

Abstract
Chronic ethanol consumption disrupts G protein-dependent signaling pathways in rat adipocytes. Because lipolysis in adipocytes is regulated by G protein-mediated cAMP signal transduction, we hypothesized that cAMP-regulated lipolysis may be vulnerable to long-term ethanol exposure. Male Wistar rats were fed a liquid diet containing ethanol as 35% of total calories or pair-fed a control diet that isocalorically substituted maltose dextrins for ethanol for 4 wk. Lipolysis was measured by glycerol release over 1 h with or without agonists in adipocytes isolated from epididymal fat. Chronic ethanol feeding decreased β-adrenergic receptor-stimulated lipolysis, but had no effect on basal lipolysis. In response to β-adrenergic activation, the early peak of cAMP accumulation was suppressed after ethanol feeding, although the basal cAMP concentration in adipocytes did not differ between pair- and ethanol-fed rats. The suppression in cAMP accumulation caused by ethanol feeding was associated with increased activity of phosphodiesterase 4. Chronic ethanol feeding also decreased β-adrenergic receptor-stimulated protein kinase A activation and phosphorylation of its downstream proteins, perilipin A and hormone-sensitive lipase, the primary lipase-mediating lipolysis. In conclusion, these data suggest that chronic ethanol feeding increased phosphodiesterase 4 activity in adipocytes, resulting in decreased accumulation of cAMP in response to β-adrenergic activation and a suppression of β-adrenergic stimulation of lipolysis.
CHRONIC HEAVY ETHANOL consumption, which is associated with hepatic, pancreatic, and myocardial diseases, as well as insulin resistance and type 2 diabetes, is one of the most common health problems in the United States with high morbidity and mortality (1). Although the exact mechanisms by which chronic ethanol contributes to these pathophysiological conditions are unknown, the disruption of lipid homeostasis by ethanol is a likely contributor to disease progression. For example, chronic ethanol administration causes excess accumulation of fat in liver with the eventual development of hepatic steatosis in both humans and animal models (2). Chronic ethanol feeding to rats induces hyperlipidemia coupled with elevated plasma cholesterol, triglyceride, and free fatty acid concentrations (3). Adipose tissue, the biggest storage pool of lipids, plays an important role for maintaining whole-body lipid homeostasis. However, the effects of chronic ethanol feeding on lipid metabolism in adipose tissue are unknown.
Lipolysis is defined as hydrolysis of triglycerides, the major form of stored energy in adipose tissue. Tight regulation of lipolysis is critical because mobilization of free fatty acid and glycerol from adipose tissue supplies other tissues with metabolites and energy substrates during fasting and in response to infection and inflammation (4). Moreover, free fatty acids released during lipolysis, acting as ligands for transcription factors, regulate expression of genes involved in lipid and energy metabolism during fasting and stress (5). Lipolysis in adipocytes is regulated by a number of hormones, such as ACTH, epinephrine, norepinephrine, and insulin (6). Catecholamine-induced lipolysis is well characterized, initiated by stimulation of β-adrenergic receptors, which are coupled to activation of adenylyl cyclase by the heterotrimeric Gαs protein, which in turn converts ATP to cAMP. cAMP-dependent protein kinase A (PKA) then phosphorylates two main targets, hormone-sensitive lipase (HSL), the primary lipase responsible for hydrolysis of triglycerides, as well as perilipin A, the coating protein of lipid droplets. In unstimulated adipocytes, perilipin A functions as a barrier to lipolysis because of its location on the surface of lipid droplets, preventing the interaction of HSL with the lipid droplet (7). In response to β-adrenergic activation, phosphorylated perilipin A undergoes a conformational change, which is essential for proper translocation of HSL from the cytosol to the surface of lipid droplets and subsequent attachment to triglycerides, leading to initiation of triglyceride hydrolysis (7, 8).
Chronic ethanol exposure disrupts receptor-activated signal transduction in a variety of cell types (9). One target of ethanol is the G protein-mediated signaling pathways (9). The effect of ethanol on G protein-dependent responses is cell type specific. In adipocytes, ethanol feeding for 4 wk causes a sensitization to stimulation by the β-adrenergic agonist isoproterenol as well as an increase in the quantity of immunoreactive Gαs protein (10).
Because lipolysis in adipocytes is regulated by a G protein-dependent signaling pathway known to be affected by chronic ethanol, we hypothesized that β-adrenergic receptor regulation of lipolysis may also be susceptible to long-term ethanol exposure. In the present work, we report that 4-wk ethanol feeding to rats suppressed β-adrenergic receptor-mediated activation of lipolysis in adipocytes isolated from epididymal fat. The suppression of lipolysis in response to β-adrenergic activation was associated with a reduction of intracellular cAMP accumulation, PKA activation, as well as phosphorylation of perilipin A and HSL.
Materials and Methods
Materials
Male Wistar rats (150–160 g) were purchased from Harlan Sprague Dawley (Indianapolis, IN). The Lieber-DeCarli high-fat ethanol diet was from Dyets (Bethlehem, PA). Antibodies were obtained from the following sources: anti- phosphodiesterase 4 (PDE4) and anti-PDE4B, from Abcam Inc. (Cambridge, MA); anti-phospho-(Ser/Thr) PKA substrate, from Cell Signaling Technology (Beverly, MA); anti-perilipin A/B, from Research Diagnostics, Inc. (Flanders, NJ); anti-caveolin, from BD Transduction Laboratories (San Jose, CA); and anti-ERK, from Upstate (Charlottesville, VA). Maltose dextrins were purchased from BioServ (French-town, NJ); Complete, EDTA-free protease inhibitor cocktail tablets and enhanced chemiluminescence reagents, from Roche (Indianapolis, IN); Calyculin A, cilostamide, and Ro20-1724, a PDE4 selective inhibitor, from BIOMOL (Plymouth Meeting, PA). cAMP enzyme immunoassay Biotrak (EIA) system was from Amersham Pharmacia Biotech (Piscat-away, NJ); PKA assay kit was purchased from Upstate; [γ-32P]ATP was from PerkinElmer Life Sciences (Boston, MA); [3H]cAMP was from Amersham Pharmacia Biotech; and all other reagents were from Sigma (St. Louis, MO).
Animal care and feeding
Rats were allowed ad libitum access to the Lieber-DeCarli ethanol diet or pair-fed a control diet as previously described (11). Briefly, rats were housed in individual wire-bottom cages under controlled temperature and humidity with 12-h light, 12-h dark cycle. After arrival, rats were acclimatized for 3 d on rat chow and water. The rats were then matched by weight and randomly assigned to the ethanol-fed or pair-fed groups, and allowed free access to control liquid diet for 2 d (12). The ethanol-fed group had ad libitum access to a liquid diet with 17% total calories as ethanol for 2 d and then 35% total calories as ethanol for 4 wk. Controls were pair-fed a liquid diet that was identical to the ethanol diet, except that maltose dextrins were isocalorically substituted for ethanol. Pair-fed rats were given the same amount of food as their ethanol-fed counter-parts consumed in the preceding 24 h. All procedures involving animals were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University.
Isolation of adipocytes
After 4 wk of feeding, rats were anesthetized by ip injection of 0.075 ml for ethanol-fed rats or 0.12 ml for pair-fed rats per 100 g body weight of a cocktail containing 10 mg/ml acepromazine, 100 mg/ml ketamine, and 20 mg/ml xylazine. The use of a lower dose of anesthetic for ethanol-fed rats was due to an increased sensitivity of rats to the anesthetic cocktail after ethanol feeding. These concentrations of ketamine and xylazine have either no effect or an equivalent effect on lipolysis (13, 14). Under anesthesia, rat epididymal fat pads were removed. Epididymal fat pads were selected as the depot for this study because epididymal adipose tissue is more sensitive to isoproterenol stimulation of lipolysis than sc fat (15). Adipocytes were then isolated by collagenase digestion following the method of Rodbell (16) as modified by Honnor et al. (17), and as described before (10). Briefly, fat pads were weighed, washed twice with Hanks' balanced salt solution containing 25 mm HEPES, minced by scissors, and incubated at 37 C in a shaking water bath [80 revolutions per min (rpm)] for 50 min in 10 ml Hanks' balanced salt solution containing 10 mg collagenase (type II), 10 mg/ml BSA (RIA grade), 25 mm HEPES, and 200 nm adenosine, pH 7.4. Adipocytes were then filtered through a 250-μm nylon mesh and washed twice with 15 ml PBS containing 1 mm sodium pyruvate and 200 nm adenosine, pH 7.4 (wash buffer). Adipocytes were then counted, and cell concentrations adjusted to 1 × 106 cells/ml.
