

Abstract
Increased interest in the fate, transport and toxicity of polybrominated diphenyl ethers (PBDEs) over the past few years has led to a variety of studies reporting different methods of analysis for these persistent organic pollutants. Because PBDEs encompass a range of vapor pressures, molecular weights and degrees of bromine substitution, various analytical methods can lead to discrimination of some PBDE congeners. Recent improvements in injection techniques and mass spectrometer ionization methods have led to a variety of options to determine PBDEs in environmental samples. The purpose of this paper is therefore to review the available literature describing the advantages and disadvantages in choosing an injection technique, gas chromatography column and detector. Additional discussion is given to the challenges in measuring PBDEs, including potential chromatographic interferences and the lack of commercial standards for higher brominated congeners, which provides difficulties in examining degradation and debromination of BDE congeners, particularly for BDE 209.
Keywords: Polybrominated diphenyl ethers, Brominated flame retardants, Methods, Analysis, Review
Introduction
Over the past 5 years there has been a surge in the number of studies investigating polybrominated diphenyl ethers (PBDEs) in the environment. In 1981 the first report on PBDEs was made in biota samples from a Swedish river [1], which was followed by a handful of studies throughout the 1980s and 1990s. Then, in 2000 Noren and Meironyte [2] published a paper on organohalogen temporal trends in human breast milk from Sweden that generated rapid interest in the fate of PBDEs. This study demonstrated that PBDE levels appeared to be increasing at an exponential rate, and since then the number of studies investigating PBDEs has increased as well.
PBDEs are used as brominated flame retardant additives which are incorporated into a number of polymers and resins found in a majority of consumer products. PBDEs are the most widely used additive flame retardant and, until recently, almost 70,000 t was produced every year, half of which was used in products sold in the USA and Canada. In 2004 the European Union phased out the use of two of the three PBDE commercial mixtures, PentaBDE and OctaBDE. Following this action, and owing to growing concerns in some US state governments, the sole US chemical manufacturer voluntarily agreed to stop manufacturing these two PBDE commercial mixtures beginning in January 2005 [3]. Presently, the only remaining unregulated PBDE mixture in production is DecaBDE, with an annual global market demand of 56,000 t (http://www.bsef.com). DecaBDE is composed almost exclusively (more than 97%) of decabromodiphenyl ether (BDE 209), a fully brominated BDE congener.
Much of the concern regarding PBDEs has been focused on the elevated levels measured in human tissues within the US population [4]. The BDE congeners typically measured in human tissues are associated primarily with the PentaBDE mixture, and to some extent with the OctaBDE mixtures. While production of PentaBDE and OctaBDE has been halted, there are still a number of products left on the market and in use today that contain these mixtures. On the basis of their persistence and current reservoirs, the fate of PBDEs will most likely need to be monitored for years to come.
One of the greatest challenges to measuring PBDEs in environmental samples has been developing methods to accurately quantify BDE 209. While analytical methods are readily available for quantifying tribrominated through heptabrominated congeners found in the PentaBDE and OctaBDE mixtures, the analysis of higher brominated compounds (i.e., congeners with eight or more bromines) has proven to be difficult. In 1999–2000, the first worldwide interlaboratory comparison on PBDE measurements was conducted. Laboratory agreements were acceptable for most of the lower brominated congeners; however, reported values for BDE 209 varied over almost 2 orders of magnitude [5]. Over the past few years, improved methods have been developed to measure BDE 209 with increased accuracy and precision, and with the use of these methods, more data on BDE 209 have been generated.
A number of papers have described analytical methods for extracting and quantifying environmental samples for the determination of PBDEs [6–11]. In 2003 Covaci et al. [9] published an excellent review on extraction methods, cleanup techniques and analytical detection methods for brominated flame retardants. Since the publication of that review, several interesting papers have been published which highlight improvements in injection techniques, analyte resolution and selectivity, particularly for BDE 209. When choosing a method one must find the best compromise in considering cost, reproducibility, chromatographic and mass resolution, sensitivity, and analysis time. The aim of this paper is to review and disseminate the latest information available and discuss the advantages and disadvantages found when using different analytical techniques to determine PBDEs in environmental and human samples.
The physical-chemical properties of PBDEs are very similar to those of PCBs [12], and as such, PBDEs are typically extracted using nonpolar solvents (e.g., hexane, dichloromethane) in methods typically employed for PCB analysis. In this paper all PBDE congeners are labeled and numbered according to the same scheme developed by Ballschmitter and Zell [13], for PCB congeners. A variety of extraction and cleanup methods have been reported [6, 9, 14–16], but for the purpose of this paper, we will focus on instrumental methods of analysis for PBDE congeners.
Injection techniques
Owing to their vapor pressures and polarity, gas chromatography (GC) has become a standard analytical separation method employed for the analysis of PBDEs. The injection of samples into the GC system is an important and crucial step for the accurate and optimal determination of compounds with relatively high boiling points like PBDEs. Various injection methods have been reported which introduce sample analytes into the GC analytical column. The three most common injection techniques for PBDE analysis are split/splitless injection, on-column injection and programmable temperature vaporization (PTV) injection. All three methods possess advantages and disadvantages that are dictated primarily by availability, price, acceptable detection limits and discrimination of congeners on the basis of molecular weight. Split/splitless injection is the most routinely used, and is capable of analyzing dirty samples; however, this method can be limited by small injection volumes (typically 1–3 μl) and high injection temperatures (250–300 °C). During split/splitless injection the analytes are immediately vaporized in the inlet liner and are transferred to the column in the gas phase. In some cases, the high inlet temperature can lead to thermal degradation and discrimination of higher molecular weight PBDEs, particularly the fully brominated BDE 209. Evaporation of the sample can begin inside the injection needle or on the tip, and differences in evaporation rates can lead to discrimination of the higher molecular weight congeners. Inefficient transfer of the sample to the column can be difficult for analytes with high boiling points. Furthermore, active sites on dirty liners can result in thermal degradation; therefore, the temperature of the injection port and the time spent in the injection port prior to transfer are important mediating factors in the response of the PBDE congeners. A detailed study by Björklund et al. [17] found that the optimal injector temperature and splitless time should be kept as high as possible, in their case, 325 °C and 4 min, respectively (Fig. 1). Using these optimal setpoints, Björklund et al. demonstrated that the BDE 209 response increased by almost 25% when compared with average settings used in an interlaboratory comparison [5]. However, they also noted a significant decrease in the precision of higher molecular weight congeners, particularly BDE 209, compared with the case for on-column techniques, which are less prone to discrimination effects.
