

Summary
PGC1β is a transcriptional coactivator that potently stimulates mitochondrial biogenesis and respiration of cells. Here, we have generated mice lacking exons 3 to 4 of the Pgc1β gene (PGC1β E3,4-/E3,4- mice). These mice express a mutant protein that has reduced coactivation activity on a subset of transcription factors, including ERRα, a major target of PGC1β in the induction of mitochondrial gene expression. The mutant mice have reduced expression of OXPHOS genes and mitochondrial dysfunction in liver and skeletal muscle as well as elevated liver triglycerides. Euglycemic-hyperinsulinemic clamp and insulin signaling studies show that PGC1β mutant mice have normal skeletal muscle response to insulin, but have hepatic insulin resistance. These results demonstrate that PGC1β is required for normal expression of OXPHOS genes and mitochondrial function in liver and skeletal muscle. Importantly, these abnormalities do not cause insulin resistance in skeletal muscle but cause substantially reduced insulin action in the liver.
Introduction
Type 2 diabetes mellitus (DM2) is one of the most common metabolic disorders worldwide, and is a major cause of blindness, renal failure, limb gangrene and cardiovascular disease (Kahn, 1998; Zimmet et al., 2001). The primary cause of DM2 remains to be understood. It is clear, however, that insulin resistance is an early feature of DM2 (Shulman, 2000). Insulin resistance is consistently found in DM2 subjects and may be detected decades before the onset of DM2 (Lillioja et al., 1988; Lillioja et al., 1993; Warram et al., 1990). Moreover, in subjects with a family history of DM2, insulin resistance is the best predictor for the development of DM2 (Lillioja et al., 1993; Warram et al., 1990).
Insulin resistance is the failure of target tissues to respond properly to insulin. Liver and skeletal muscle are among the major sites of insulin action. Insulin reduces liver glucose production and stimulates glucose uptake into skeletal muscle. The development of insulin resistance in these tissues accounts for major alterations in glucose metabolism seen in DM2 subjects. Various factors have been implicated as causally related to insulin resistance (Lazar, 2005; Lowell and Shulman, 2005); nevertheless the underlying cellular mechanisms of insulin resistance in these tissues remain to be fully elucidated.
Recently, two independent gene expression profile studies identified a set of genes consistently downregulated in skeletal muscle of DM2 patients: oxidative phosphorylation (OXPHOS) genes (Mootha et al., 2003; Patti et al., 2003). Among them, there were several genes encoding subunits of Complex I to V of the respiratory chain, which were modestly (20%) but coordinately reduced. Moreover, the same set of genes were also found to be reduced in pre-diabetics (with a DM2 family history) (Patti et al., 2003), demonstrating that reduction of OXPHOS gene expression was present very early during the development of the disease and may have a causal role in the onset of DM2. Consistent with the downregulation of OXPHOS genes expression, other studies have reported an impairment of mitochondrial oxidative phosphorylation function in skeletal muscle of diabetic and pre-diabetic subjects (Kelley et al., 2002; Petersen et al., 2003; Petersen et al., 2004; Ritov et al., 2005). For instance, young lean insulin resistant offspring of parents with DM2 were found to have a 30% reduction in rates of mitochondrial ATP synthesis in skeletal muscle assessed by 31P NMR, which was associated with increases in intramyocellular lipid content (Petersen et al., 2004). Furthermore healthy, lean, elderly subjects were found to have severe skeletal muscle insulin resistance which was associated with increased intramyocellular and hepatic triglyceride content and reduced rates of mitochondrial oxidative phosphorylation activity compared to younger, activity- and body-weight matched volunteers (Petersen et al., 2003). Finally, recent studies in muscle biopsy samples obtained from patients with DM2 have found a direct correlation between electron transport chain activity or mitochondria area to the rate of insulin stimulated glucose disposal (Kelley et al., 2002; Ritov et al., 2005). While all of these studies have implicated mitochondrial dysfunction in the pathogenesis of insulin resistance in humans it remains to be determined whether mitochondria dysfuntion precedes or follows the development of insulin resistance.
Of note, peroxisome proliferator activator receptor γ coactivator 1-α (PGC1α) and 1-β (PGC1β) were also significantly less expressed in insulin resistant skeletal muscle (Mootha et al., 2003; Patti et al., 2003). In contrast, Morino et al. (2005) have recently shown that PGC1α and PGC1β expression in skeletal muscle of young, lean, insulin resistant offspring of parents with DM2 was not altered. PGC1α is a transcriptional coactivator, which has been shown to modulate the expression of many genes in different biological programs (Puigserver and Spiegelman, 2003) including mitochondrial oxidative metabolism in vitro and in vivo. It promotes mitochondrial biogenesis and cellular respiration in several cell types, and also when it is expressed transgenically in mice (Lehman et al., 2000; St-Pierre et al., 2003; Wu et al., 1999). Moreover, DNA microarray analysis following adenoviral-mediated PGC1α expression in C2C12 muscle cells has shown increased expression of a subset of OXPHOS genes that greatly overlap with those downregulated in skeletal muscle of diabetic/pre-diabetic humans (Mootha et al., 2004). Consistent with this, a large number of OXPHOS genes were shown to be reduced by 20–50% in skeletal muscle of PGC1α null mice (Arany et al., 2005).
PGC1β was recently identified as the closest homolog to PGC1α (Lin et al., 2002). PGC1β has a wide tissue distribution, but it is most highly expressed in tissues of high oxidative metabolism such as brown adipose tissue (BAT), heart and skeletal muscle (Lin et al., 2002). It coactivates a broad range of nuclear receptors/transcriptional factors (Kressler et al., 2002; Lin et al., 2002; Meirnaghae et al., 2003), which partially overlaps with PGC1α’s target transcription factors. There are also transcriptional factors that are exclusively coactivated by PGC1β but not by PGC1α, such as the SREBP family (Lin et al., 2004). PGC1β has been far less studied than PGC1α; nevertheless there is evidence that PGC1β also regulates mitochondrial metabolism. Adenoviral-mediated PGC1β expression in muscle cells, primary rat hepatocytes and rat liver induces the expression of genes of mitochondrial oxidative metabolism, such as cytochrome C and β-ATPase (Lin et al., 2003; St-Pierre et al., 2003). In addition, PGC1β strongly promotes mitochondrial proliferation and cell respiration in muscle cells, (St-Pierre et al., 2003). These results strongly suggest that PGC1β is a major regulator of mitochondrial metabolism.
In the present study, we have generated mice lacking exons 3 to 4 of the Pgc1β gene (Pgc1β E3, 4 -/ E3, 4 – mice) and used these animals to investigate the function of PGC1β in mitochondrial gene expression and function in liver and skeletal muscle. We have also directly addressed the question of whether a mutation in PGC1β causes insulin resistance in mice.
Results
Generation of Pgc1β mutant mice
Mice homozygous for Pgc1β allele lacking sequences from exon 3 to exon 4 (Pgc1β E3, 4 -/ E3, 4 -) (Figure 1A) were generated. Our strategy was originally designed in attempt to generate Pgc1β null mice. The sequence from exon 3 to 4, containing two out of the three LXXLL motifs present in mouse PGC1β, was replaced by a neomycin cassette such that the splicing acceptor sequences of exon 3 were retained and the splicing donor sequences of intron 4 were deleted. Our expectation was that the neomycin cassette would be retained in the mature mRNA and thus disrupts the reading frame of PGC1β. However, this cassette was spliced out of the mature mRNA and a mutant transcript lacking the sequence from exon 3 to exon 4 was generated. Heterozygous mice (Pgc1β+/ E3, 4-) were mated and wild type (WT), heterozygous and homozygous offspring (Figure 1B) were obtained at the ratio consistent with Mendelian transmission (1:2:1), thus indicating that deletion of exons 3 to 4 of Pgc1β did not cause lethality. The absence of transcripts containing exons 3 and 4 was verified by Northern blot analyses. Total RNA samples from BAT and heart were analyzed with a probe spanning exons 3 to 4. Both PGC1β ‘s transcripts (~9 and 4 kb) were detected in Pgc1β +/+ RNA samples but were absent in Pgc1β E3, 4 -/ E3, 4 – RNA samples (Figure 1C), indicating the absence of mRNA containing exons 3 and 4 in Pgc1β E3, 4 -/ E3, 4 – mice. In addition, total RNA of BAT was analyzed with a probe spanning exon 5 of Pgc1β, which is outside of the deleted region. Again, two Pgc1β transcripts were observed in the Pgc1β +/+ BAT RNA sample. However, two Pgc1β transcripts were also observed in the Pgc1β E3, 4 -/ E3, 4 – BAT RNA sample. These were approximately 0.3kb smaller than the former (Figure 1D). The combined length of exons 3 and 4 is 330 nucleotides and a splicing event from exon 2 to exon 5 in Pgc1β heterogeneous nuclear RNA was a possible explanation for the occurrence of such transcripts in Pgc1β E3, 4 -/ E3, 4 – mice. This possibility was tested by RT-PCR analysis of BAT, liver and muscle total RNA, using primers spanning exons 2 and 5. Amplicons of ~1.1kb and ~0.8kb were observed in Pgc1β +/+ and Pgc1β E3, 4 -/ E3, 4 – samples, respectively (Figure 1E). The splicing event from Pgc1β exon 2 to exon 5 in Pgc1β E3, 4 -/ E3, 4 – mice was confirmed by sequencing of the latter (0.8kb) amplicon. The nucleotide sequence of transcripts lacking exons 3 and 4 is in frame with PGC1β’s start codon and the predicted amino acid sequence would lack 110 amino acids (from AA 85 to AA 194). The presence of this protein was confirmed in BAT lysates by Western blot analyses using antisera raised against the N-terminal region of PGC1β (AA 1 to AA 350) (Figure 1F). Of note, the amount of PGC1β mutant (MT) detected was substantially reduced. However, this is due to decreased antigen reactivity because of the lack of protein encoded by exons 3 and 4, since we have demonstrated that the PGC1β antisera detects the MT protein at an efficiency of ~25–50% compared to the WT protein (Figure 1F and Figure 4S). Thus, expression of the mutant protein is approximately equal to expression of the WT protein.