Lipolysis assay and cAMP concentration
Adipocytes were isolated as described above, except that adenosine was not added during digestion and washing steps unless indicated in the figure legend. In addition, 1 mg/ml BSA (RIA grade) was included in wash buffer. Then 200-μl aliquots of cells (1 × 106 cells/ml) were placed into 5-ml polypropylene tubes, and a number of agents were added: 1) isoproterenol (10−10 to 10−5 m) with or without preincubation of cells with 10 μm Ro20-1724 for 3 min; 2) adenosine deaminase (ADA) (0.4 U/ml) and (−)-N6-(2-phenylisopropyl)adenosine (R-PIA) (10 nm); and 3) dibutyryl-cAMP (0.05–1 mm). Adipocytes were incubated for 1 h at 37 C in a shaking water bath (100 rpm), and lipolysis was measured as glycerol release (18). After incubation, cells were centrifuged briefly, and the cell medium was collected for analysis of glycerol concentration using glycerol reagent (GPO trinder reagent) in a flat-bottom 96-well plate. OD at 540 nm was measured on a Maxline microplate reader. Control experiments showed that glycerol release from isolated adipocytes was dependent on incubation time (from 20 to 90 min) and cell concentration (from 0.5 to 1.8 × 106 cells/ml) in cells from both pair- and ethanol-fed rats. For cAMP assay, 100 μl of cell suspension was removed to a 96-well plate containing 10 μl of 2% Nonidet P-40 in 1 n HCl after 0.5–15 min of incubation. The samples were frozen at −20 C until analysis of cAMP concentration by ELISA using the cAMP enzyme immunoassay Biotrak (EIA) system.
PDE activity
One-milliliter aliquots of isolated adipocytes were incubated with or without 1 μm isoproterenol in the presence of 1 U/ml ADA for 5 or 10 min at 37 C in a shaking water bath (100 rpm) and then immediately placed on ice and homogenized in 500 μl ice cold TSE buffer (20 mm Tris, 255 mm sucrose, 1 mm EDTA, 40 μl/ml protease inhibitor cocktail, 10 μl/ml activated sodium vanadate) with 60 nm calyculin A. Homogenates were then spun briefly, and the fluid fraction was collected. PDE activity was assayed by the method of Ahmad et al. (19). Briefly, 20-μg samples were incubated with 0.1 μmol/liter [3H]cAMP for 10 min at 30 C in the presence or absence of 1 μm cilostamide (PDE3B inhibitor) or 10 μm Ro20-1724 (PDE4 inhibitor), and the formed [3H]AMP was degraded to [3H]adenosine by 0.3 mg/ml 5′-nucleotidase of rattlesnake venom. [3H]Adenosine was separated from the reaction mixture by chromatography on QAE Sephadex A-25 columns, and the radioactivity in the eluate was measured by liquid scintillation counting. PDE3B activity was expressed as the difference between activity with and without cilostamide, and PDE4 activity was expressed as the difference between activity with and without Ro20-1724.
PKA activity
PKA activity was measured using the Kemptide assay (20). One-milliliter aliquots of isolated adipocytes were treated with or without 1 μm isoproterenol for 2–10 min in the presence of 1 U/ml ADA, and cell homogenates were prepared in TSE buffer as described above. The homogenates were then centrifuged at 5500 × g for 10 min at 4 C, and the fluid fraction was collected. Protein content was measured, and 20 μg protein was used for assessing PKA activity. The activity of PKA was measured over 10 min in the presence of 0.33 μm protein kinase C inhibitor peptide and 3.3 μm calmodulin-dependent protein kinase (CaMK) inhibitor peptide. Maximal PKA activity was assessed by the addition of 1.67 μm cAMP.
Western blotting
Quantification of immunoreactive PDE4
One-milliliter aliquots of isolated adipocytes, treated with 1 U/ml ADA, were homogenized in the modified radioimmunoprecipitation buffer [50 mm Tris (pH 7.4), 1% Nonidet P-40, 150 mm NaCl, 1 mm EDTA, 40 μl/ml protease inhibitor cocktail, 1 mm Na3VO4, 1 mm NaF]. After removal of the fat layer by centrifugation at 4000 × g for 3 min, the fluid fraction was prepared in the sodium dodecyl sulfate (SDS) sample buffer and applied to a 6% SDS-polyacrylamide gel. Immunoreactive PDE4 isoforms were probed using both PDE4 (detecting all known PDE4 A and D variants) and PDE4B (detecting all known PDE4B proteins) antibodies.
Quantification of immunoreactive perilipin A/B and phospho-perilipin A and HSL
One-milliliter aliquots of isolated adipocytes were treated with or without 1 μm isoproterenol or 0.5 mm dibutyryl-cAMP for 5 or 10 min in the presence of 1 U/ml ADA, and cell homogenates were prepared in TSE buffer. The homogenates were then centrifuged at 5500 × g for 10 min at 4 C. After removal of the fluid fraction, 1 ml chloroform/methanol (1:1) mixture was added to the fat layer. Samples were vortexed thoroughly and spun for 10 min at 16,000 × g at 4 C to precipitate fat layer-associated proteins. Protein pellets were then dissolved in 1% SDS, and protein concentration was measured. Both cell homogenates and fat layer-associated protein samples were prepared in SDS sample buffer and applied to an 8% SDS-polyacrylamide gel. Proteins were then transferred to polyvinylidene fluoride membrane for Western blotting. Total perilipin A/B was probed using an anti-perilipin A/B antibody. Phospho-perilipin A and phospho-HSL were probed with a phospho(Ser/Thr) PKA substrate antibody. Protein expression of ERK1 in adipocyte homogenates was not different between pair-fed (2.15 ± 0.3 arbitrary units of density, n = 4) and ethanol-fed rats (1.93 ± 0.3 arbitrary units of density, n = 4); therefore, ERK1 was used as a loading control of the cell homogenate samples (see Figs. 7 and 8). ERK1 was probed using an anti-ERK antibody. Caveolin is found in lipid droplets (21); therefore, we used caveolin as a control for equal protein extraction from fat samples. Caveolin was probed with anticaveolin in a 12% duplicate gel of fat layer-associated protein samples.
Fig. 7.
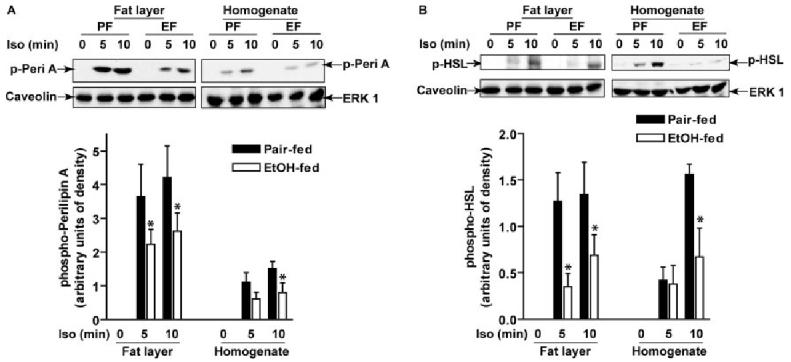
Chronic ethanol feeding reduced β-adrenergic receptor-stimulated phosphorylation of perilipin A and HSL. Adipocytes isolated from pair- and ethanol-fed rats were treated with or without 1 μm isoproterenol for 5 or 10 min, and fat layer-associated protein samples or whole cell homogenates were prepared as indicated. Phospho-perilipin A (p-Peri A; panel A) and phospho-HSL (p-HSL; panel B) were immunoblotted using a phospho-(Ser/Thr) PKA substrate antibody. A, A representative immunoblot of phospho-perilipin A is shown; the image shown for homogenate is from a longer exposure compared with the fat layer. With equal exposure times, phospho-perilipin A was 3.8 arbitrary units of density in fat layer compared with 1 in homogenate. B, A representative immunoblot for phospho-HSL is shown with equal exposure times for the fat layer and homogenate. Caveolin was used as a control for equal protein extraction from fat samples; ERK1 was used as a loading control in the cell homogenates. Values represent means ±sem (n = 4–6). *, P < 0.05, pair-fed vs. EtOH-fed. PF, Pair-fed; EF, ethanol-fed.