Fig. 1.
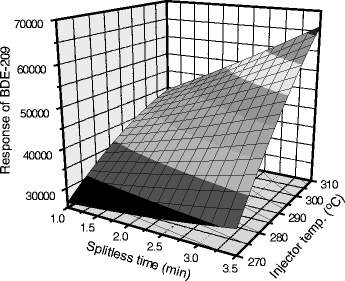
Detector response for BDE 209 as a function of splitless time and injector temperature, with pressure held constant at 2.3 bar. BDE brominated diphenyl ether. (Reprinted from Björklund et al. [17] with permission)
On-column injection has become a popular method for introducing samples into the gas chromatograph for PBDE analysis [18–20] and is comparable in cost to split/splitless injectors. The design is much simpler relative to a split/splitless injection port and involves direct injection of the sample, dissolved in a carrier solvent, onto the head of the column. This reduces the potential for thermal degradation and discrimination experienced in split/splitless injectors. In the same study mentioned previously, Björklund et al. [17] observed the highest precision in PBDE measurements, particularly for BDE 209, when using on-column injection compared with split/splitless and PTV injectors. However, when using on-column injection, care must be employed to ensure a clean sample, to prevent coextracted compounds from building up in the retention gap and column leading to increased noise, peak tailing, retention shifts and reduced lifetimes of the columns. On-column injections are also limited by injection volumes (typically 1–2 μl) to prevent excess solvent from overwhelming the detectors.
PTV inlets have become a more popular choice for injection over the past 5 years, particularly for the analysis of PBDEs [21–23]. The primary advantage in using PTV injectors is an increase in injection volumes (up to 125 μl), which can drastically improve detection limits, and can be useful in measuring low-level samples such as human serum [14, 24–26]. With this type of injection, multiple injections are made into a liner while the solvent is vented off, trapping the analytes and increasing the mass of analytes present in the liner. Transfer of the analytes to the head of the column occurs after the solvent vent is closed, and rapid heating of the injection port (200–700 °C/min) aids the carrier gas in transferring the analytes to the column. Several studies have demonstrated that the PTV injectors must be optimized prior to use as they are more complicated than the conventional split/splitless injector. Analyte discrimination can result if the following are not optimized: injection rate or flow, injection temperature, vent flow, solvent elimination time, injection volume, transfer time and transfer temperature.
Tollbäck et al. [25] published a review on optimization parameters for PTV injection and their optimal values are reported in Table 1. In general, it is important that the rate of injection is equivalent to the rate of solvent evaporation to avoid flooding the injector. Care must also be taken to avoid discriminating low molecular weight compounds through solvent vent losses. The rate of injection will influence the solvent introduction mode (i.e., drops versus spray), which can discriminate congeners of different masses on the basis of the point (vertical position) of solvent evaporation within the injection liner. BDE 209 responses can be particularly sensitive to injection flow rate. To balance the solvent removal from the injector, the temperature and vent flow must also be optimized. At elevated injector temperatures, low molecular weight compoundss can be lost through solvent venting and at high flow rates the risk of analyte loss increases. The type of liner used in the injector can also influence PBDE responses. Some liners contain activated sites, which can lead to irreversible adsorption or catalysis of thermal degradation of BDEs, particularly the high molecular weight congeners. Tollbäck et al. [25] recommended adding 0.1–0.35% (v/v) of dodecane with multibaffled liners to trap low molecular weight congeners and changing the liner every 200 injections. Transfer of analytes to the GC column is accomplished by rapid heating of the injector port after the solvent vent is closed. PBDE responses are not significantly affected by the analyte transfer temperature ramp; therefore, steep ramps (700 °C/min) should be used to increase the rate of transfer to GC columns. A final injector temperature of 325 °C was found to be a good compromise to balance transfer efficiency of high molecular weight analytes relative to the risk of their thermal degradation in the injector.
Table 1.
Optimized injector parameters for polybrominated diphenyl ether (PBDE) analysis using programmable temperature vaporization as reported by Tollbäck et al. [25]
| Parameter | Domain |
|---|---|
| Injection rate | 300 μl/min |
| Injection temperature | 80 °C |
| Vent flow | 150 ml/min |
| Temperature rate | 700 °C/min |
| Transfer temperature | 325 °C |
| Transfer time | 0.3–0.6 min |
| Solvent elimination time | 0.1 min |
| Carrier gas pressure | 650 kPa |
While split/splitless, on-column and PTV injections are the most common methods used in PBDE analysis, other injection techniques have been reported. Large volume injections up to 500 μl have been reported using both loop-type [26] and automated rotary valves [27]. Loop-type injectors can easily be constructed and used but require day-to-day checks for stability and reproducibility owing to problems with the injection pressure and temperature which can result in peak distortion. Björklund et al. [27] used a stainless steel high-performance liquid chromatography (LC) automated rotary valve to inject PBDEs into a GC system. This technique reduces discrimination of higher molecular weight congeners and thermal degradation similar to on-column injection, but offers the advantage of injecting larger volumes (up to 50 μl). However, care must be taken to incorporate a solvent vent system for certain detector systems, particularly mass spectrometry (MS). Lastly, Sjodin [28] used a septum-equipped programmable injector (SPI), which resulted in reproducible results with minimal congener discrimination; however, these injectors are no longer produced by the manufacturer.
GC column selection
The next crucial step in the method development is the selection of an appropriate column system. The optimal chromatographic column is one that optimizes resolution and discrimination of congeners with the greatest sensitivity in the shortest amount of time. Of the three primary injection methods described earlier, both on-column and PTV injections require the use of a retention gap or guard column to reduce column deterioration and to aid in focusing the initial band of analytes. Most guard columns are either composed of untreated fused silica with active silanol groups or deactivated fused silica that ranges in polarity. Björklund et al. [17] recommends using a Siltek deactivated guard column, which the authors found had the best precision (measured by peak response) and displayed the least degradation of higher molecular weight congeners. In contrast, a nonpolar fused silica guard column resulted in a 50% reduction in sensitivity for the higher molecular weight PBDE congeners.