Figure 1.
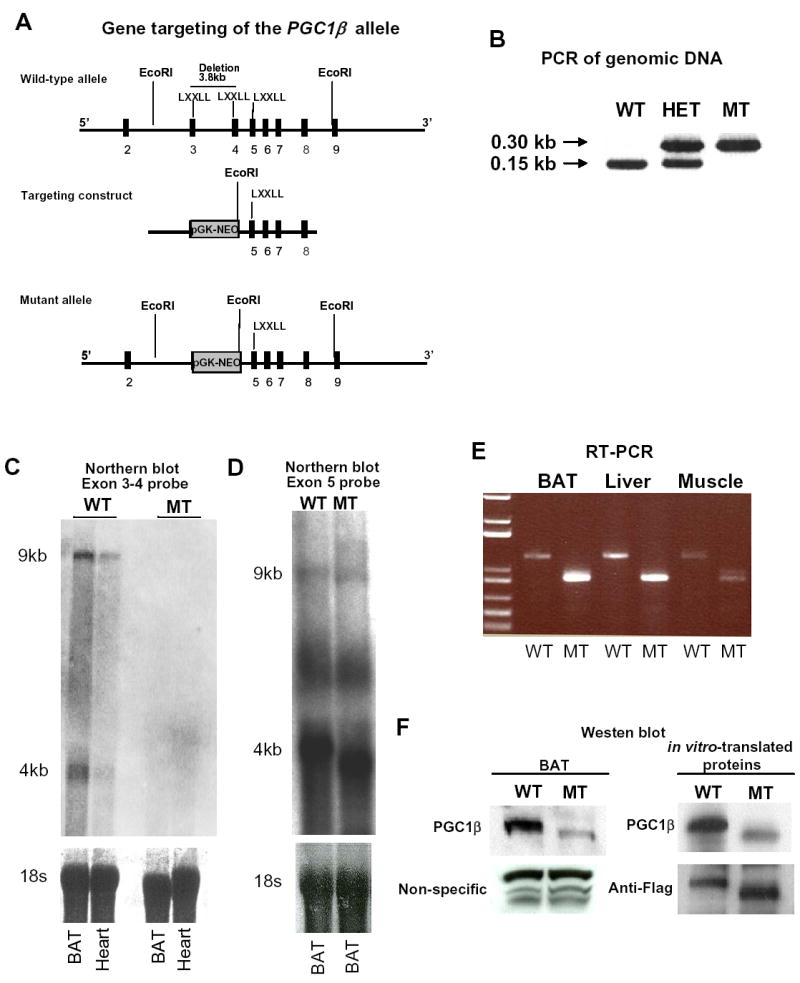
Generation of Pgc1β exon 3 to 4 null mice (Pgc1βE3,4-/E3,4- mice). (A) Schematic representation of the wild-type Pgc1β allele, the targeting construct and the mutant Pgc1β allele after homologous recombination. Exons are represented by filled boxes. EcoRI restriction sites and the probe (represented by an open box) used for Southern blot for genotyping of ES clones are shown. The deletion of exon 3 to 4 includes two out of the three canonical LXXLL motifs present in the wild-type Pgc1β gene. (B) Genotyping by PCR analysis of tail genomic DNA of offsprings of heterozygous (HET) crosses. The 150 bp and 300 bp amplicons for the wild-type (WT) and the mutant Pgc1β (MT) alleles, respectively, are indicated. (C) Northern blot analysis of brown fat (BAT) and heart total RNA. A probe spanning Pgc1β exons 3 and 4 was used. Pgc1β mRNA signal was detected as two bands at ~9 and ~4 kb in Pgc1β +/+ BAT and heart samples. (D) Northern blot analysis of BAT total RNA. A probe spanning Pgc1β exon 5 was used. Pgc1β mRNA was detected as two bands at ~9 and ~4 kb in Pgc1β +/+ BAT mRNA sample. The MT Pgc1β mRNA was detected as two bands at ~8.7 and ~3.7 kb in Pgc1β E3,4-/E3,4- BAT mRNA sample (E). RT-PCR analysis of BAT, liver and skeletal muscle total RNA using primers located in Pgc1β exon 2 and exon 5. The amplicons were 650 bp and 350 bp for WT (left lanes) and MT (right lanes) mRNAs, respectively. (F) Western blot analysis using rabbit polyclonal antisera against the N-terminal (AA 1-350) of the mouse PGC1β, as the primary antibody. (Left panel) The specific signal for PGC1β was detected at ~160 kD in Pgc1β +/+ BAT lysate. The PGC1β MT signal was detected at ~150 kD in Pgc1β E3,4-/E3,4- BAT lysate. (Right panel) In vitro-translated WT or MT PGC1β proteins were used as controls. As loading controls, it was used rabbit polyclonal antisera against YY1 (left panel) and mouse monoclonal Anti-Flag antibody (right panel). Note the reduced affinity of the antisera against PGC1β MT.
Figure 4.
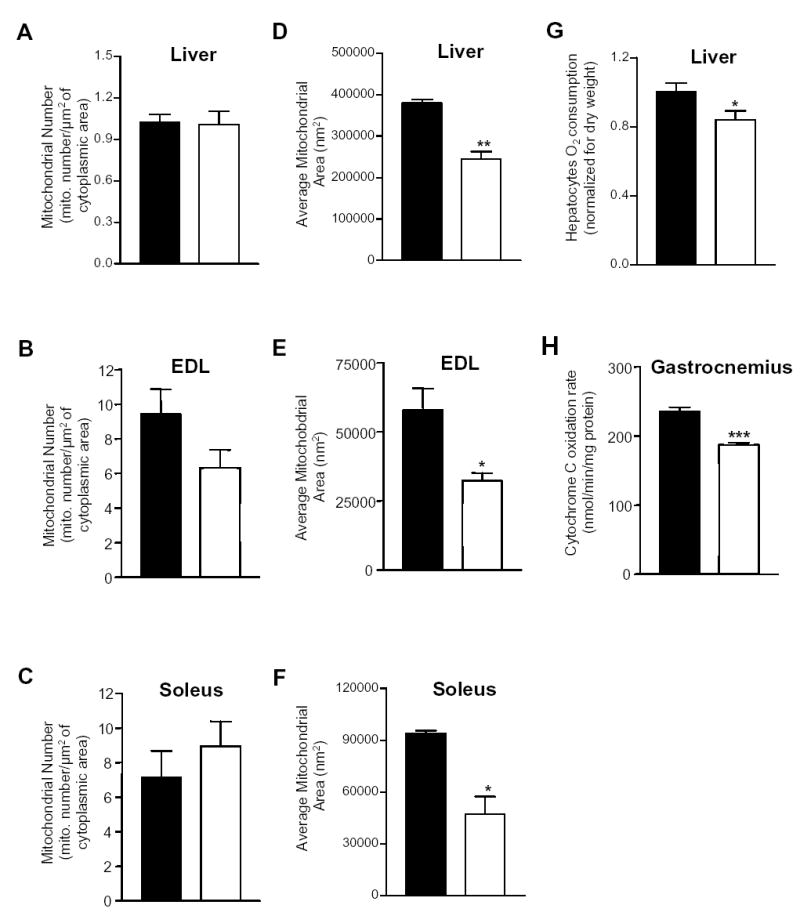
Reduced mitochondrial area and function in liver and skeletal muscle of Pgc1β E3, 4 -/ E3, 4 – mice. Mitochondria number on electron micrographs of (A) liver, (B) extensor digitorum longus (EDL) and (C) soleus skeletal muscles from Pgc1β +/+ and Pgc1β E3, 4 -/E3, 4 – mice. Average mitochondrial area was obtained by computer-assisted morphometry on electron micrographs of (D) liver, (E) EDL and (F) soleus skeletal muscles from Pgc1β +/+ and Pgc1β E3, 4 -/E3, 4 – mice. (G) Basal O2 consumption was measured in hepatocytes of Pgc1β +/+ and Pgc1β E3, 4 -/E3, 4 – mice. (H) Cytochrome C activity of gastrocnemius skeletal muscle samples of Pgc1β +/+ and Pgc1β E3, 4 -/E3, 4 – mice. Bars represent samples derived from Pgc1β +/+ mice (black) and Pgc1β E3, 4 -/E3, 4 – mice (white). Results are expressed as mean ± SEM (n=3–8). *P < 0.05; **P < 0.01; ***P < 0.001.