Fig. 8.
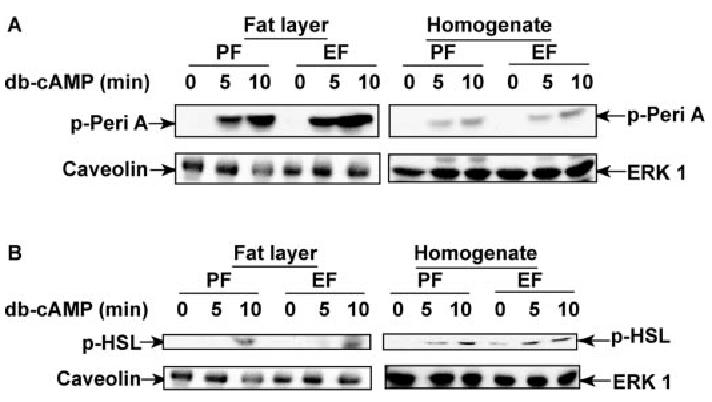
Chronic ethanol feeding did not affect dibutyryl-cAMP-stimulated phosphorylation of perilipin A or HSL. Adipocytes isolated from pair- and ethanol-fed rats were treated with or without 0.5 mm dibutyryl-cAMP (db-cAMP) for 5 or 10 min, and fat layer-associated protein samples or whole cell homogenates were prepared as indicated. Phospho-perilipin A (p-Peri A; panel A) and phospho-HSL (pHSL; panel B) were immunoblotted using a phospho-(Ser/Thr) PKA substrate antibody. A representative immunoblot from four experiments is shown. PF, Pair-fed; EF, ethanol-fed.
Statistical analyses
Data are expressed as means ± sem. Dose response curves of adipocytes to isoproterenol were estimated by nonlinear regression (GraphPad Prism 4; GraphPad Software, Inc., San Diego, CA). Statistical analyses were performed using the general linear model procedure on SAS for personal computers (SAS Institute, Cary, NC). Differences between groups were determined by least square means.
Results
We have previously reported that chronic ethanol feeding increases Gαs expression associated with an increase in β-adrenergic receptor activation in adipocytes (10). Therefore, we first asked if chronic ethanol feeding increased lipolysis in response to β-adrenergic receptor activation. Basal rates of lipolysis did not differ between adipocytes isolated from pair-fed (0.13 ± 0.02 μmol/106 cells; n = 8) and ethanol-fed (0.14 ± 0.02 μmol/106 cells; n = 8) rats. Lipolysis in adipocytes from both pair- and ethanol-fed rats displayed a sigmoidal dose response to isoproterenol (Fig. 1A). EC50 values for isoproterenol-stimulated lipolysis were not different between pair-fed (94.0 nm) and ethanol-fed (81.0 nm) rats. However, maximal stimulation of lipolysis by isoproterenol in adipocytes isolated from ethanol-fed rats was reduced to 29% of that observed in pair-fed rats (Fig. 1A). This attenuation in isoproterenol-stimulated lipolysis was not influenced by extracellular adenosine because, in the presence of ADA (0.4 U/ml) and R-PIA (an A1 receptor agonist, 10 nm), isoproterenol (1 μm)-stimulated lipolysis was still lower in adipocytes from ethanol-fed rats compared with pair-fed rats (Fig. 1B). Additionally, reduced isoproterenol-stimulated lipolysis was not due to a reduced total capacity of lipolysis in adipocytes, because maximal rate of lipolysis, measured in the presence of ADA (22), did not differ between adipocytes from pair-fed (6.6 ± 0.9 μmol/106 cells; n = 4) and ethanol-fed (6.2 ± 0.7 μmol/106 cells; n = 4) rats.
Fig. 1.
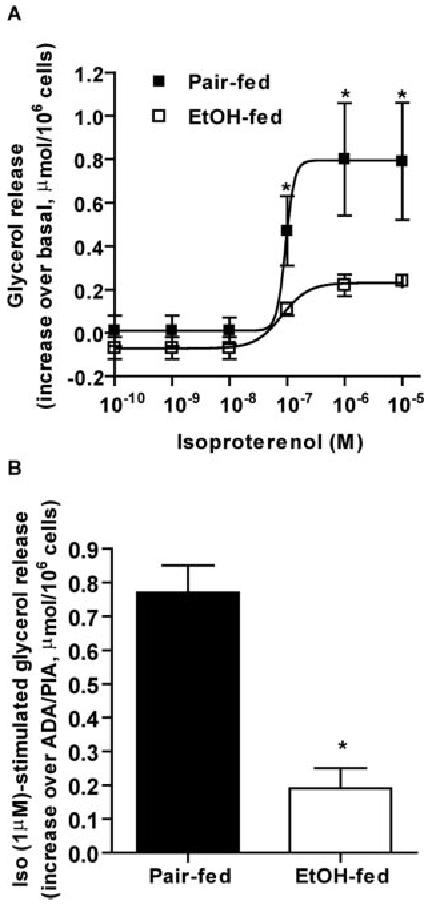
Chronic ethanol feeding decreased β-adrenergic receptor-stimulated lipolysis in adipocytes isolated from epididymal fat. A, Adipocytes isolated from pair- and ethanol-fed rats were treated with or without increasing concentrations of isoproterenol. Basal lipolysis, measured as glycerol release over 1 h, did not differ in adipocytes from pair-fed (0.13 ± 0.02 μmol/106 cells; n = 8) and ethanol-fed (0.14 ± 0.02 μmol/106 cells; n = 8) rats. Values are the increase in glycerol release in response to isoproterenol over basal. Values represent means ± sem (n = 3) and were graphed using non-linear regression. B, Lipolysis in adipocytes was measured as described in Materials and Methods except that adenosine was included in the digestion and washing steps during adipocyte isolation. Adipocytes were treated with or without 1 μm isoproterenol in the presence of ADA (0.4 U/ml) and R-PIA (an A1 receptor agonist, 10 nm). Values are the increase in glycerol release in response to isoproterenol over release in the presence of ADA and R-PIA, and represent means ± sem (n = 4). *,P < 0.05, pair-fed vs. EtOH-fed. Iso, Isoproterenol; EtOH, ethanol.