A variety of different columns have been used for the determination of PBDEs in environmental samples. When choosing a column one must consider the column stationary phase (polarity), column length, film thickness and inner diameter, all of which will influence the response towards PBDEs. Generally speaking, the most sensitive method for measuring the entire range (low to high bromine substitution) of PBDE congeners is found when using short (10–15-m) nonpolar DB columns with thin (0.1-μm) stationary phases [17]. Most GC/MS methods employed for PCB quantification use a 60-m column to increase the resolution power; however, longer columns are not well suited for PBDE analysis, particularly for the higher molecular weight congeners. Longer columns result in longer residence times for the analytes, initiating degradation of the higher brominated congeners (noticeably for heptaBDE, octaBDE, nonaBDE and decaBDE congeners) in the column. Björklund et al. [17] determined that the DB-5MS column (Agilent Technologies, Palo Alto, CA, USA) produced the lowest discrimination and the highest precision compared with five other columns tested (Fig. 2). Columns that contained similar stationary phases but that were produced by two different manufacturers also resulted in different column performance. An HP-1 column from Agilent Technologies significantly degraded nonaBDEs and BDE 209, whereas a comparable DB-1 column from J & W Scientific (Folsom, CA, USA) did not. Narrow-bore columns (inner diameters 0.1 mm or less) have been used recently in combination with PTV injectors, resulting in faster and more efficient separation of congeners [24, 25]. Narrow-bore columns produce extremely narrow peaks, with average peak widths at half peak height of less than 1 s, compared with almost 2 s using columns with 0.25-mm film thickness.
Fig. 2.
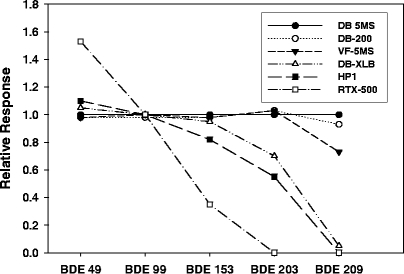
The relative response of five selected BDE congeners using different gas chromatography capillary columns. The response for DB-5MS was set to 1. Mean values are plotted (n=5) and the error bars correspond to the standard deviation. (Reprinted from Björklund et al. [17] with permission)
Another consideration in choosing a GC column is its resolving power. Korytár et al. [29] recently published a retention time database for 126 PBDE congeners using seven different capillary columns with varying film thicknesses (Table 2). This database is useful for determining the most suitable column for quantitative, congener-specific PBDE analysis. Of the seven columns tested, the DB-XLB column (30 m × 0.25 mm × 0.25 μm, J & W Scientific) was found to have the fewest number of coelutions with other BDE congeners and with other brominated flame retardants, followed closely by the DB-1 column (30 m × 0.25 mm × 0.25 μm, J & W Scientific). However, the DB-XLB column demonstrated greater degradation of the higher molecular weight congeners relative to the DB-1 column. Korytár et al. [29] compared the relative retention times (RRTs) from their study to a GC-RRT model developed by Rayne and Ikonomou [30] which was based on the number of ortho-, meta- and para-bromine substituents, dipole moments and the molecular weight of the compounds. In general the RRTs of lower molecular weight PBDE congeners compared very well between the two studies. In contrast, the higher molecular weight congeners, noticeably the heptaBDE and octaBDE congeners were not tightly correlated, most likely owing to a lack of commercially available standards for heptaBDE and octaBDE congeners when the GC-RRT model was developed. Additional standards are now available and it will be useful to update these types of models in the future.
Table 2.
An assessment of PBDE coelution from seven gas chromatography columns as tested and reported by Korytár et al. [63]
| Column | DB-1 | DB-5 | HT-5 | DB-17 | DB-XLB | HT-8 | CP-Sil 19 |
|---|---|---|---|---|---|---|---|
| Dimension (m × mm × μm) | 30 × 0.25 × 0.25 | 30 × 0.25 × 0.25 | 30 × 0.25 × 0.10 | 30 × 0.25 × 0.25 | 30 × 0.25 × 0.25 | 25 × 0.22 × 0.25 | 17 × 0.15 × 0.30 |
| Number of coeluting BDEs | 62 | 63 | 66 | 67 | 56 | 62 | 72 |
| Number of coelutions with flame retardants | 24 | 26 | 27 | 30 | 22 | 26 | 29 |
| Co-elution with major BDE congeners | |||||||
| Major BDE | |||||||
| 28 | 16, 33 | 16, 33 | 16, 33, 38 | 16, 33, 38 | 16, 33 | ||
| 47 | |||||||
| 49 | 68, 80 | 68 | 68 | 62 | 42, 48, 68, 71 | 68 | 51, 75 |
| 85 | 155 | 114 | |||||
| 99 | 116 | 127 | |||||
| 100 | 109 | 101 | 109, 120 | ||||
| 138 | 166 | HBCD | 166 | ||||
| 153 | HBCD | 168 | |||||
| 154 | MTBBP-A, BB-153 | MTBBP-A, BB 153 | 105 | 126 | BB 153 | ||
| 183 | BB 169 | BB 169 | |||||
MTBBP-A dimethylated tetrabromobisphenol-A, HBCD hexabromocyclododecane
Chromatographic interferences
Coelution of compounds can be an important consideration when using GC/electron capture detection (ECD) and GC/electron capture negative ionization (ECNI) MS techniques to quantify environmental samples, especially as more PBDE congeners become available for analysis. The database of Korytár et al. [29] found that at a minimum, 56 of the 126 PBDE congeners tested were coeluted on the GC columns. The majority of these coelutions were observed for congeners not typically observed in environmental samples; however, a few major congeners (i.e., BDE 28, BDE 49 and BDE 154) can often be composed of different BDE congeners and other brominated flame retardants. Because ECD and ECNI-MS techniques are not as selective as electron ionization (EI) MS for the analysis of lower brominated congeners, coextracted halogenated compounds can sometimes be mistaken for PBDE congeners. Several papers have reported the coelution of 2,2′,4,4′,5,5′-hexabromobiphenyl (BB 153) with BDE 154 and of tetrabromobisphenol-A with BDE 153 [6, 9, 29], typically on 15- and 30-m capillary columns. PCB congeners can also be coeluted if not specifically separated during the extraction process. Alaee et al. [31] reported ten possible interferences between PBDEs and organochlorines. Of particular notice was the interference of BDE 47 with CB-180, and that of BDE 99 with CB-205. The additional mass accuracy of high resolution MS (HRMS) may not be adequate to distinguish certain PCBs and BDEs. Alaee et al. [31] found that the isotopic cluster of [M-Cl2]+ from heptachlorinated biphenyls contains the same mass fragments found in tetrabrominated diphenyl ethers [M-Br2]+ and resolving powers of 25,000 (m/Δm) were required for discriminating the two.