Reduced coactivation of a subset of nuclear receptors by PGC1β mutant protein
The LXXLL sequence is a very important ligand-dependent nuclear hormone receptor-interacting motif of nuclear receptors (e.g. Savkur and Burris, 2004). The human PGC1β contains two LXXLL motifs and the LXXLL motif encoded by exon 4 has been shown to be crucial for interaction with ERα (Kressler et al., 2002). Thus, it seemed likely that the amino acids encoded by exons 3 and 4 of PGC1β, which includes two of three LXXLL motifs present in mouse PGC1β, would be required for its coactivation activity toward some nuclear receptors. To test this, we transiently co-transfected H2.35 hepatoma cells with plasmids encoding nuclear receptors or other transcription factors, and equal amounts of plasmids encoding the PGC1β WT or PGC1β MT protein. ERRα has been shown to be a critical factor in the earliest stages of mitochondrial biogenesis induced by PGC1β (Mootha, et al., 2004; Schreiber et al., 2004). As shown in Fig 2A, ERRα transactivates its own promoter and PGC1β increases its transcriptional activity by ~14 fold; PGC1β MT is substantially reduced in this activity, increasing reporter gene expression by only ~3 fold. NRF1 is also a crucial factor in mitochondrial gene expression induced by the PGC-1s (Mootha, et al, 2004). NRF-1 binds to NRF1 recognitions sites and PGC1β WT activates its transcriptional activity by nearly 3 fold. In contrast, activation by PGC1β MT is negligible. Similarly, HCF fused to a Gal 4 DNA binding domain binds to GAL 4 DNA binding motifs (UAS) and PGC1β WT activates its transcriptional activity by ~20 fold, whereas PGC1β MT increases it by only ~5 fold (Figure 2B and 2C). These results suggest that amino acids encoded by exons 3 and 4 are required for full coactivation of the transcriptional factors ERRα, NRF1 and HCF. In sharp contrast, the mutant protein has nearly normal activity on several other transcription factors. SREBP1c fused to a Gal 4 DNA binding domain binds to a Gal 4 luciferase reporter plasmid and the WT activates its transcriptional activity by more than 3 fold (Figure 2D). PGC1β MT activates its transactivation activity more than 2 fold. Similar results were also obtained using SREBP1c on a fatty acid synthase promoter luciferase reporter plasmid (Supplemental Figure 1A). Coactivation of LXRα and PPARγ by PGC1β WT or PGC1β MT was also tested (data not shown) in the absence and presence of their respective ligands, T0901317 and rosiglitazone respectively. PGC1β WT and PGC1β MT were found to similarly activate those nuclear receptors in the absence and presence of their ligands. These results suggests that coactivation of SREBP1c, LXRα and PPARγ is not greatly affected by deletion of amino acids encoded by exons 3 and 4. Taken together, this data suggests that amino acids 85 to 194 of PGC1β are required for coactivation of ERRα, NRF1 and HCF, but likely not for coactivation of SREBP1c, LXRα and PPARγ.
Figure 2.
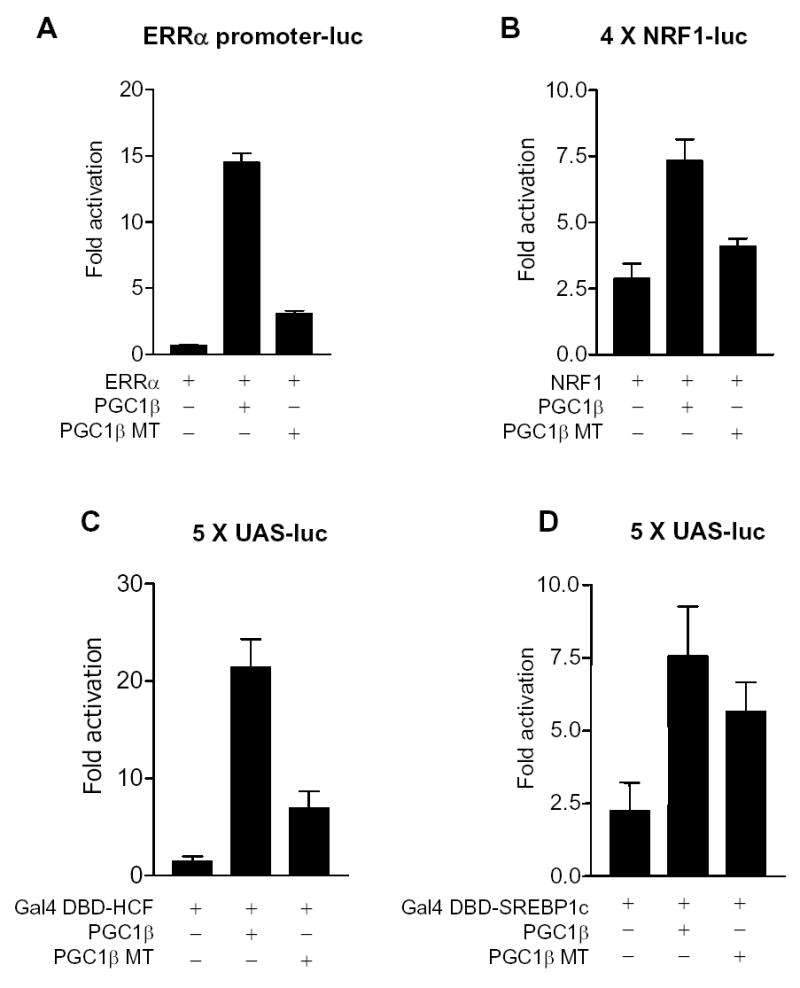
Reduced coactivation of a subset of nuclear receptors/transcription factors by PGC1β mutant protein. Mouse H2.35 hepatoma cells were transiently cotransfected with either (A) ERRα-promoter luciferase reporter and ERRα plasmids, (B) 4xNRF1-Luciferase reporter and NRF1 plasmids, (C) UAS-luciferase reporter and gal-DBD-HCF plasmids, (D) UAS-luciferase reporter and gal-DBD-SREBP1c plasmids along with pCATCH-PGC1β or pCATCH-PGC1β mutant (MT) plasmid. pCMV-βGal was added for transfection efficiency normalization. Cells were harvested 48 hours after cotransfection and assayed for luciferase and β-galactosidase assays. The results represent the means ± SEM of at least three independent experiments.
Reduced expression of OXPHOS genes in liver and skeletal muscle of PGC1β mutant mice
To investigate whether this mutation in PGC1β results in alterations in gene expression and several physiological functions, we first performed transcriptional microarray analysis on RNA samples obtained from liver and skeletal muscle. In liver, clustering analysis of the microarray data revealed that expression of many genes of mitochondrial metabolism were downregulated by 30–40% in Pgc1β E3, 4 -/ E3, 4 – mice compared to Pgc1β +/+ mice (Figure 3A). These genes include Ndufv1, Ndufs1 and Ndufs7 (Complex I subunits), Uqcrc1 (complex III subunit), Coq7 (ubiquinone synthesis) Aco2 and Dlst (Krebs cycle) and Sod2 (superoxide removal). G6pc and CYP7A expression were also found to be reduced. To validate the reduced expression of these genes, we performed Northern blot analysis (Figure 3B). Consistent with the microarray data, the Northern blots confirmed a reduction in the expression of these genes. Importantly, all of the genes previously described as targets of PGC1β-SREBP1c interaction (Fasn, Scd1 and Hmgcr) (Lin et al., 2005) were equally expressed in the livers of PGC1β+/+ and PGC1βE3, 4 -/ E3, 4 – mice. The LXRα target genes (Pltp, Abca1, Abcg1, Abcg8, Add1) were similarly expressed in the two groups, with the exception of CYP7A, which was markedly reduced in liver of PGC1βE3, 4 -/ E3, 4 –mice. Quantitative PCR demonstrated that Coq7 and Ndufs1 mRNA levels were normal in heterozygous mice, suggesting no dominant negative or gain-of-function effect of the mutant allele. Ndufv1 mRNA levels were intermediate between wild-type and homozygous mice which is consistent with a gene dosage effect (Supplemental Figure 1B). Finally, expression of Pgc1α mRNA (as assessed by q-PCR) was not altered in Pgc1β E3, 4 -/ E3, 4 – mice, indicating that PGC1α did not compensate for decreased PGC1β function in liver (data not shown).
