. The effect of kindling of different nuclei in the left and right amygdala on anxiety in the rat. Physiol Behav. 1994;55(1):1–12. doi: 10.1016/0031-9384(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 39.Nieminen SA, Sirvio J, Teittinen K, Pitkanen A, Airaksinen MM, Riekkinen P. Amygdala kindling increased fear-response, but did not impair spatial memory in rats. Physiol Behav. 1992;51(4):845–9. doi: 10.1016/0031-9384(92)90125-l. [DOI] [PubMed] [Google Scholar]
- 40.Keele NB, Randall DR. Altered modulation of excitatory neurotransmission in the amygdala by serotonin in an animal model of impulsive-aggression. Ann NY Acad Sci. 2003;985:528–532. [Google Scholar]