Natural brominated compounds have recently been identified in some marine algae, mammals and birds [32–34]. Because GC/ECNI-MS methods rely upon selective ion monitoring (SIM) of Br− ions [79Br and 81Br], other brominated compounds can produce the same fragment ion and confound analysis of PBDEs. Methoxylated PBDEs (MeO-BDEs) are often extracted with PBDEs in environmental samples and can cause interferences when using both GC/EI-MS and GC/ECNI-MS methods. As seen in Fig. 3, the BDEs and MeO-BDEs are eluted very closely together using common techniques and differentiation must be based solely on retention time. Two MeO-BDEs, (6-methoxy-2,2′,4,4′-tetrabromodiphenyl ether and 2′-methoxy-2,3′,4,5′-tetrabromodiphenyl ether) have been identified in several marine species [32, 34] and are eluted between BDE 47 and BDE 100 with identical Br− ion ratios. Other studies have also identified a brominated bipyrrole known as 1,1′-dimethyl-tetrabromo-dichloro-2,2′-bipyrrole (DBP-Br4Cl2) in marine species [35–37]. DBP-Br4Cl2 is also eluted very closely to BDEs and MeO-BDEs using common methods of analysis (Fig. 4).
Fig. 3.
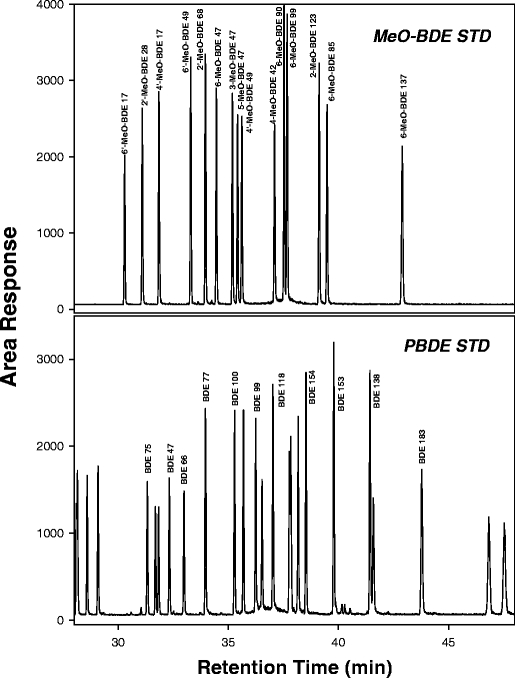
Comparison of elution order for methoxylated BDEs (MeO-BDE) and BDE congeners on a 30-m DB-5MS capillary column using gas chromatography (GC)/electron capture negative ionization (ECNI) mass spectrometry (MS). PDBE polyBDE
Fig. 4.
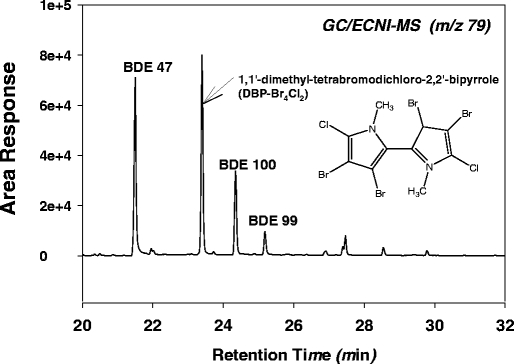
GC/ECNI-MS chromatogram of sea lion blubber containing both PBDEs and a marine natural product, DBP-Br4Cl2
Calibration and quantification standards
PBDE calibration standards are commercially available from a few suppliers, including Accustandard (New Haven, CT, USA), Cambridge Isotope Laboratories (Andover, MA, USA), Wellington Laboratories (Guelph, ON, Canada) and Chiron Co. (Trondheim, Norway). All of these suppliers provide the suite of major PBDE congeners typically identified in environmental samples. To the author’s knowledge, about 160 of the 209 possible BDE congeners are currently available commercially. There are still many hexaBDE, heptaBDE and octaBDE congeners that are not available and it can sometimes be difficult to identify unknown peaks in chromatograms that may be those of BDE congeners. For example, octaBDE congeners have been identified as degradation products of BDE 209 through apparent debromination mechanisms [38–40]; however, the bromine substitution of these congeners is unclear owing to a lack of commercially available octaBDE congeners for comparison. Hopefully in the future, more standards will be available for comparison and will provide more information on the fate of higher brominated congeners.
Another facet to consider in measuring PBDEs is the choice of an appropriate internal quantification standard. Ideally, isotopically labeled (13C) PBDE standards would be the best choice for quantifying PBDEs. Unfortunately, for some detection methods (see later), 13C PBDEs cannot be distinguished from native PBDEs and cannot be used. Several studies have used alternative internal standards such as 13C-labeled bromobiphenyls and chlorinated diphenyl ethers [41], polychlorinated biphenyls [42], or unlabeled BDE congeners [5, 20] that are not typically identified in samples such as BDE 15 and BDE 75. Chiron Co. recently announced the availability of fluorinated BDEs for use as internal standards. For these standards one fluorine atom is substituted for hydrogen on a BDE congener, resulting in a slightly different vapor pressure. These fluorinated BDEs fragment in a manner similar to that of native BDEs, but have slightly different retention times from their nonfluorinated analogues (Fig. 5), making them very useful as internal standards in GC/ECNI-MS methods when 13C-labeled BDE congeners cannot be used.
Fig. 5.