Because cAMP is the key signal to increase HSL-dependent lipolysis, we asked whether the suppression of β-adrenergic receptor-stimulated lipolysis after chronic ethanol feeding was associated with decreased cAMP accumulation. Basal cAMP concentration in adipocytes did not differ between pair- and ethanol-fed rats (Fig. 2A). In response to isoproterenol, cAMP concentration increased rapidly in adipocytes from pair-fed rats, with a peak at 1 min, followed by a rapid decline. In contrast, this peak of isoproterenol-stimulated cAMP accumulation was suppressed after ethanol feeding. After stimulation by isoproterenol for 5–15 min, cAMP concentration remained elevated over basal but was not different between pair- and ethanol-fed rats (Fig. 2A). cAMP accumulation is a balance between its synthesis from ATP by activated adenylyl cyclase and its degradation to AMP by activated PDEs (6). The reduction of chronic ethanol in the early peak of β-adrenergic receptor-dependent cAMP accumulation could be due to either a decrease in cAMP synthesis or an increase in cAMP degradation. To measure cAMP synthesis independent of degradation, adipocytes were pretreated with Ro20-1724, a PDE4-selective inhibitor. In the presence of Ro20-1724, β-adrenergic receptor-stimulated cAMP production was higher in adipocytes from both pair- and ethanol-fed rats compared with the accumulation observed in the absence of Ro20-1724 (Fig. 2, B vs. A). In contrast to the inhibition of the early peak of cAMP in the absence of Ro20-1724, cAMP production in the presence of Ro20-1724 was higher in ethanol-fed rats after isoproterenol stimulation for 5–15 min compared with pair-fed rats (Fig. 2B). These data suggest that chronic ethanol feeding decreased cAMP accumulation primarily through increased cAMP degradation via PDE4, rather than decreased cAMP synthesis.
Fig. 2.
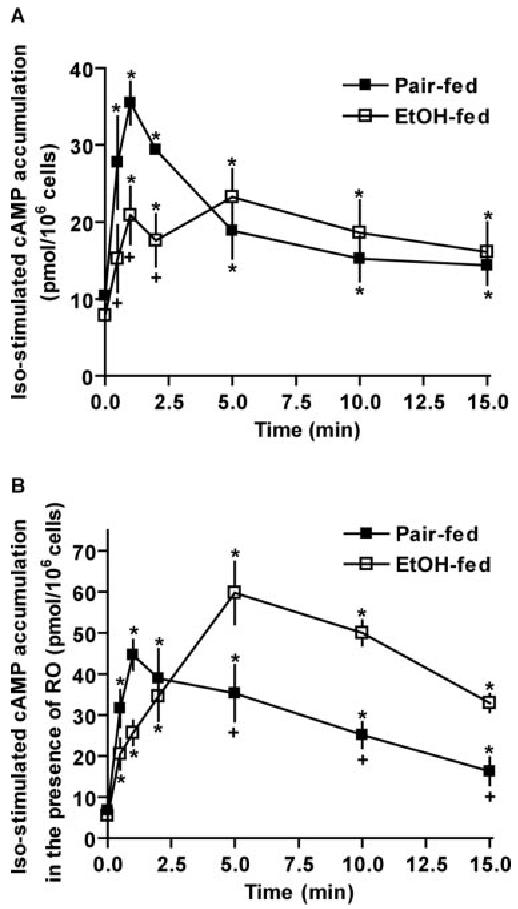
Chronic ethanol feeding suppressed β-adrenergic receptor-stimulated cAMP accumulation by increasing cAMP degradation via PDE4. Adipocytes isolated from pair- and ethanol-fed rats were pre-treated without (A) or with (B) 10 μm Ro20-1724 (RO) for 3 min, then 1 μm isoproterenol was added except the basal samples, and incubation was continued for another 0.5–15 min. Intracellular cAMP concentration was determined by cAMP enzyme immunoassay Biotrak system. Data are means ± sem (n = 3–7). *, P < 0.05 compared with 0 min within dietary group; +, P < 0.05 pair-fed vs. EtOH-fed. Iso, Isoproterenol; EtOH, ethanol.
To further clarify the involvement of PDE4 in chronic ethanol-reduced cAMP accumulation, PDE activity was measured. Adipocytes predominantly express two PDE isoforms, PDE3B and PDE4 (23, 24). Basal activity of PDE4 was significantly higher after chronic ethanol feeding compared with pair feeding (Fig. 3A). Isoproterenol increased PDE4 activity in cell homogenates from pair-fed rats, but did not further increase PDE4 activity in cell homogenates from ethanol-fed rats (Fig. 3A). In contrast to the increase in basal activity of PDE4, chronic ethanol feeding had no effect on either basal or isoproterenol-stimulated activation of PDE3B (Fig. 3B).
Fig. 3.
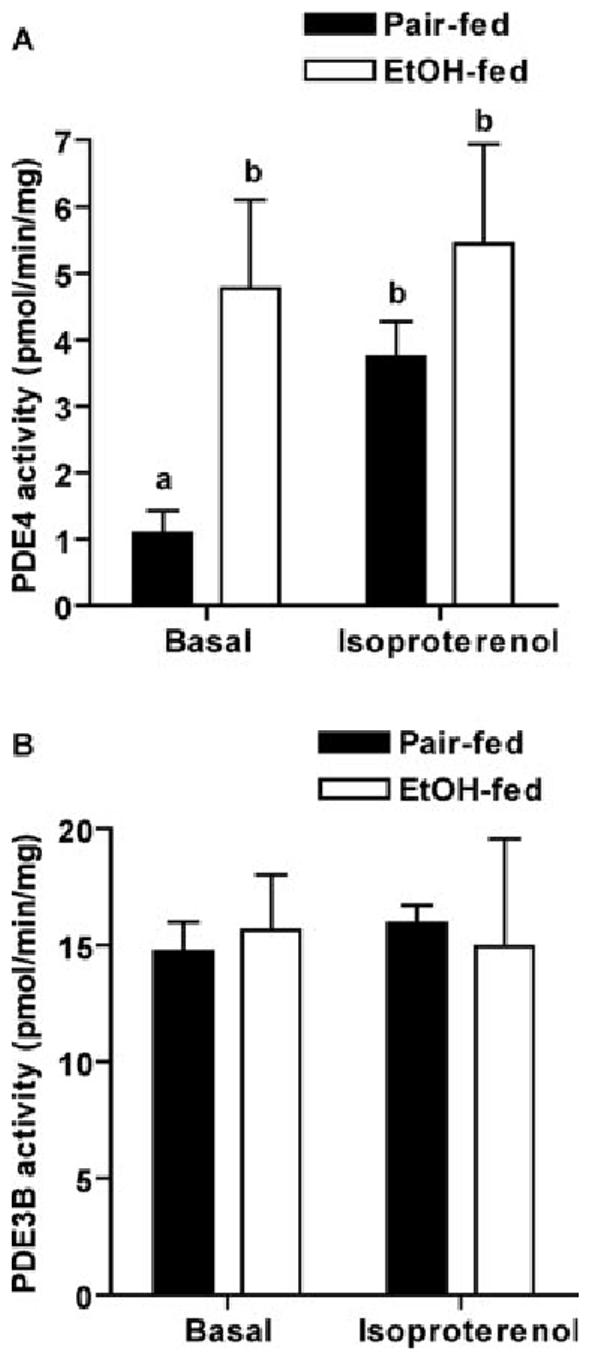
Chronic ethanol feeding increased basal activity of PDE4. Adipocytes isolated from pair- and ethanol-fed rats were incubated with or without 1 μm isoproterenol for 5 min. Cell homogenates were used to analyze PDE activity as described in Materials and Methods. Data represent means ± sem (n = 8 for basal samples or 4 for isoproterenol-treated samples). Values with different letters are significantly different (P < 0.05). EtOH, Ethanol.
PDE4 is a multigene family. Four genes (PDE4A, PDE4B, PDE4C, and PDE4D) encode over 18 different PDE4 isoforms (25). Because chronic ethanol feeding increased the activity of PDE4, we asked whether this increase was due to the up-regulation of expression of one or more of PDE4 family members. Western blot analysis of PDE4 using a PDE4 antibody (detecting all known PDE4 A and D variants) and a PDE4B antibody (detecting all known PDE4B variants), showed that the quantity of immunoreactive PDE4A, PDE4B, or PDE4D isoforms was not increased by chronic ethanol; indeed, the expression of some isoforms was even decreased in adipocytes from ethanol-fed rats compared with pair-fed rats (Fig. 4).
Fig. 4.