Figure 3.
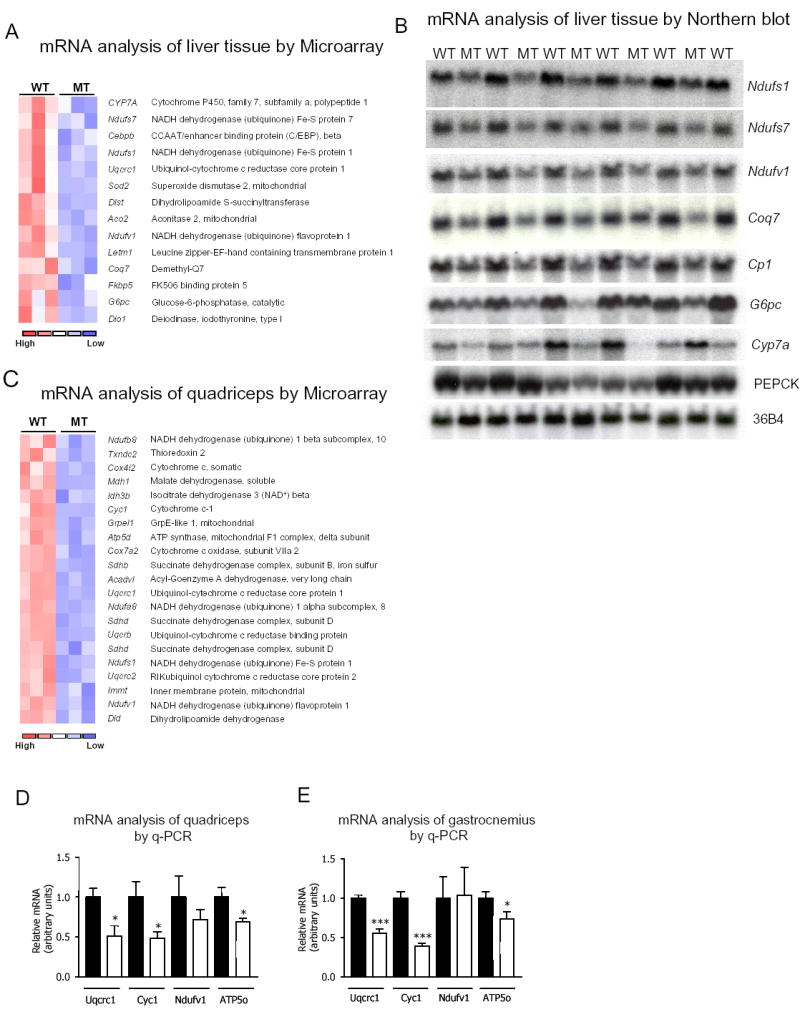
Reduced expression of OXPHOS genes in liver and skeletal muscle of Pgc1βE3, 4 -/ E3, 4-mice. (A) Clustering analysis of liver microarray data of Pgc1β +/+ (WT) and Pgc1β E3, 4 -/ E3, 4 – (MT) mRNA samples. Genes that were reduced in MT samples are indicated. (B) Northern blot analysis of WT and MT liver total RNA samples for validation of the microarray data. A probe specific for ribosomal protein 36B4 was included as a loading control. (C) Clustering analysis of quadriceps skeletal muscle microarray data from Pgc1β +/+ (WT) and Pgc1β E3, 4 -/ E3, 4 – (MT) mRNA samples. Genes reduced in MT samples are indicated. (D) TaqMan quantitative real-time PCR (Q-PCR) analysis of WT and MT quadriceps total RNA samples for validation of the microarray data. (E) The expression of the same genes as in (D) was assessed by Q-PCR analysis of WT and MT gastrocnemius total RNA. The mRNA content of specific genes was normalized to cyclophylin mRNA content. The mRNA levels are expressed relative to the WT values. Black bars represent WT; white bars represent MT. Results are expressed as mean ± SEM (n=8–11). *P < 0.05; ***P < 0.001.
Clustering analysis of the microarray from quadriceps muscle revealed that expression of several genes of mitochondrial oxidative metabolism were reduced in Pgc1β E3, 4 -/E3, 4 – RNA samples. These genes were reduced by 20–30% and most of them were OXPHOS genes encoding subunits of Complexes I to V. Of note, Ndufv1, Ndufa8, Ndufs1, Ndufb10 (complex I subunits), Sdhb, Sdhd (complex II subunits), Uqcr, Uqcrc1, Uqcrc2 (complex III subunits), Cox7a2 (complex IV subunit), ATP5o, ATP5d (Complex V) and cytochrome C were all reduced in quadriceps muscle of Pgc1β E3, 4 -/ E3, 4 – mice (Figure 3C). Q-PCR assays were performed for selected OXPHOS genes to confirm the expression pattern shown by the microarray data. These studies revealed mRNA content to be reduced by ~30–50% in quadriceps muscle of Pgc1β E3, 4 -/ E3, 4 – mice (Figure 3D). Quadriceps muscle is composed mostly of type II fibers (white, glycolitic), and we asked whether a similar reduction in those OXPHOS genes would be present in a muscle such as gastrocnemius, which contains both type I (red, oxidative) and II fibers. The q-PCR assays revealed a marked reduction (30–60%) in mRNA content of OXPHOS genes in gastrocnemius of Pgc1β E3, 4 -/ E3, 4 –mice (Figure 3E). Also, we performed in gastrocnemius samples q-PCR assays for genes encoding proteins characteristic of type I fibers, such as troponin I (slow) and myoglobin, which are under PGC1α’s regulation (Lin et al., 2002). No differences in the expression of these genes were observed comparing samples of Pgc1β E3, 4 -/ E3, 4– and Pgc1β+/+ mice (data not shown). Taken together, these results demonstrate conclusively that the amino acids encoded by exons 3 and 4 of PGC1β are required for normal expression of many genes of mitochondrial oxidative metabolism in liver, and skeletal muscle.
Reduced mitochondrial area and function in liver and skeletal muscle of PGC1β mutant mice
PGC1β has been shown to strongly stimulate mitochondrial biogenesis and cellular respiration in hepatocytes and muscle cells, and this suggested that PGC1β is a critical regulator of mitochondrial proliferation and function. To test this genetically, we first performed morphometric analysis of liver and skeletal muscle (extensor digitorum longus (EDL) and soleus) by quantitative electron microscopy. Liver and both types of skeletal muscles, EDL (white, glycolitic) and soleus (red, oxidative) of Pgc1β E3, 4 -/ E3, 4 – mice had a similar number of mitochondria as observed in samples derived from PGC1β+/+ mice (Figure 4A–4C). However, in liver, EDL and soleus, mitochondria area was greatly reduced in Pgc1β E3, 4 -/ E3, 4 – samples compared to those of PGC1β +/+ mice (Figure 4D–4F).
We next assessed mitochondrial function as measured by O2 consumption of isolated hepatocytes. A significant reduction of basal O2 consumption (15%) was observed (Figure 4G). We also assessed Complex IV (COX) activity, which is a critical component of the electron transport chain, in gastrocnemius muscle homogenates. COX activity of Pgc1β E3, 4 -/ E3, 4 – muscle homogenate was markedly reduced compared to that of PGC1β+/+ mice (Figure 4H). These results show that PGC1β is required to maintain normal mitochondria function in liver and skeletal muscle.
Increased hepatic triglyceride content in the liver of PGC1β mutant mice
Despite clear data that this is an early correlate of insulin resistance in humans, it is unclear whether reduced expression of the PGC-1 coactivators and OXPHOS genes is a causal factor in insulin resistance. Thus, we investigated several parameters involved in the insulin/glucose homeostasis in Pgc1β E3, 4 -/ E3, 4 – mice. As shown in Table 1, both genotypes had similar blood glucose and fatty acids serum levels in both the fed and fasted states. Serum triglycerides levels tended to be lower in fed Pgc1β E3, 4 -/ E3, 4 – mice, but similar in both genotypes in fasted mice (Table 1). Cholesterol and insulin serum levels were normal in fed Pgc1β E3, 4 -/ E3, 4 – mice. However, a trend towards elevated insulin serum levels were observed in fasted Pgc1β E3, 4 -/ E3, 4 – mice (Table 1). Serum adiponectin levels, total body fat content, and subscapular brown fat pad weights were normal in Pgc1β E3, 4 -/ E3, 4 – mice (Table 1). Impairment of mitochondrial oxidative metabolism may cause lipid accumulation, which has been strongly associated with insulin resistance in liver and skeletal muscle (Shulman, 2000). Thus, we measured triglycerides content in liver and skeletal muscle. Triglycerides were significantly increased in Pgc1β E3, 4 -/ E3, 4 – livers, compared to Pgc1β+/+ livers, but not in quadriceps, gastrocnemius or soleus muscles (Table 1). Of note, liver triglyceride levels were normal in heterozygous mice (Supplemental Figure 1C).