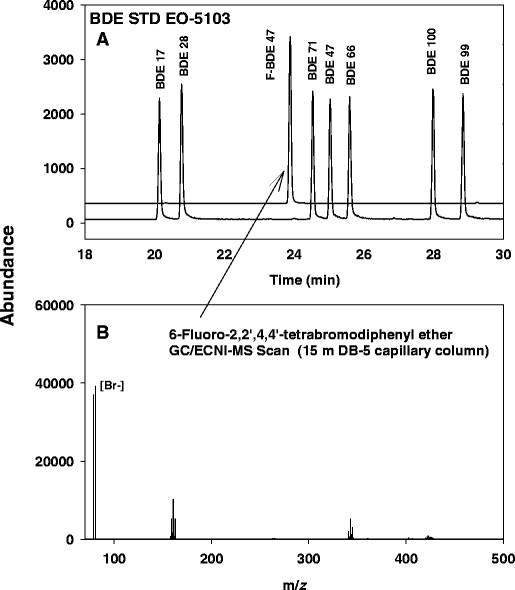
GC/ECNI-MS chromatogram (a) illustrating the elution order of 6-fluro-BDE 47 relative to the PBDE congeners and its comparable ion fragmentation pattern (b)
Detection techniques
Determination of PBDEs in environmental samples is sometimes conducted using ECD, but more commonly mass spectrometers are used, the latter being classified into low-resolution (LR) and HR instruments. ECD is advantageous because is relatively inexpensive, user-friendly and sensitive for measurements of halogenated organics; however, ECD is limited in selectivity because detection and identification is based solely on retention time and halogenated interferences can lead to misidentification [31]. Determination of PBDEs using LR instruments is typically made with the instruments operated in either EI-MS or ECNI-MS modes. In contrast, HR instruments are used almost exclusively in EI mode (EI-HRMS). HRMS offers the best selectivity for PBDE measurements, with a mass spectrometric resolution (m/Δm) of approximately 10,000, resulting in fewer coeluting interferences and facilitating the use of isotopically labeled internal standards. However, HR instruments are more expensive relative to LR instruments. In addition, HRMS is labor-intensive, requiring trained personnel to maintain the instruments and keep them actively running. A brief review of LRMS and HRMS techniques are given next.
A variety of papers have reported PBDE measurements in environmental samples using both GC/LR-EI-MS [43–45] and GC/LR-ECNI-MS [19, 46, 47]. In general, LR-EI-MS is more selective than LR-ECNI-MS methods because molecular fragments (typically [M-Br2]+) are monitored for each homologue group, in contrast to the bromide ions ([Br−]−, m/z 79 and 81) monitored for all homologue groups in LR-ECNI-MS methods. However, LR-ECNI-MS is a much more sensitive method with lower limits of detection (LOD) relative to LR-EI-MS, even when using SIM mode. The LOD reported for LR-EI-MS range from 0.53 to 32.09 pg, whereas for LR-ECNI-MS the LOD is an order of magnitude lower, ranging from 30 fg to 1.72 pg [48], which is particularly useful for the analysis of low-concentration samples such as human serum and plasma [14, 33, 49]. Additionally, there is a large drop in sensitivity when measuring BDE congeners with more than six bromine atoms using quadrupoles in LR-EI-MS; therefore, one must prioritize selectivity versus sensitivity when choosing a reliable detection method. However, recent work conducted by Ackerman et al. [50] suggests that selectivity can be retained when using LR-ECNI-MS under optimized conditions. The authors reported that optimization of the electron energy, emission current, source temperature and system pressure increased the relative abundance of molecular fragment ions [M-xH-yBr]−, which can be monitored for each homologue group in place of the nonspecific bromide ions. With this method one could use isotopically labeled BDE congeners as internal standards, which cannot be used when monitoring bromide ions (79Br and 81Br) in typical LR-ECNI-MS methods. The exception is BDE 209, in which 13C-labeled BDE 209 can be used in all LR-ECNI-MS applications, since fragmentation produces a higher abundance of the [C6Br5O]− ion relative to bromide [51].
Different reagent gases have also been used to increase PBDE response in LR-ECNI-MS. Typical methods use methane gas as a buffering reagent in the ionization process, but ammonia and isobutane can also be used. Eljarrat et al. [48] found that ammonia and methane gas provided similar LOD for PBDEs, but required significantly different ion source temperatures and system pressure. Ackerman et al. [50] found that isobutane resulted in a 20% increase in the abundance of molecular fragments relative to methane; however, isobutane appeared to contaminate the ion source very quickly, resulting in decreased sensitivity after only 12 h of use and was therefore not recommended for use.
HRMS offers both good sensitivity and optimum selectivity. Alaee et al. [52] described a GC/HRMS method for the determination of PBDEs in fish. HRMS instruments have a dual magnetic and electrostatic sector, which increases sensitivity for higher molecular weight analytes such as the higher brominated congeners. This is particularly useful for quantifying heptaBDE through decaBDE congeners with greater confidence. Additionally, HRMS allows one to use isotope dilution with 13C-labeled BDE standards, which are ideal for accurate and precise measurements. In a comparison between GC/HR-EI-MS and GC/LR-ECNI-MS, Thomsen et al. [49] found that both methods provided similar LOD and repeatability. And, as stated previously, HRMS methods will limit the number of potential mass interferences. Therefore, if an instrument and funds are available, HRMS methods provide the best standards for selectivity and sensitivity.
Additional ionization methods and detectors have also been used to measure PBDEs in environmental samples. Ikonomou [53] used GC/HRMS with metastable atom bombardment with nitrogen gas as an alternative to electron impact ionization. This method provided less fragmentation of the parent BDE congeners and was useful for identifying the degree of bromination on unknown BDE congeners relative to conventional GC/HR-EI-MS methods. Tandem mass spectrometers using ion traps have also been reported for the analysis of PBDEs [16, 54]. Ion traps offer the advantage of increased selectivity at a low mass resolution because the analytes are fragmented twice. This type of detection minimizes the chance of isobaric interferences and significantly reduces background noise. Ion traps typically use a collision-induced dissociation (CID) cell to fragment a precursor ion, forming a secondary fragment ion. For PBDEs, the precursor ion is typically a molecular ion [M]+or [M-Br2]+ and the secondary fragment formed from the CID is a [M-COBr]+ ion. Wang et al. [54] optimized a GC/ion trap method for 20 BDE congeners and noted that the relative abundance of the fragment ions was dependent upon the number of ortho-substituted bromines on the congener. They also pointed out that coelution could be a potential problem for higher brominated compounds that could produce similar precursor and fragment ions and contribute to additional signals to the MS/MS channel monitored. More work is needed to determine if other brominated compounds would be isobaric using this method.