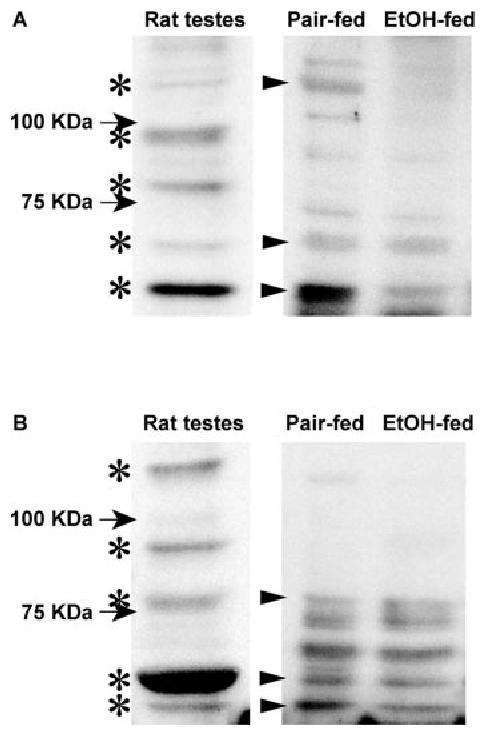
Chronic ethanol (EtOH) feeding did not increase the quantity of immunoreactive PDE4A, PDE4B, or PDE4D isoforms. Adipocytes isolated from pair- and ethanol-fed rats were homogenized in radio-immunoprecipitation buffer. After removal of the fat layer by centrifugation at 4000 × g for 3 min, the fluid fraction was prepared in SDS sample buffer and applied to a 6% SDS-polyacrylamide gel. PDE4 was immunoblotted using a PDE4 antibody (detecting all known PDE4 A and D variants) (A) and a PDE4B antibody (detecting all known PDE4B variants) (B). Rat testes were used as positive controls. A representative immunoblot from six experiments is shown. Asterisks indicate the immunoreactive PDE4 isoforms detected in rat testes, and the arrowheads indicate the corresponding bands detected in rat adipocytes.
Because chronic ethanol feeding decreased β-adrenergic receptor-stimulated intracellular cAMP accumulation, we next asked whether the activity of PKA, a downstream target of cAMP, was also impaired by chronic ethanol feeding. Basal activity of PKA was not affected by chronic ethanol feeding (Fig. 5, A and B). Isoproterenol dose-dependently increased PKA activity in cell homogenates from pair-fed rats. However, PKA activation in response to isoproterenol was inhibited by chronic ethanol feeding (Fig. 5A). In the presence of 1.67 μm cAMP, maximal activation of PKA did not differ between pair feeding and ethanol feeding (Fig. 5B).
Fig. 5.
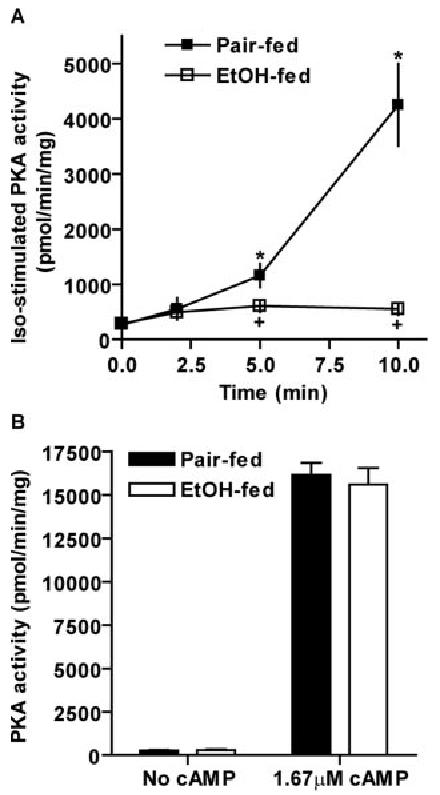
Chronic ethanol (EtOH) feeding decreased β-adrenergic receptor-stimulated PKA activity, but not maximal PKA activity. Adipocytes isolated from pair- and ethanol-fed rats were incubated with (A) or without (B) 1 μm isoproterenol (Iso) for 0–10 min. Cell homogenates were used to analyze PKA activity as described in Materials and Methods. Maximal PKA activity was assessed by the addition of 1.67 μm cAMP (B). Data represent means ± sem (n = 3– 8). *, P < 0.05 compared with own basal; +, P < 0.05, pair-fed vs. EtOH-fed.
Upon activation, PKA phosphorylates multiple substrates in adipocytes, but two particular substrates have emerged as important in the regulation of lipolysis, perilipin A and HSL (6). Because chronic ethanol feeding decreased isoproterenol-stimulated PKA activity, we next investigated whether isoproterenol-stimulated phosphorylation of perilipin A and/or HSL was also decreased by ethanol. To investigate further the subcellular distribution of these proteins, we isolated the fat layer from cell homogenates and extracted fat layer-associated proteins. Using caveolin, a protein resident in the lipid droplet as a marker, we found equivalent recovery of lipid droplet protein from adipocytes from both pair- and ethanol-fed rats (see Fig. 7). Adipocytes express two forms of perilipin, A and B. Western blot analysis showed that both forms of perilipin were predominantly localized to the fat layer and their expression was not affected by chronic ethanol feeding (perilipin A, 2.42 ± 0.53 arbitrary units of density in pair-fed, 2.13 ± 0.47 in ethanol-fed; perilipin B, 0.40 ± 0.15 arbitrary units of density in pair-fed, 0.35 ± 0.14 in ethanol-fed; n = 3).
To assess the phosphorylation of perilipin A and HSL, Western blot analysis with a phospho-(Ser/Thr) PKA substrate antibody was used. Phosphorylation of a 62-kDa protein, which migrated coincident with immunoreactive perilipin A, was identified as phospho-perilipin A (Fig. 6A). Phosphorylation of an 83-kDa protein, the exclusive protein in the molecular mass range of 75 to 200 kDa detected by phospho-(Ser/Thr) PKA substrate antibody in adipocyte homogenates, which had the same molecular mass as HSL, was identified as phospho-HSL (Fig. 6B). Phospho-perilipin A was not detectable at baseline in either the fat layer or cell homogenates from pair- and ethanol-fed rats (Fig. 7A). However, treatment of adipocytes from pair- and ethanol-fed rats with isoproterenol for 5 or 10 min induced a strong and rapid phosphorylation of perilipin A; this response was more pronounced in the fat layer (Fig. 7A). Concomitant with decreased isoproterenol-stimulated PKA activation, phosphorylation of perilipin A was inhibited by chronic ethanol feeding in the fat layer (Fig. 7A). Similarly, phosphorylated HSL was not detectable in nonstimulated cells but was induced in response to isoproterenol (Fig. 7B). Phosphorylation of HSL was decreased in both the fat layer and cell homogenates from ethanol-fed rats compared with pair-fed rats after stimulation with isoproterenol. In contrast to the abundance of phospho-perilipin A in the fat layer, phospho-HSL was distributed in both the fat layer and nonfat fraction (Fig. 7B).
Fig. 6.
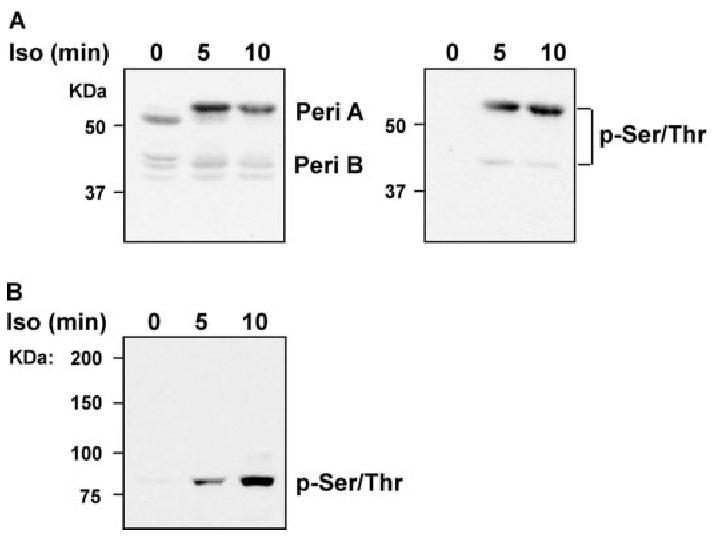
Phosphorylation of perilipin A and HSL were detected by a phospho-(Ser/Thr) PKA substrate antibody. Adipocytes isolated from pair-fed rats were treated with or without 1 μm isoproterenol for 5 or 10 min, and fat layer-associated protein samples or whole cell homogenates were prepared as indicated. A, Fat layer-associated protein samples were subjected to SDS-PAGE and immunoblotted with antibodies against perilipin A/B (Peri A/B) or phospho-(Ser/Thr) PKA substrate (p-Ser/Thr). B, Cell homogenates were subjected to SDS-PAGE and immunoblotted with phospho-(Ser/Thr) PKA substrate antibody.