Table 1.
Body and blood composition of PGC1β+/+ (WT) and Pgc1βE3,4-/E3,4- (MT) male mice.
| WT | MT | |
|---|---|---|
| Glucose (mg/dL) | ||
| Fed | 129 ± 7 (10) | 127 ± 6 (11) |
| Fasted | 63 ± 3 (13) | 66 ± 2 (13) |
| Insulin (ng/mL) | ||
| Fed | 1.41 ± 0.13 (14) | 1.62 ± 0.13 (15) |
| Fasted | 0.28 ± 0.07 (13) | 0.36 ± 0.05 (16) |
| Adiponectin (μg/mL) | ||
| Fed | 8.69 ± 0.99 (6) | 8.98 ± 1.04 (6) |
| Fatty acids (mM) | ||
| Fed | 0.67 ± 0.05 (11) | 0.77 ± 0.07 (10) |
| Fasted | 1.38 ± 0.09 (14) | 1.28 ± 0.09 (13) |
| Triglycerides (mg/dL) | ||
| Fed | 90.3 ± 12.7 (9) | 64.8 ± 8.0 (9) |
| Fasted | 109.8 ± 23.2 (6) | 84.7 ± 24.9 (6) |
| Cholesterol (mg/dL) | ||
| Fed | 148.2 ± 11.3 (5) | 158.8 ± 12.5 (5) |
| Liver triglycerides (mg/g tissue) | ||
| Fed | 3.82 ± 0.58 (13) | 7.76 ± 0.75 (14) *** |
| Muscle triglycerides (mg/g tissue) | ||
| Quadriceps | 6.61 ± 2.54 (4) | 2.39 ± 1.21 (4) |
| Gastrocnemius | 4.11 ± 0.81 (10) | 3.81 ± 1.28 (8) |
| Soleus | 10.07 ± 2.31 (6) | 11.86 ± 2.45 (6) |
| White adipose tissue (WAT) mass | ||
| Tissue mass (g) | 4.85 ± 0.88 (5) | 4.99 ± 0.59 (5) |
| WAT mass/to body mass (%/g) | 20.58 ± 2.35 (5) | 21.76 ± 2.94 (5) |
| Brown adipose fat pad weight (mg) | ||
| Subscapular | 87.9 ± 15.29 (8) | 83.5 ± 14.60 (6) |
Parameters were assessed in mice at 9–12 weeks of age. All data represent the mean ± SEM.
The number of animal per group is shown in parentheses.
Hepatic insulin resistance in PGC1β mutant mice
To further investigate insulin sensitivity and whole body glucose homeostasis, euglycemic-hyperinsulinemic clamp studies were performed (Figure 5A–5D). Glucose infusion rate during the clamps was similar in Pgc1β E3, 4 -/ E3, 4 – mice and in PGC1β+/+ mice (Figure 5A–5B). Whole body glycolysis and glycogen synthesis were also similar in both genotypes (Figure 5C). Skeletal muscle glucose uptake was also similar (Figure 5C), indicating absence of skeletal muscle insulin resistance in Pgc1β E3, 4 -/ E3, 4 – mice. However, while insulin significantly reduced hepatic glucose production rates in PGC1β+/+ mice during the euglycemic-hyperinsulinemic clamp, it had no effect on hepatic glucose production rates in Pgc1β E3, 4 -/ E3, 4 – mice (Figure 5D). These results indicate the presence of striking insulin resistance in the liver of the Pgc1β mutant mice.
Figure 5.
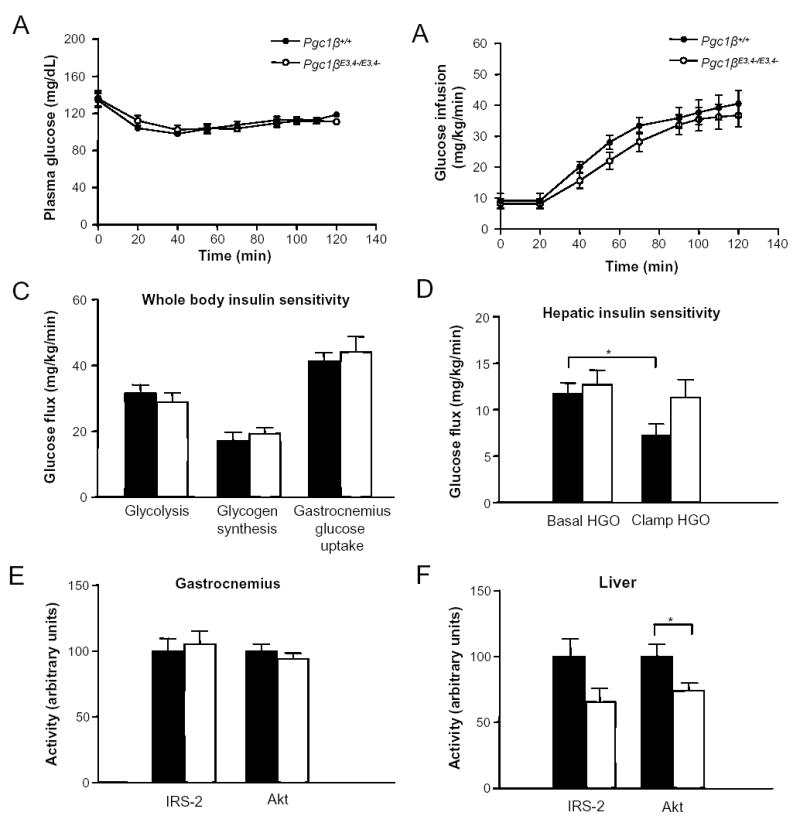
Hyperinsulinemic-euglycemic clamp study and intracellular insulin signaling in Pgc1βE3, 4 -/ E3, 4 - mice. Hyperinsulinemic-euglycemic clamp studies were performed in (A–D) on chow-fed 18–20 weeks-old Pgc1β +/ + and Pgc1β E3, 4 -/ E3, 4 – male mice. (A) Plasma glucose levels and (B) glucose infusion rate during clamp study are shown. (C) Whole body glycolysis and glycogen synthesis, gastrocnemius glucose uptake and (D) hepatic insulin sensitivity parameters are shown. (E–F) IRS-1- or IRS-2-associated PI3-kinase activity and Akt activity in (E) gastrocnemius and (F) liver. Mice were injected with 2.5 mU/kg/min of insulin. Bars represent Pgc1β +/ + (black) and Pgc1β E3, 4 -/ E3, 4 – (white) mice. Results are expressed as mean ± SEM (n=6–11). *P < 0.05.
To further explore the mechanism for this, we measured the ability of insulin to activate the PI3K-Akt pathway in liver and muscle of the Pgc1β mutant mice. Specifically, we assessed IRS-associated PI3-kinase activity and AKT activity in liver and muscle in response to the hyperinsulinemic conditions of the clamp (Figure 5E and 5F). Insulin-stimulated IRS-1-associated PI3-kinase activity and Akt activity was similar in WT and mutant samples of gastrocnemius samples. In liver of Pgc1β E3, 4 -/ E3, 4 – mice, insulin-stimulated IRS-2-associated PI3-kinase activity tended to be reduced and Akt activity was significantly reduced compared to WT samples. These results demonstrate reduction of biochemical actions of insulin in liver of Pgc1β E3, 4 -/ E3, 4 – mice, but no alterations in skeletal muscle. Together, these results suggest that insulin resistance in the liver of Pgc1β E3, 4 -/ E3, 4 – mice is explained, at least in part, by decreased insulin-stimulated PI3K-Akt activation.
Finally, to assess whether this liver insulin resistance would cause whole body glucose intolerance and/or insulin resistance, intraperitoneal insulin tolerance tests (ITT) and glucose tolerance tests (GTT) were performed on mice of both genotypes. At both 4 and 9 months of age, PGC1β+/+ and Pgc1β E3, 4 -/ E3, 4 – mice had similar blood glucose concentrations during GTT (data not shown). Also, according to the ITT in 4 and 9 months old mice, a similar suppression of blood glucose levels was observed in both PGC1β+/+ and Pgc1β E3, 4 -/ E3, 4 – mice (Figure 6A and 6B). These results indicate that although Pgc1β E3, 4 -/ E3, 4 – mice have quite significant hepatic insulin resistance, there is sufficient compensation by other tissues and/or physiological systems to prevent whole body glucose intolerance and/or insulin resistance. Finally, to see if Pgc1β E3, 4 -/ E3, 4 – mice have altered sensitivity to high fat diet-induced insulin resistance, we fed mice a high fat diet for 12 weeks. Glucose levels in the fed state were equal in wild-type and Pgc1β E3, 4 -/ E3, 4 –mice (data not shown). Also, insulin tolerance tests were similar in high fat fed wild-type versus Pgc1β E3, 4 -/ E3, 4 – mice (Supplemental Figure 2A).