HR time-of-flight (TOF) mass spectrometers have also been used to determine PBDEs in environmental samples [55, 56], with detection limits comparable to those of most other MS methods. TOF methods offer the advantage of acquiring spectral data across a wide mass range without compromising sensitivity, which is often the case with quadrupole mass filters. TOF-MS can be operated in both electron impact and ECNI modes, similar to quadrupole methods. Operation of a TOF mass spectrometer in ECNI mode results in the same type of fragmentation pattern observed in quadrupole MS, and therefore offers little improvement and/or advantage in spectral data because the bromide ions, and not molecular fragments, are found in greatest abundance. HRTOF instruments offer increased mass selectivity and also permit the estimation of elemental composition; however, like most high mass resolution instruments, TOF-MS can be expensive and require high maintenance. Additionally, Cajka et al. [56] found that concentrated samples can result in inaccurate mass determination owing to saturation of the instrument’s time-to-digital converter. Therefore, TOF mass spectrometers have a limited linear range, and dilution and reanalysis would be necessary to quantify concentrated samples.
Alternate analytical techniques
While a gas chromatograph connected to a mass spectrometer is generally the method of choice for PBDE measurements, alternate methods have been investigated. Despite the limited chromatographic resolving power of LC, methods employing LC/MS and LC/MS-MS offer promise for PBDE determination. Debrauwer et al. [57] recently investigated the use of atmospheric pressure photoionization (APPI) using LC/MS-MS. Most traditional LC/MS-MS methods use electrospray ionization (ESI) or atmospheric chemical ionization; however, PBDEs do not ionize well with either of these two techniques. Using APPI however, PBDEs will ionize in both negative and positive modes, depending on the degree of bromine substitution. Positive ion mode was more sensitive towards diBDE through pentaBDE, whereas negative ion mode was more sensitive towards pentaBDE through decaBDE. APPI appears to be a softer ionization technique relative to EI as M+ ions are the most intense ions produced by the interaction of PBDEs with charged dopants (charged by photons), compared with the [M-Br2]+ ions formed in EI. Multiple reaction monitoring in the MS-MS system follows the M+ to [M-Br2]+ transition.
The use of ESI may be limiting for PBDEs, but the analysis of PBDE metabolites can be conducted using ESI-LC/MS methods. Hydroxylated BDEs (OH-BDEs) have been identified in environmental samples in several studies [58–60] and have been observed in serum samples of rats dosed with PBDEs [61], suggesting they are products of PBDE metabolism. A method for measuring OH-BDEs using LC/MS-MS has recently been reported [62]. LC/MS-MS holds much promise for investigating other potential metabolic and breakdown products of PBDEs and other brominated flame retardants in general.
The development of comprehensive two dimensional GC (GC × GC) has improved the GC resolving power for organohalogen determination and GC × GC can be used to sidestep some of the coelution problems encountered in standard GC/MS methods. The use of two columns with different separation characteristics increases the chromatographic resolving power and has been proven successful in separating PBDEs from other halogenated compounds. Korytár et al. [63] used a DB-1 column in combination with 65% phenylmethylspolysiloxane (007-65HT) to efficiently separate PBDEs from polychlorinated alkanes. Focant et al. [55] used GC×GC coupled to a TOF mass spectrometer to resolve 58 different halogenated compounds, including PBDEs, PCBs, bromobiphenyls and organochlorine pesticides.
Challenges in PBDE analyses
Increase in the fate and transport of PBDEs in the environment has led to the development of a number of different analytical methods for measuring this class of persistent halogenated contaminants. An interlaboratory comparison exercise for the determination of PBDEs in marine sediment conducted during 2004–2005 [64] found that laboratory agreement in measurements of PBDEs have improved since the first PBDE intercomparison exercise was conducted in 2001 [5]. Of particular note was the improvement of BDE 209 measurements among laboratories. The development of methods to limit degradation of BDE 209 in the injection port and column of GC systems has drastically improved the accuracy and precision of measurements. With this advance, an increasing number of studies are reporting BDE 209 measurements in environmental samples [65, 66], particularly in human samples [67, 68], suggesting that BDE 209 is more diffuse and ubiquitous in the environment than originally observed.
Theoretically, there are 209 different BDE congeners that can potentially be found in the environment. As stated previously, commercial standards are not currently available for all 209 different congeners and thus one of the major challenges in PBDE analysis is the lack of data on the prevalence of higher brominated BDE congeners in the environment. Several laboratory studies have demonstrated that PBDEs, particularly BDE 209, may debrominate both through abiotic [69, 70] and biotic [38, 39, 71–73] pathways, resulting in the formation of pentaBDE through nonaBDE congeners. The lack of standards has made it difficult to determine the products and elucidate the mechanisms of selective bromine loss. Very little is known about the potential for debromination in the ambient environment and few laboratories routinely measure octaBDE and nonaBDE congeners. The identification of octaBDE and nonaBDE congeners in human samples [40, 74] has led to many questions about the origin of these congeners, such as if they are a result of selective uptake of impurities in the technical mixtures, or if they are a result of debromination. Studies have attempted to identify all major and minor BDE congeners present in the commercial mixtures [29, 75]; however, the lack of standards has made congener identification difficult.
With the implementation of the European Union’s directive on waste electrical and electronic equipment and on the restriction of the use of certain hazardous substances (including PBDEs) in electrical and electronic equipment (RoHS; 2002/95/EC), fast and reliable methods for determining PBDE content in electrical equipment will be needed to ensure compliance. The use of X-ray fluorescence (XRF) portable analyzers is a promising method that would allow measurement of the total bromine content of a plastic component by scanning its surface with the handheld device. Obviously there will be difficulties in determining which type of brominated flame retardants are present in materials analyzed with this instrument. However, the XRF may be useful for determining if PBDE degradation occurs after exposure to light and it may be possible to determine the percentage of bromine lost through this pathway in an easy and fast method. Future directions in PBDE research may also wish to examine the fate of the bromine atoms that could be lost from PBDEs via the aforementioned degradation/debromination processes.