Decreased β-adrenergic receptor-stimulated phosphorylation of perilipin A and HSL after ethanol feeding could result from decreased cAMP accumulation or a combination of decreased cAMP accumulation and a direct impact of ethanol on protein phosphorylation. To ask whether ethanol directly impaired phosphorylation of these proteins, independent of changes in cAMP concentration, we determined phosphorylation of perilipin A and HSL in the presence of dibutyryl-cAMP, a PDE-resistant cAMP analog. Treatment of adipocytes with dibutyryl-cAMP for 5 or 10 min stimulated phosphorylation of both perilipin A and HSL (Fig. 8). In contrast to the inhibition of isoproterenol-stimulated phosphorylation of perilipin A and HSL after chronic ethanol feeding, dibutyryl-cAMP-stimulated phosphorylation of perilipin A and HSL did not differ between pair- and ethanol-fed rats in either the fat layer or cell homogenates (Fig. 8).
Because treatment of adipocytes with dibutyryl-cAMP restored ethanol-reduced phosphorylation of proteins, we then asked whether suppressed lipolysis could also be restored by the addition of dibutyryl-cAMP. Treatment of adipocytes with dibutyryl-cAMP increased lipolysis in adipocytes isolated from both pair- and ethanol-fed rats in a dose-dependent manner (Fig. 9A). However, dibutyryl-cAMP-stimulated lipolysis in adipocytes from ethanol-fed rats was consistently lower than that in adipocytes from pair-fed rats at all doses tested (Fig. 9A). Furthermore, because decreased cAMP accumulation after ethanol was restored in the presence of PDE4 inhibitor, Ro20-1724 (Fig. 2B), we examined whether the decrease in isoproterenol-stimulated lipolysis after ethanol feeding was also restored when PDE4 activity was inhibited. Although pretreatment of adipocytes with Ro20-1724 did not change isoproterenol-stimulated lipolysis in adipocytes from pair-fed rats, it elevated lipolysis in adipocytes from ethanol-fed rats. However, adipocyte lipolysis in ethanol-fed rats was still lower compared with pair-fed rats in the presence of Ro20-1724 (Fig. 9B).
Fig. 9.
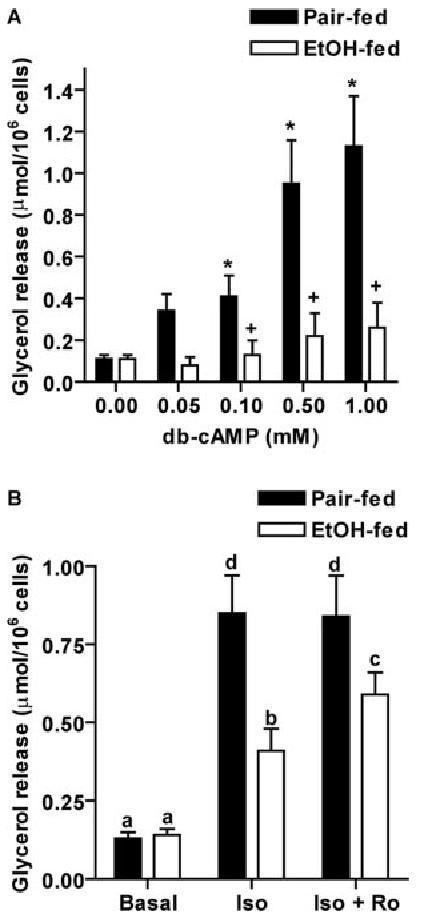
Chronic ethanol feeding impaired signaling downstream of phosphorylation of perilipin A and HSL. Adipocytes isolated from pair- and ethanol-fed rats were treated with or without increasing concentrations of dibutyryl-cAMP (db-cAMP; panel A) or pretreated with or without 10 μm Ro20-1724 (Ro) for 3 min before stimulation with 1 μm isoproterenol (Iso; panel B). Lipolysis was determined as glycerol release over 1 h. Values represent means ± sem (n = 3–8). *, P < 0.05, compared with own basal; +, P < 0.05, pair-fed vs. EtOH-fed. Values with different letters are significantly different (P < 0.05).
Discussion
Adipose tissue is the major site for storage of triglycerides; mobilization of free fatty acids and glycerol via lipolysis provides a rapid source of fuel for other organs in response to fasting, infection, and inflammation (4). During chronic ethanol exposure, whole-body lipid homeostasis is disrupted, with the eventual development of hepatic steatosis (2). Although it is clear that the regulation of lipolysis in adipocytes plays an important role in maintaining lipid homeostasis, the influence of chronic ethanol feeding on the regulation of lipolysis in adipocytes has not been investigated. Here, we report that chronic ethanol feeding to rats decreased β-adrenergic receptor-stimulated lipolysis. This suppression of β-adrenergic activation of lipolysis was associated with inhibition of intracellular cAMP accumulation and PKA activation, as well as decreased phosphorylation of perilipin A and HSL. cAMP signaling is known to be an important target of acute and chronic ethanol exposure in a number of systems (9). In adipocytes, chronic ethanol feeding impaired β-adrenergic signaling in at least two sites: first, chronic ethanol feeding increased PDE4 activity, leading to decreased cAMP accumulation; and second, impaired PKA-mediated phosphorylation of perilipin A and HSL, two proteins localized to the lipid droplet of adipocytes. These data thus are the first to identify an association between impaired PKA-mediated phosphorylation of proteins at the lipid droplet with decreased lipolysis during a pathophysiological response to diet, such as chronic ethanol feeding. Taken together, these data provide evidence that chronic ethanol feeding results in a complex dysregulation of β-adrenergic regulation of lipolysis and suggest that impaired regulation of lipolysis in adipose tissue may contribute to the pathophysiological effects of ethanol on lipid homeostasis.
Chronic ethanol feeding to rats had no effect on basal rates of lipolysis in adipocytes isolated from epididymal adipose tissue, but diminished β-adrenergic receptor-stimulated lipolysis compared with pair feeding (Fig. 1). These data are consistent with a previously reported decrease in adrenaline-induced lipolysis in adipocytes from rats provided ethanol in their drinking water for 2 wk (26). Inhibition of β-adrenergic receptor-stimulated lipolysis after chronic ethanol feeding was due, at least in part, to decreased accumulation of intracellular cAMP (Fig. 2A). Chronic ethanol-induced desensitization of β-adrenergic receptor-cAMP signaling has been reported in a number of other cell types, primarily associated with changes in expression of heterotrimeric G proteins (9). In adipocytes, we have previously reported that chronic ethanol feeding increases expression of Gαs′, and this increase is associated with an increase in β-adrenergic receptor-stimulated cAMP synthesis, when measured in the presence of a PDE4 selective inhibitor (Fig. 2B) (10). However, despite these increased rates of cAMP production, accumulation of cAMP by adipocytes after chronic ethanol feeding in response to β-adrenergic activation was actually decreased (Fig. 2A), suggesting an increase in cAMP degradation via PDE4 after chronic ethanol feeding. Direct measurement of PDE activity showed increased activity of PDE4 in adipocytes from ethanol-fed rats, with no effect of ethanol feeding on activity of PDE3B (Fig. 3). This increase in PDE4 activity was not due to increased expression of PDE4A, PDE4B, or PDE4D (Fig. 4). Of the 11 known isoforms of PDE, PDE4 and PDE3 are the predominant isoforms expressed in the adipocytes (23). However, these two isoforms are differentially localized and regulated within rat adipocytes. PDE4, originally found in the cytosolic fraction and recently shown to interact with β-arrestin and thus be recruited to plasma membrane, is regulated by PKA- and MAPK-mediated phosphorylation (27), whereas PDE3, localized in the microsomal fraction, is activated by insulin (28). In this study, PDE4 activity was specifically increased after chronic ethanol exposure, functionally countering the ethanol-induced increase in Gαs/cAMP synthesis, and decreasing cAMP accumulation. Although one other report has described an increase in PDE4 activity by acute ethanol in bovine bronchial epithelial cells (29), this is the first report of a change in PDE activity after chronic ethanol feeding and suggests that there is a pathophysiological relationship between the regulation of PDE4 and the impact of ethanol on cellular processes.