Figure 6.
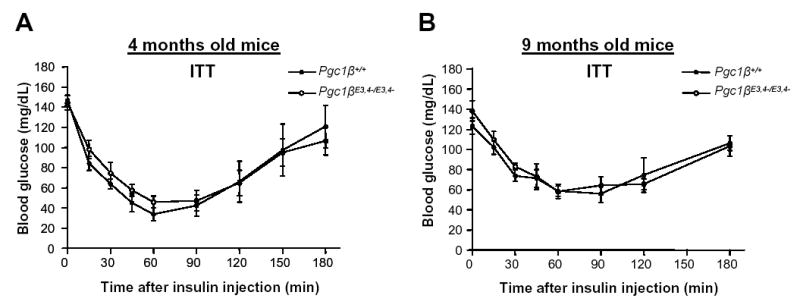
Insulin tolerance tests in aged Pgc1β +/ + and Pgc1β E3, 4 -/ E3, 4 – male mice. (A–B) Intraperitoneal insulin tolerance test (ITT) was performed in male (E) 4 and (F) 9-month-old Pgc1β +/ + and Pgc1β E3, 4 -/ E3, 4 – mice. Following 4 hours without food, mice were injected with insulin 1.5 U/Kg of body weight. Results are expressed as mean ± SEM (n=5–8).
Discussion
Several reports have suggested that reduced expression of genes encoding mitochondrial enzymes of oxidative phosphorylation (OXPHOS) in skeletal muscle could play a critical role in causing insulin resistance. However, whether these alterations precede and cause the development of insulin resistance is presently unknown (Lowell and Shulman, 2005). In this report, we were able to address this question in mice. To study the function of the transcriptional coactivator PGC1β, which has been suggested to regulate the expression of genes of mitochondrial oxidative metabolism, we have generated mice lacking exons 3–4 of Pgc1β gene (Pgc1β E3, 4 -/ E3, 4 – mice). These mice have reduced expression of several OXPHOS genes as well as smaller and functionally impaired mitochondria in liver and skeletal muscle. The data presented here show conclusively an important role for PGC-1β as a regulator of mitochondrial biology in vivo. Furthermore, the Pgc1β E3, 4 -/ E3, 4 – mice were used as a model to test whether a mutation in PGC-1β and subsequent mitochondrial dysfunction causes insulin resistance. Importantly, Pgc1β E3, 4 -/ E3, 4 – mice are not hyperactive (Supplemental Figure 2A) and have similar body weight and fat mass to Pgc1β +/+ littermates, making it feasible to assess whether a primary reduction of OXPHOS genes is sufficient to cause insulin resistance in muscle or liver. Similar rates of skeletal muscle glucose uptake in response to insulin were observed in both genotypes of mice, indicating similar degrees of skeletal muscle insulin sensitivity in Pgc1β E3, 4 -/ E3, 4 – and Pgc1β +/+ mice. Furthermore, skeletal muscle insulin signaling, as assessed by Akt and IRS-1 associated PI3-kinase activity, were similar in Pgc1β E3, 4 -/ E3, 4 – and Pgc1β +/+ mice. Finally, a similar suppression of blood glucose levels was also observed in ITTs of both PGC1β +/+ and Pgc1β E3, 4 -/ E3, 4 – mice fed a high fat diet (Supplemental Figure 2B). These results suggest that Pgc1β mediated reductions in expression of OXPHOS genes per se does not cause insulin resistance in skeletal muscle.
It is possible, however, that downregulation of OXPHOS genes leads to the development of skeletal muscle insulin resistance, but only in the presence of other mitochondrial alterations which are absent in Pgc1β E3, 4 -/ E3, 4 – mice. Despite the fact that skeletal muscle mitochondria were functionally abnormal, we did not observe any associated increases in intracellular lipid content, which would have been anticipated if mitochondrial fatty acid oxidation was also impaired. Intramyocellular triglyceride accumulation, and more importantly increases in intracellular diacylglycerol content has been shown to cause insulin resistance in skeletal muscle and it is likely that its presence is required for the development of insulin resistance (Dresner et al., 1999; Griffin et al., 1999; Yu et al., 2002). On the other hand, it is also plausible that reduced expression of PGC1α or PGC1β is causally implicated in skeletal muscle insulin resistance, but independently of the expression of OXPHOS genes. Indeed, PGC1α has been shown to regulate the expression of Glut4 in muscle cells (Michael et al., 2001) and has also been shown to control the formation of Type1 fibers (Lin, et al., 2002), the most insulin sensitive type of skeletal muscle fibers. Further studies with specific deletion of Pgc1α and Pgc1β in skeletal muscle are required to definitively address these questions.
In contrast, this mutation in Pgc1β clearly causes dysfunction in the liver, including the induction of insulin resistance. PGC1β has been recently shown to regulate the expression of lipogenic genes and genes involved in lipid secretion by liver via the coactivation of SREBP1c, and likely LXRα, respectively (Lin et al., 2005). We show here that PGC1β is also required, in liver, for normal expression of genes encoding mitochondrial proteins, including OXPHOS genes. However, according to the liver transcriptional profile data, PGC1β-SREBP1c target genes were not reduced in Pgc1βE3, 4 -/ E3, 4 – mice. This may be explained by the fact that PGC1β MT coactivates SREBP1c normally in vitro. Indeed, this data is consistent with the known SREBP1c-docking site, being located between amino acids 350–530 of PGC1β, downstream of the region deleted in the mutant PGC1β (AA 85 to AA 194) (Lin et al., 2005).
The Pgc1β E3, 4 -/ E3, 4 – mice also show reduced mitochondrial substrate oxidation, as assessed by oxygen consumption of isolated hepatocytes, as well as an accumulation of triglycerides in the liver. Since this hypomorphic mutation causes a deficiency in expression of mitochondrial genes and mitochondrial function, it seems very likely that this is largely a consequence of the poor coactivation of ERRα and NFR-1 by the mutant PGC-1β. However, loss of coactivation of other transcription factors as a contributing factor to the phenotype of these mice also seems likely.
To examine the possibility that the Pgc1β E3, 4 -/ E3, 4– mutant allele could be functioning as a dominant negative and/or gain-of-function mutant, or alternatively as a simple loss of function mutant, we studied heterozygous animals. If the mutant was functioning in a dominant negative and/or gain-of-function manner, heterozygotes would be predicted to have abnormalities similar to those observed in homozygotes. We assessed Coq7, Ndufv1 and Ndufs1 mRNA expression in liver as well as triglyceride levels in liver, four parameters that were markedly abnormal in homozygous mice. Of note, in heterozygous mice, Coq7 and Ndufs1 mRNA levels, as well as triglyceride levels were unchanged compared to wild-type mice. Ndufv1 mRNA levels were intermediate between wild-types and homozygous mice which is consistent with a gene-dosage effect. Overall, these findings strongly suggest that abnormalities observed in Pgc1β E3, 4 -/ E3, 4 – mice are due to loss of PGC-1β function and are not due to dominant negative and/or gain-of-function effects.
An association between intrahepatic lipid accumulation and insulin resistance was observed in several previous studies of other animal models (Kim et al., 2000; Neschen et al., 2005; Samuel et al., 2004). Of course, other explanations for the intrahepatic lipid accumulation include increased hepatic lipogenesis, or reduced lipoprotein secretion. Consistent with the possibility of reduced lipoprotein secretion, serum TG levels tended to be reduced in Pgc1β E3, 4 -/ E3, 4 – mice. Related to this, it was recently shown that PGC-1β co-activates Foxa2 and induces expression of microsomal transfer protein (MTP), facilitating the secretion of VLDLs (Wolfrum and Stoffel, 2006). Given this, it is possible that MTP expression could be decreased in Pgc1β E3, 4 -/ E3, 4 – mice and, as such, could contribute to hepatic steatosis. This scenario, however, is unlikely since Mtp mRNA expression was not altered in Pgc1β E3, 4 -/ E3, 4 – mice (Supplemental Figure 1D).
In summary, these results demonstrate that Pgc1β is required for normal expression of OXPHOS genes and mitochondrial function in the liver, and suggest that mitochondria dysfunction may be a cause of hepatic insulin resistance. An understanding of the full range of the physiological effects of deletion of PGC1β must await creation of mice carrying a null mutation.