Conclusions
In conclusion, a variety of methods are now available to accurately and precisely measure a suite of PBDE congeners in environmental samples. The choice of method selection will involve a compromise between cost, selectivity and sensitivity, particularly since different congeners require different instrumental optimization techniques, especially BDE 209. The Environmental Protection Agency has outlined a protocol for measuring PBDEs (EPA method 1614), but to the author’s knowledge, this method is still in draft form.
Acknowledgements
The author wishes to thank Dr. Rob Letcher and Dr. Göran Marsh for providing MeO-BDE standards and Dr. Jon Johansen from Chiron Co. for the generous gift of the fluoro-BDE standards. Additional thanks are extended to Dr. Nathan Dodder for reviewing a draft of the manuscript.
References
- 1.Andersson O, Blomkvist G (1981) Chemosphere 10:1051–1060 [DOI]
- 2.Norén K, Meironyte D (2000) Chemosphere 40:1111–1123 [DOI] [PubMed]
- 3.Tullo A (2003) Chem Eng News 81:13
- 4.Betts K S (2002) Environ Sci Technol 36:50A–52A [DOI] [PubMed]
- 5.de Boer J, Cofino WP (2002) Chemosphere 46:625–633 [DOI] [PubMed]
- 6.de Boer J, Allchin C, Law R, Zegers B, Boon JP (2001) Trac Trends Anal Chem 20:591–599 [DOI]
- 7.de la Cal A, Eljarrat E, Barcelo D (2003) J Chromatogr A 1021:165–173 [DOI] [PubMed]
- 8.Hyotylainen T, Hartonen K (2002) Trac Trends Anal Chem 21:13–29 [DOI]
- 9.Covaci A, Voorspoels S, de Boer J (2003) Environ Int 29:735–756 [DOI] [PubMed]
- 10.Bayen S, Lee HK, Obbard JP (2004) J Chromatogr A 1035:291–294 [DOI] [PubMed]
- 11.Pepich BV, Prakash B, Domino MM, Dattilio TA, Munch DJ, Price EK (2005) Environ Sci Technol 39:4996–5004 [DOI] [PubMed]
- 12.Tittlemier SA, Halldorson T, Stern GA, Tomy GT (2002) Environ Toxicol Chem 21:1804–1810 [DOI] [PubMed]
- 13.Ballschmiter K, Zell M (1980) Fresenius’ Z Anal Che 302:20–31 [DOI]
- 14.Covaci A, Voorspoels S (2005) J Chromatog B 827:216–223 [DOI] [PubMed]
- 15.Rieck RH (2004) LC-GC 22:914–925
- 16.Polo M, Gomez-Noya G, Quintana JB, Llompart M, Garcia-Jares C, Cela R (2004) Anal Chem 76:1054–1062 [DOI] [PubMed]
- 17.Björklund J, Tollbäck P, Hiarne C, Dyremark E, Östman C (2004) J Chromatogr A 1041:201–210 [DOI] [PubMed]
- 18.Huwe JK, Lorentzsen M, Thuresson K, Bergman Å (2002) Chemosphere 46:635–640 [DOI] [PubMed]
- 19.Stapleton HM, Dodder NG, Offenberg JH, Schantz MM, Wise SA (2005) Environ Sci Technol 39:925–931 [DOI] [PubMed]
- 20.Sjödin A, Patterson DG, Bergman Å (2001) Environ Sci Technol 35:3830–3833 [DOI] [PubMed]
- 21.Voorspoels S, Covaci A, Schepens P (2003) Environ Sci Technol 37:4348–4357 [DOI] [PubMed]
- 22.Voorspoels S, Covaci A, Maervoet J, Schepens P (2004) J Environm Monit 6:914–918 [DOI] [PubMed]
- 23.Covaci A, Gheorghe A, Voorspoels S, Maervoet J, Steen Redeker E, Blust R, Schepens P (2005) Environ Int 31:367–375 [DOI] [PubMed]
- 24.Covaci A, de Boer J, Ryan JJ, Voorspoels S, Schepens P (2002) Anal Chem 74:790–798 [DOI] [PubMed]
- 25.Tollbäck P, Björklund J, Östman C (2003) J Chromatogr A 991:241–253 [DOI] [PubMed]
- 26.Björklund J, Tollbäck P, Östman C (2003) J Sep Sci 26:1104–1110
- 27.Björklund J, Tollbäck P, Dyremark E, Östman C (2003) J Sep Sci 26:594–600 [DOI]
- 28.Sjödin A (1999) Environmental chemistry. PhD thesis, Stockholm University
- 29.Korytár P, Covaci A, de Boer J, Gelbin A, Brinkman UAT (2005) J Chromatogr A 1065:239–249 [DOI] [PubMed]
- 30.Rayne S, Ikonomou MG (2003) J Chromatogr A 1016:235–248 [DOI] [PubMed]
- 31.Alaee M, Backus S, Cannon C (2001) J Sep Sci 24:465–469 [DOI]
- 32.Vetter W (2001) Anal Chem 73:4951–4957 [DOI] [PubMed]
- 33.Jakobsson K, Thuresson K, Rylander L, Sjödin A, Hagmar L, Bergman A (2002) Chemosphere 46:709–716 [DOI] [PubMed]
- 34.Teuten EL, Xu L, Reddy CM (2005) Science 307:917–920 [DOI] [PubMed]
- 35.Stapleton HM, Dodder NG, Kucklick JR, Reddy CM, Schantz MM, Becker PR, Gulland F, Porter BJ, Wise SA (2006) Mar Pollut Bull (in press) [DOI] [PubMed]
- 36.Reddy CM, Xu L, O'Neil GW, Nelson RK, Eglinton TI, Faulkner DJ, Norstrom R, Ross PS, Tittlemier SA (2004) Environ Sci Technol 38:1992–1997 [DOI] [PubMed]
- 37.Tittlemier SA, Simon M, Jarman WM, Elliott JE, Norstrom RJ (1999) Environ Sci Technol 33:26–33 [DOI]
- 38.Gerecke AC, Hartmann PC, Heeb NV, Kohler HPE, Giger W, Schmid P, Zennegg M, Kohler M (2005) Environ Sci Technol 39:1078–1083 [DOI] [PubMed]