In addition to the decrease in isoproterenol-stimulated cAMP accumulation, signaling downstream of cAMP, PKA activation and PKA-dependent phosphorylation of perilipin A and HSL in response to isoproterenol were also decreased by chronic ethanol feeding (Figs. 5 and 7). Perilipin is a lipid droplet-associated protein in the PAT protein family, including perilipin, adipocyte differentiation-related protein (ADRP), and TIP47 (30). These structurally and functionally similar PAT proteins play a critical role in lipid metabolism in a variety of cell types (31-33). Two forms of perilipin are expressed in rat adipose tissue, perilipin A and B. Perilipin A, the dominant isoform in adipocytes, not only acts as a protective barrier in the absence of stimulation but also is necessary to facilitate HSL translocation from the cytosol to the surface of lipid droplet when it is phosphorylated by PKA (34). By contrast, perilipin B fails to protect basal lipolysis or elicit an activation response (35). In the present study, total quantity of perilipin A and B was not affected by chronic ethanol feeding; however, phosphorylation of perilipin A in response to isoproterenol was abolished in adipocytes from ethanol-fed rats compared with pair-fed rats (Fig. 7A). Upon lipolytic stimulation of adipocytes, a redistribution of perilipin from the surface of lipid droplet to the cytosol has been reported (36, 37). This redistribution of perilipin may be subject to regulation. Clifford et al. (36) found no movement of perilipin from the lipid droplet to the cytosol in isoproterenol-stimulated adipocytes isolated from young rats (180–220 g); however, there was a significant movement of perilipin away from the lipid droplet in adipocytes isolated from more mature rats (230–280 g). In contrast, in our rat model using mature rats (280–340 g), both unphosphorylated and phosphorylated forms of perilipin were predominantly localized to the fat layer of adipocytes (Fig. 7A, and data not shown). Thus, chronic ethanol feeding impaired the phosphorylation of perilipin A, rather than its distribution within the adipocyte.
Phosphorylation of perilipin A and HSL in response to isoproterenol was decreased after chronic ethanol feeding; however, phosphorylation of these proteins per se was not affected by ethanol because dibutyryl-cAMP-stimulated phosphorylation of perilipin A and HSL was not different between pair feeding and ethanol feeding (Fig. 8). Despite normal phosphorylation of perilipin A and HSL in response to dibutyryl-cAMP, lipolysis in adipocytes treated with dibutyryl-cAMP was still decreased by chronic ethanol feeding (Fig. 9A). Additionally, lipolysis in adipocytes pretreated with a PDE4 inhibitor, Ro20-1724, was lower in ethanol-fed rats than pair-fed rats (Fig. 9B). These results suggest that, in addition to reduced cAMP accumulation and cAMP-dependent PKA activation and phosphorylation of perilipin A and HSL, chronic ethanol feeding impaired the signaling pathway from phosphorylation of perilipin A and HSL to the eventual stimulation of lipolysis. Upon phosphorylation, HSL translocates from the cytosol to the surface of lipid droplets, initiating the hydrolysis of triglycerides (38). Although the target of ethanol is not known, impaired translocation of HSL from the cytosol to the surface of lipid droplets is a possible site of action.
In summary, the current investigations demonstrate that chronic ethanol feeding to rats decreased β-adrenergic receptor-stimulated lipolysis in adipocytes isolated from epididymal adipose tissue. This reduction was associated with inhibited intracellular cAMP accumulation and coincident repression of cAMP-dependent PKA activation and phosphorylation of perilipin A and HSL. Furthermore, decreased β-adrenergic receptor-stimulated cAMP accumulation was attributable to increased PDE4 activity after chronic ethanol feeding. These data suggest that the disruption of β-adrenergic receptor regulation of lipolysis may contribute to changes in whole-body lipid homeostasis seen after chronic ethanol exposure.
Acknowledgments
This work was supported by National Institutes of Health Grant AA 11876.
Abbreviations
- ADA
Adenosine deaminase
- HSL
hormone-sensitive lipase
- PDE
phosphodiesterase
- PKA
protein kinase A
- R-PIA
(−)-N6-(2-phenylisopropyl)adenosine
- rpm
revolutions per minute
- SDS
sodium dodecyl sulfate
References
- 1.Room R, Babor T, Rehm J. Alcohol and public health. Lancet. 2005;365:519–530. doi: 10.1016/S0140-6736(05)17870-2. [DOI] [PubMed] [Google Scholar]
- 2.Baraona E, Lieber CS. Effects of ethanol on lipid metabolism. J Lipid Res. 1979;20:289–315. [PubMed] [Google Scholar]
- 3.Balasubramaniyan V, Nalini N. The potential beneficial effect of leptin on an experimental model of hyperlipidemia, induced by chronic ethanol treatment. Clin Chim Acta. 2003;337:85–91. doi: 10.1016/j.cccn.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR, Grunfeld C. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res. 2004;45:1169–1196. doi: 10.1194/jlr.R300019-JLR200. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura MT, Cheon Y, Li Y, Nara TY. Mechanisms of regulation of gene expression by fatty acids. Lipids. 2004;39:1077–1083. doi: 10.1007/s11745-004-1333-0. [DOI] [PubMed] [Google Scholar]
- 6.Holm C, Osterlund T, Laurell H, Contreras JA. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Annu Rev Nutr. 2000;20:365–393. doi: 10.1146/annurev.nutr.20.1.365. [DOI] [PubMed] [Google Scholar]
- 7.Tansey JT, Sztalryd C, Hlavin EM, Kimmel AR, Londos C. The central role of perilipin a in lipid metabolism and adipocyte lipolysis. IUBMB Life. 2004;56:379–385. doi: 10.1080/15216540400009968. [DOI] [PubMed] [Google Scholar]
- 8.Londos C. Hormone-sensitive lipase and the control of lipolysis in adipocytes. In: LeRoith D, Taylor SI, Olefsky JM, editors. Diabetes mellitus: a fundamental and clinical text. Lippincott-Raven; Philadelphia: 1996. pp. 223–227. [Google Scholar]
- 9.Diamond I, Gordon AS. Cellular and molecular neuroscience of alcoholism. Physiol Rev. 1997;77:1–20. doi: 10.1152/physrev.1997.77.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Wilkes JJ, DeForrest LL, Nagy LE. Chronic ethanol feeding in a high-fat diet decreases insulin-stimulated glucose transport in rat adipocytes. Am J Physiol. 1996;271:E477–E484. doi: 10.1152/ajpendo.1996.271.3.E477. [DOI] [PubMed] [Google Scholar]