Experimental procedures
Generation of PGC1β mutant mice
The targeting plasmid was constructed using a BAC clone containing genomic DNA derived from Sv129 mouse strain. Replacement of Pgc1β gene exon 3 to 4 by a neomycin cassette, as well as cloning of DNA in pGEM-T easy vector (Promega, Madison, WI) for transfection of ES cells, was performed using homologous recombination in EL250 bacteria strain, as described by Lee et al. (2001). ES cell electroporation, selection and screening were performed using standard techniques. Neomycin-resistant ES cell clones were screened for 5′ and 3′ end recombination. Screening of 3′ end recombination was performed by Southern blot (Supplemental Figure 3A). The DNA of ES cell clones was digested with EcoRI and analyzed with a probe outside of the construct. Screening for 5′ end recombination was performed by PCR (Supplemental Figure 3B). The ES clones with recombination of both ends (5′ and 3′ end) were injected into blastulas. The chimeric founders were bred to wild-type C57/Bl6 mice and heterozygous mice were bred to obtain mice homozygous for the wild-type Pgc1β allele (PGC1β +/+) and for the mutant Pgc1β allele (PGC1β E3,4-/E3,4).
Animal Care
All the experiments were performed in accordance with Beth Israel Deaconess Medical Center and Yale University Institutional Animal Care and Use Committee (IACUC) guidelines. Mice were housed in groups of two to four at 22–24°C in a 14 hr light/10 hr dark cycle, and fed chow diet (Teklad F6 Rodent Diet 8664, Harlan Teklad, Madison, Wisconsin) and water ad libitum. A high-fat, high-sucrose diet (Research Diets, D-12331) was fed for 12 weeks, where indicated. All studies were performed with male mice 9–12 weeks of age, unless otherwise specified.
Transient transfections
pCATCH-FLAG-PGC1β was cloned as previously described (Lin et al., 2001). To clone pCATCH-FLAG-PGC1β mutant, the region spanning exon 1 to exon 5 was first amplified by RT-PCR using BAT mRNA and the following primers: sense, 5′ cctggatccGCGGGGAACGACTGCGGCGC 3′ and antisense, 5′ GCC TTT GTT TCC GTT GGC AGA G 3′ (non-PGC1β specific sequence is shown in lower case for the sense primer which includes a BamHI site). The amplified product was digested with BamHI and cloned into BamHI sites of pCATCH-FLAG-PGC1β. pCATCH-FLAG-PGC1β mutant clone (MT) was verified by DNA sequencing.
H2.35 hepatoma cell line (ATCC, Manassas, VA) were maintained in DMEM containing 4.5 g/L glucose and supplemented with 200nM dexamethasone and 4% FBS. Transient transfections were performed using SuperFect Reagent (Promega, Madison, WI) according to the manufacturer’s instructions. Cells were transiently co-transfected with the indicated luciferase reporter (100ng) and trancriptional factor/nuclear receptor (10–20ng) plasmids and equal amounts of PGC1β WT or PGC1β MT plasmids (250–800ng). pCMV-βGal (50ng) was also included as a control for transfection efficiency normalization. pCATCH without inserted PGC1β was added if necessary for transfection of equal amounts of DNA. Cells were harvested after 48 hours of transfection for Luciferase and β-galactosidase assays (Tropix Inc., Bedford, MA).
Gene transcription profile
Total RNA was extracted from liver and skeletal muscle using RNAeasy kit (Qiagen, Valencia, CA), according to the manufacturer’s instructions. Synthesis of cRNA, hybridization and scanning of the Affymetrix Murine 430 2.0 chip was performed by Dana Farber Cancer Institute Microarray Core Facility. The microarray data was analyzed by Clustering Analysis using the d-Chip software (Li and Wong, 2001). Microarray data was validated by Northern blot or q-PCR. For Northen blots, 20μg of total RNA/sample was separated on a formaldehyde gel, transferred to nylon membrane, and then hybridized with gene-specific probes. For q-PCRs, cDNA was synthesized using SuperScript™ First Strand Synthesis System for RT-PCR (Invitrogen Corp., Carlsbad, CA) and the amplifications were performed using Taqman gene expression assays (Applied Biosystems, Foster City, CA). Mtp primer (forward: 5′ATGATCCTCTTGGCAGTGCTT3′; reverse: 5′TGAGAGGCCAGTTGTGTGAC3′) and probe (5′FAM TCTCTGCTTCTTCTCCTCCTACTCTGCTTC BHQ-1 3′) where synthesized according to (Biosearch Tecnologies, Inc., Novato, CA).
Electromicrograph morphometry
Tissues were dissected and fixed in 4% paraformaldehyde in 0.1M phosphate buffer (pH 7.4). Samples were subsequently dehydrated and embedded in plastic (JB-4, Electron Microscopy Sciences). Thin sections were obtained with an MTX ultramicrotome (RMC, Tucson, AZ) and examined with a transmission electron microscope (CM10, Philips, Eindhoven, The Netherlands). Ten sections per sample were analyzed. Mitochondrial number represents the average of a total number of ~500 mitochondria analyzed per genotype.
O2 consumption and Complex IV activity
To isolate hepatocytes, mice were anesthetized and liver was perfused for 3 and 4 min with pre-warmed (42oC) Liver Perfusion and Liver Digestion media (Invitrogen Corp., Carlsbad, CA), respectively. After perfusion, liver was quickly removed, minced and put in DMEM supplemented with 20% FBS. The cell suspension was gently put through a cell strainer (70μM) and collected in a Falcon tube. The media volume was brought to 40 ml and the cells were pelleted. The pellets were resuspended in 30ml of media. Cells were washed in media 3 times in total. At the end, the cells were resuspended in a small volume of DMEM for O2 consumption assay. O2 consumption was corrected by dry weight of cells. Complex IV activity was performed as previously described (Rustin et al., 1994). Gastrocnemius muscle samples were homogenized in PBS buffer pH 7.0 (1:10 w/v) using a Teflon homogenizer. The homogenates were centrifuged (800×g, 10min) and the supernatants were diluted in Seth buffer (250 mM sucrose, 2 mM EDTA, 10 mM Tris-HCl and 50 U/mL of Heparin) at pH 7.4 for determination of cytochrome C oxidase activity. Activity was normalized by protein content.
Body composition analysis
Body composition was assessed by Dual-Energy X-ray Absorptiometry (MEC LunarCorp. Minster, OH). Brown adipose tissue mass was assessed by harvesting and weighing subscapular brown-adipose fat pads.
Serum Metabolites
Tail vein blood was collected from fed or 24 hour-fasted mice, 9–12 weeks old. Blood was assayed for glucose level using OneTouch FastTake glucometer (LifeScan Inc., Milpitas, CA) and serum was collected after blood centrifugation. Insulin levels were determined by Rat insulin ELISA (Crystal Chem. Inc., Downers Grove, Il). Serum adiponectin levels were measured by Mouse Adiponectin ELISA (Linco Research, St. Charles, Missouri). Serum fatty acids, triglycerides and cholesterol were assayed using NEFA C, L-Type TG H and Cholesterol CII kit, respectively (Wako Chemicals USA Inc., Richmond, VA).
Tissue triglycerides
To measure triglyceride content in liver and skeletal muscle, triglycerides were extracted using chloroform/MeOH. Approximately 100 mg liver, quadriceps, and gastrocnemius muscle biopsies, and ~9 mg of soleus muscle were homogenized for 1 min in 500 μl of buffer containing 50 mM Tris-HCl (pH 7.5), 5 mM EDTA, 300 mM Mannitol and 1 mM PMSF. Samples were kept in ice. 200 μl of this mix was transferred into a new tube, and then 5 μl of KOH and 800 μl of chloroform/MeOH (2:1) were added. Samples were vortexed vigorously, left for 5 min at room temperature and centrifuged at 10.000×g for 10 min. 500 μl of the bottom layer was taken and mixed to an equal volume of chloroform/MeOH/H2O (3:48:47). Samples were vortexed vigorously and spun at 10.000×g for 10 min. 180 μl of the bottom layer was transferred to a new tube and dried completely overnight. To the dried samples, 50 μl of butanol/[Triton-X114:MeOH (2:1)] (30:20) was added and TG levels were assayed using L-Type TG H kit (Wako Chemicals USA Inc., Richmond, VA).