- 39.Stapleton HM, Alaee M, Letcher RJ, Baker JE (2004) Environ Sci Technol 38:112–119 [DOI] [PubMed]
- 40.Thuresson K, Bergman Å, Jakobsson K (2005) Environ Sci Technol 39:1980–1986 [DOI] [PubMed]
- 41.Stapleton HM, Harner T, Shoeib M, Keller JM, Schantz MM, Leigh SD, Wise SA (2006) Anal Bioanal Chem 384:791–800 [DOI] [PubMed]
- 42.Hale RC, La Guardia MJ, Harvey EP, Mainor TM, Duff WH, Gaylor MO (2001) Environ Sci Technol 35:4585–4591 [DOI] [PubMed]
- 43.Dodder NG, Strandberg B, Hites RA (2002) Environ Sci Technol 36:146–151 [DOI] [PubMed]
- 44.Stapleton HM, Baker JE (2003) Arch Environ Contam Toxicol 45:227–234 [DOI] [PubMed]
- 45.Tuerk KJS, Kucklick JR, Becker PR, Stapleton HM, Baker JE (2005) Environ Sci Technol 39:692–698 [DOI] [PubMed]
- 46.Thomsen C, Lundanes E, Becher G (2001) J Environ Monit 3:366–370 [DOI] [PubMed]
- 47.Rice CP, Chernyak SM, Begnoche L, Quintal R, Hickey J (2002) Chemosphere 49:731–737 [DOI] [PubMed]
- 48.Eljarrat E, Lacorte S, Barceló D (2002) J Mass Spectrom 37:76–84 [DOI] [PubMed]
- 49.Thomsen C, Haug LS, Leknes H, Lundanes E, Becher G, Lindstrom G (2002) Chemosphere 46:641–648 [DOI] [PubMed]
- 50.Ackerman LK, Wilson GR, Simonich SL (2005) Anal Chem 77:1979–1987 [DOI] [PubMed]
- 51.Björklund J, Tollbäck P, Östman C (2003) J Mass Spectrom 38:394–400 [DOI] [PubMed]
- 52.Alaee M, Sergeant DB, Ikonomou MG, Luross JM (2001) Chemosphere 44:1489–1495 [DOI] [PubMed]
- 53.Ikonomou MG, Rayne S (2002) Anal Chem 74:5263–5272 [DOI] [PubMed]
- 54.Wang DL, Cai ZW, Jiang GB, Wong MH, Wong WK (2005) Rapid Commun Mass Spectrom 19:83–89 [DOI] [PubMed]
- 55.Focant JF, Sjödin A, Patterson DG (2003) J Chromatogr A 1019:143–156 [DOI] [PubMed]
- 56.Čajka T, Hajšlová J, Kazda R, Poustka J (2005) J Sep Sci 28:601–611 [DOI] [PubMed]
- 57.Debrauwer L, Riu A, Jouahri M, Rathahao E, Jouanin I, Antignac JP, Cariou R, Le Bizec B, Zalko D (2005) J Chromatogr A 1082:98–109 [DOI] [PubMed]
- 58.Verreault J, Gabrielsen GV, Chu SG, Muir DCG, Andersen M, Hamaed A, Letcher RJ (2005) Environ Sci Technol 39:6021–6028 [DOI] [PubMed]
- 59.Malmberg T, Athanasiadou M, Marsh G, Brandt I, Bergman Å (2005) Environ Sci Technol 39:5342–5348 [DOI] [PubMed]
- 60.Marsh G, Athanasiadou M, Bergman Å, Asplund L (2004) Environ Sci Technol 38:10–18 [DOI] [PubMed]
- 61.Marsh GA M, Athanassiadis I, Sandholm A (2005) Chemosphere 62 (in press) [DOI] [PubMed]
- 62.Hua WY, Bennett ER, Letcher RJ (2005) Environ Int 31:621–630 [DOI] [PubMed]
- 63.Korytár P, Leonards PEG, de Boer J, Brinkman UAT (2005) J Chromatogr A 1086:29–44 [DOI] [PubMed]
- 64.Stapleton HM, Keller JM, Schantz MM, Kucklick JR, Wise SA (2005) In: NIST inter-comparison exercise program for polybrominated diphenyl ethers (PBDEs) in marine sediment: description and results of the 2004 inter-comparison exercise. National Institute of Standards and Technology, Gaithersburg, pp 1–81
- 65.Schecter A, Päpke O, Tung KC, Staskal D, Birnbaum L (2004) Environ Sci Technol 38:5306–5311 [DOI] [PubMed]
- 66.Christensen JR, Macduffee M, Macdonald RW, Whiticar M, Ross PS (2005) Environ Sci Technol 39:6952–6960 [DOI] [PubMed]
- 67.Schecter A, Vuk MP, Päpke O, Ryan JJ, Birnbaum L, Rosen R (2003) Environ Health Perspect 111:1723–1729 [DOI] [PMC free article] [PubMed]
- 68.Schecter A, Päpke O, Tung KC, Joseph J, Harris TR, Dahlgren J (2005) J Occup Environ Med 47:199–211 [DOI] [PubMed]
- 69.Söderström G, Sellström U, De Wit CA, Tysklind M (2004) Environ Sci Technol 38:127–132 [DOI] [PubMed]
- 70.Eriksson J, Green N, Marsh G, Bergman Å (2004) Environ Sci Technol 38:3119–3125 [DOI] [PubMed]
- 71.Kierkegaard A, Balk L, Tjärnlund U, De Wit CA, Jansson B (1999) Environ Sci Technology 33:1612–1617 [DOI]
- 72.Tomy GT, Palace VP, Halldorson T, Braekevelt E, Danell R, Wautier K, Evans B, Brinkworth L, Fisk AT (2004) Environ Sci Technol 38:1496–1504 [DOI] [PubMed]
- 73.Stapleton HM, Letcher RJ, Baker JE (2004) Environ Sci Technol 38:1054–1061 [DOI] [PubMed]
- 74.Thuresson KHP, Hagmar L, Sjödin A, Bergman Å, Jakobsson K (2006) Environ Health Perspect 114:179–181 [DOI] [PMC free article] [PubMed]
- 75.La Guardia MJ, Hale RC, Harvey E (2005) Paper presented at the Society of Environmental Toxicology and Chemistry. Abstract vol 26. Society of Environmental Toxicology and Chemistry, Baltimore, p 80