- 11.Poirier LA, Rachdaoui N, Nagy LE. GLUT4 vesicle trafficking in rat adipocytes after ethanol feeding: regulation by heterotrimeric G-proteins. Biochem J. 2001;354:323–330. doi: 10.1042/0264-6021:3540323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lieber CS, DeCarli LM. The feeding of alcohol in liquid diets: two decades of applications and 1982 update. Alcohol Clin Exp Res. 1982;6:523–531. doi: 10.1111/j.1530-0277.1982.tb05017.x. [DOI] [PubMed] [Google Scholar]
- 13.Mersmann HJ. Effect of anesthetic or analgesic drugs on lipogenic and lipolytic adipose tissue activities. Proc Soc Exp Biol Med. 1983;172:375–378. doi: 10.3181/00379727-172-41573. [DOI] [PubMed] [Google Scholar]
- 14.Ambrisko TD, Hikasa Y. Neurohormonal and metabolic effects of medetomidine compared with xylazine in beagle dogs. Can J Vet Res. 2002;66:42–49. [PMC free article] [PubMed] [Google Scholar]
- 15.Portillo MP, Villaro JM, Torres MI, Macarulla MT. In vivo lipolysis in adipose tissue from two anatomical locations measured by microdialysis. Life Sci. 2000;67:437–445. doi: 10.1016/s0024-3205(00)00642-1. [DOI] [PubMed] [Google Scholar]
- 16.Rodbell M. Metabolism of isolated fat cells. I. Effects of hormones on glucose metabolism and lipolysis. J Biol Chem. 1964;239:375–380. [PubMed] [Google Scholar]
- 17.Honnor RC, Dhillon GS, Londos C. cAMP-dependent protein kinase and lipolysis in rat adipocytes. I. Cell preparation, manipulation, and predictability in behavior. J Biol Chem. 1985;260:15122–15129. [PubMed] [Google Scholar]
- 18.Foster LB, Dunn RT. Stable reagents for determination of serum triglycerides by a colorimetric Hantzsch condensation method. Clin Chem. 1973;19:338–340. [PubMed] [Google Scholar]
- 19.Ahmad F, Cong LN, Stenson Holst L, Wang LM, Rahn Landstrom T, Pierce JH, Quon MJ, Degerman E, Manganiello VC. Cyclic nucleotide phosphodiesterase 3B is a downstream target of protein kinase B and may be involved in regulation of effects of protein kinase B on thymidine incorporation in FDCP2 cells. J Immunol. 2000;164:4678–4688. doi: 10.4049/jimmunol.164.9.4678. [DOI] [PubMed] [Google Scholar]
- 20.Kemp BE, Cheng HC, Walsh DA. Peptide inhibitors of cAMP-dependent protein kinase. Methods Enzymol. 1988;159:173–183. doi: 10.1016/0076-6879(88)59018-3. [DOI] [PubMed] [Google Scholar]
- 21.Brasaemle DL, Dolios G, Shapiro L, Wang R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3–L1 adipocytes. J Biol Chem. 2004;279:46835–46842. doi: 10.1074/jbc.M409340200. [DOI] [PubMed] [Google Scholar]
- 22.Fruhbeck G, Gomez-Ambrosi J, Salvador J. Leptin-induced lipolysis opposes the tonic inhibition of endogenous adenosine in white adipocytes. FASEB J. 2001;15:333–340. doi: 10.1096/fj.00-0249com. [DOI] [PubMed] [Google Scholar]
- 23.Schmitz-Peiffer C, Reeves ML, Denton RM. Characterization of the cyclic nucleotide phosphodiesterase isoenzymes present in rat epididymal fat cells. Cell Signal. 1992;4:37–49. doi: 10.1016/0898-6568(92)90006-t. [DOI] [PubMed] [Google Scholar]
- 24.Snyder PB, Esselstyn JM, Loughney K, Wolda SL, Florio VA. The role of cyclic nucleotide phosphodiesterases in the regulation of adipocyte lipolysis. J Lipid Res. 2005;46:494–503. doi: 10.1194/jlr.M400362-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Houslay MD, Adams DR. PDE4 cAMP phosphodiesterases: modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem J. 2003;370:1–18. doi: 10.1042/BJ20021698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szkudelski T, Bialik I, Szkudelska K. Adipocyte lipolysis, hormonal and metabolic changes in ethanol-drinking rats. J Anim Physiol Anim Nutr (Berl) 2004;88:251–258. doi: 10.1111/j.1439-0396.2004.00478.x. [DOI] [PubMed] [Google Scholar]
- 27.Houslay MD, Baillie GS. The role of ERK2 docking and phosphorylation of PDE4 cAMP phosphodiesterase isoforms in mediating cross-talk between the cAMP and ERK signalling pathways. Biochem Soc Trans. 2003;31:1186–1190. doi: 10.1042/bst0311186. [DOI] [PubMed] [Google Scholar]
- 28.Degerman E, Belfrage P, Manganiello VC. Structure, localization, and regulation of cGMP-inhibited phosphodiesterase (PDE3) J Biol Chem. 1997;272:6823–6826. doi: 10.1074/jbc.272.11.6823. [DOI] [PubMed] [Google Scholar]
- 29.Forget MA, Sisson JH, Spurzem JR, Wyatt TA. Ethanol increases phosphodiesterase 4 activity in bovine bronchial epithelial cells. Alcohol. 2003;31:31–38. doi: 10.1016/j.alcohol.2003.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Londos C, Sztalryd C, Tansey JT, Kimmel AR. Role of PAT proteins in lipid metabolism. Biochimie (Paris) 2005;87:45–49. doi: 10.1016/j.biochi.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 31.Greenberg AS, Egan JJ, Wek SA, Garty NB, Blanchette-Mackie EJ, Londos C. Perilipin, a major hormonally regulated adipocyte-specific phospho-protein associated with the periphery of lipid storage droplets. J Biol Chem. 1991;266:11341–11346. [PubMed] [Google Scholar]
- 32.Servetnick DA, Brasaemle DL, Gruia-Gray J, Kimmel AR, Wolff J, Londos C. Perilipins are associated with cholesteryl ester droplets in steroidogenic adrenal cortical and Leydig cells. J Biol Chem. 1995;270:16970–16973. doi: 10.1074/jbc.270.28.16970. [DOI] [PubMed] [Google Scholar]
- 33.Miura S, Gan JW, Brzostowski J, Parisi MJ, Schultz CJ, Londos C, Oliver B, Kimmel AR. Functional conservation for lipid storage droplet association among perilipin, ADRP, and TIP47 (PAT)-related proteins in mammals, Drosophila, and Dictyostelium. J Biol Chem. 2002;277:32253–32257. doi: 10.1074/jbc.M204410200. [DOI] [PubMed] [Google Scholar]
- 34.Sztalryd C, Xu G, Dorward H, Tansey JT, Contreras JA, Kimmel AR, Londos C. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. J Cell Biol. 2003;161:1093–1103. doi: 10.1083/jcb.200210169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tansey JT, Huml AM, Vogt R, Davis KE, Jones JM, Fraser KA, Brasaemle DL, Kimmel AR, Londos C. Functional studies on native and mutated forms of perilipins. A role in protein kinase A-mediated lipolysis of triacylglycerols. J Biol Chem. 2003;278:8401–8406. doi: 10.1074/jbc.M211005200. [DOI] [PubMed] [Google Scholar]
- 36.Clifford GM, Londos C, Kraemer FB, Vernon RG, Yeaman SJ. Translocation of hormone-sensitive lipase and perilipin upon lipolytic stimulation of rat adipocytes. J Biol Chem. 2000;275:5011–5015. doi: 10.1074/jbc.275.7.5011. [DOI] [PubMed] [Google Scholar]
- 37.Souza SC, de Vargas LM, Yamamoto MT, Lien P, Franciosa MD, Moss LG, Greenberg AS. Overexpression of perilipin A and B blocks the ability of tumor necrosis factor α to increase lipolysis in 3T3-L1 adipocytes. J Biol Chem. 1998;273:24665–24669. doi: 10.1074/jbc.273.38.24665. [DOI] [PubMed] [Google Scholar]
- 38.Holm C. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem Soc Trans. 2003;31:1120–1124. doi: 10.1042/bst0311120. [DOI] [PubMed] [Google Scholar]