Hyperinsulinemic-euglycemic clamps and insulin tolerance tests
Hyperinsulinemic-euglycemic clamp studies were performed with PGC1β +/+ and PGC1β E3,4-/E3,4- littermates of 18–20 weeks of age. Body fat and lean body mass were assessed by 1H magnetic resonance spectroscopy (Bruker BioSpin, Billerica, MA). Six days before the experiment an indwelling catheter was implanted into the left jugular vein. After an overnight fast, [3-3H]-glucose (HPLC purified; Perkinelmer, Boston, MA) was infused at a rate of 0.05 mCi/min for 2 hours to assess the basal glucose turnover. Following this period, hyperinsulinemic euglycemic clamp was conducted for 120 min with a primed and continuous infusion of human insulin (105 pmol/kg prime, 15 pmol/kg/min infusion) (Novo Nordisk, Princeton, NJ), and 20% dextrose was infused at variable rates to maintain plasma glucose at basal concentrations (~6.7 mM). To estimate insulin-stimulated whole body glucose fluxes, [3-3H]-glucose was infused at a rate of 0.1 mCi/min throughout the clamps and 2-deoxy-D-[1-14C] glucose (2-[14C] DG; Perkinelmer, Boston, MA) was injected as bolus at the 75 minute of the clamp to estimate the rate of insulin-stimulated tissue glucose uptake. After clamp, liver and gastrocnemius muscle were rapidly removed and stored for later analysis of insulin signalling. Whole body and tissue glucose uptakes were calculated as previously described (Youn and Buchanan, 1993). For insulin tolerance tests, mice were fasted for 4 hours before receiving an intraperitoneal injection of insulin at 1.5 mU/g. Blood glucose levels were collected from tail before and at 15, 30, 60, 90, and 120 min after insulin infusion. Glucose levels were determined using OneTouch FastTake glucometer (LifeScan Inc., Milpitas, CA).
Immunoblotting
Brown-adipose tissue was homogenized in lysis buffer (50 mM Tris pH 7.8, 137 mM NaCl, 10 mM NaF, 1 mM EDTA, 1% Triton X, 0.2% Sarkosyl, 10% Gycerol, and protease inhibitor), solubilized by continuous stirring for 1 hr at 4 °C, and clarified by centrifugation. In vitro translated proteins were prepared from 1 μg of wild-type and mutant pCATCH-FLAG-PGC1β plasmid DNA using TnT-T7-coupled reticulocyte lysates system (Promega, Madison, WI, USA), by manufacturer protocol. Tissue lysates and in vitro translated proteins were separated by SDS-PAGE and transferred onto a nitrocellulose membrane by electroblotting. PGC1β was detected using antisera against the N-terminal (AA 1-350) of the mouse PGC1β. The in vitro translated FLAG protein was detected using monoclonal ANTI-FLAG M2 antibody (Sigma).
Determination of IRS-1 or IRS-2 associated PI3-K and Akt activity
To prepare gastrocnemius and liver lysates, 50 mg of tissue was homogenized using a polytron at half maximum speed for 1 min on ice in 500 μl buffer (20 mM Tris pH 7.5, 5 mM EDTA, 10 mM Na4P2O7, 100 mM NaF, 2mM Na3VO4) containing 1% NP-40, 1mM PMSF, 10 μg/mL aprotinin and 10 μg/mL leupeptin. Tissue lysates were solubilized by continuous stirring for 1 hr at 4 °C, and centrifuged for 10 min at 14,000×g. The supernatants were stored at −80 ° C until analysis. Tissue lysates (500 μg protein) were subjected to immunoprecipitation for 4 hr at 4 °C with either 5 μL of a polyclonal IRS-1 antibody (1:100 dilution; gift from Dr. Morris White, Joslin Diabetes Center) or 5 μL of a polyclonal IRS-2 antibody (1:100 dilution; gift from Dr. Morris White, Joslin Diabetes Center) or 2 μg of Akt antibody that recognizes both Akt1 and Akt2 (Upstate Biotechnology, Lake Placid, NY), coupled to protein A-sepharose (Sigma, St. Louis, MO) or protein G-Sepharose beads. The immune complex was washed, and PI3-K and Akt activity were determined as described previously (Kim et al., 2002).
Statistical analysis
All results are expressed as means ± SEM. Two-tailed Student’s t test was used to determine p-values.
Microarray data accession
The Affymetrix GeneChip data set is publicly available from the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) with the GEO Accession Number GSE6210.
Supplementary Material
Acknowledgments
We thank Dr. Chen-Yu Zhang for helpful discussions and Jia Yu for technical assistance in ES cells manipulation. We also thank Cleide G. da Silva and Marcelo A. Christoffolete for technical advices. This work was supported by grants DK 61562 (BMS) and DK 49569 (BBL) from the National Institute of Health, by a grant 7-05-PPG-02 (YBK) from the American Diabetes Association, by a grant KF-Nr. 31/2006 (MH) from the Köln Fortune Nachwuchsförderprogramm, and by a grant HU 1583/1-1 (MH) from the Deutsche Forschungsgemeinschaft.
References
- Arany Z, He Huamei, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, Rybkin II, Shelton JM, Manieri M, Cinti S, Schoen FJ, Bassel-Duby R, Rosenzweig A, Ingwall JS, Spiegelman BM. Transcriptional coactivator PGC-1α controls energy state and contractile function of cardiac muscle. Cell Metabolism. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, Slezak LA, Andersen DK, Hundal RS, Rothman DL, Petersen KF, Shulman GI. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. Diabetes. 1999;103:253–259. doi: 10.1172/JCI5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin ME, Marcucci MJ, Cline GW, Bell k, Barucci N, Lee D, Goodyear LJ, Kraegen EW, White MF, Shulman GI. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes. 1999;48:1270–1274. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- Kahn BB. Type 2 diabetes: When insulin secretion fails to compensate for insulin resistance. Cell. 1998;92:593–596. doi: 10.1016/s0092-8674(00)81125-3. [DOI] [PubMed] [Google Scholar]
- Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem. 2000;275:8456–8460. doi: 10.1074/jbc.275.12.8456. [DOI] [PubMed] [Google Scholar]
- Kim YB, Shulman GI, Kahn BB. Fatty acid infusion selectively impairs insulin action on Akt1 and protein kinase C lambda/zeta but not on glycogen synthase kinase-3. J Biol Chem. 2002;277:32915–22. doi: 10.1074/jbc.M204710200. [DOI] [PubMed] [Google Scholar]
- Kressler D, Schreiber SN, Knutti D, Kralli A. The PGC-1-related protein PERC is a selective coactivator of estrogen receptor alpha. J Biol Chem. 2002;277:13918–13925. doi: 10.1074/jbc.M201134200. [DOI] [PubMed] [Google Scholar]
- Lazar MA. How obesity causes diabetes: Not a tall tale. Science. 2005;307:373–375. doi: 10.1126/science.1104342. [DOI] [PubMed] [Google Scholar]
- Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc. Natl. Acad. Sci. USA. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillioja S, Mott DM, Howard BV, Bennett PW, Yki-Jarvinen H, Freymond D, Nyomba BL, Zurlo F, Swinburn B, Bogardus C. Impaired glucose tolerance as a disorder of insulin action. Longitudinal and cross-sectional studies in Pima Indians. N Engl J Med. 1988;318:1217–1225. doi: 10.1056/NEJM198805123181901. [DOI] [PubMed] [Google Scholar]
- Lillioja S, Mott DM, Spraul M, Ferraro R, Foley JE, Ravussin E, Knowler WC, Bennett PW, Bogardus C. Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus. Prospective studies of Pima Indians. N Engl J Med. 1993;329:1988–1992. doi: 10.1056/NEJM199312303292703. [DOI] [PubMed] [Google Scholar]
- Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator activated receptor γ coactivator 1β (PGC-1β), a novel PGC-1 related transcription coactivator associated with host cell factor. J Biol Chem. 2002;277:1645–1648. doi: 10.1074/jbc.C100631200. [DOI] [PubMed] [Google Scholar]
- Lin J, Tarr P, Yang R, Rhee J, Puigserver P, Newgard CB, Spiegelman BM. PGC-1beta in the regulation of hepatic glucose and energy metabolism. J Biol Chem. 2003;278:30843–30848. doi: 10.1074/jbc.M303643200. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu H, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman B. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature. 2002;428:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- Lin J, Yang R, Tarr PT, Wu PH, Pei L, Uldry M, Tontonoz P, Newgard CB, Spiegelman BM. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1β coactivation of SREBP. Cell. 2005;120:261–273. doi: 10.1016/j.cell.2004.11.043. [DOI] [PubMed] [Google Scholar]
- Lowell BB, Shulman GI. Mitochondrial dysfunction and Type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- Michael LF, Wu Z, Cheatham RB, Puigserver P, Adelmant G, Lehman JJ, Kelly DP, Spiegelman BM. Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc. Natl. Acad. Sci. USA. 2001;98:3820–3825. doi: 10.1073/pnas.061035098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirhaeghe A, Crowley V, Lenaghan C, Lelliott C, Green K, Stewart A, Hart K, Schinner S, Sethi JK, Yeo G, Brand MD, Cortright RN, O’Rahilly S, Montague C, Vidal-Puig A. Characterization of the human, mouse and rat PGC-1β (peroxisome proliferator activated receptor γ coactivator 1β) gene in vitro and in vivo. Biochem J. 2003;373:155–165. doi: 10.1042/BJ20030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman BM, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC1-α responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Med. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Handschin C, Arlow D, Xie X, Pierre J, Sihag S, Yang W, Altshuler D, Puigserver P, Patterson N, Willy PJ, Schulman GI, Heyman RA, Lander ES, Spiegelman BM. ERRα and Gabpa/b specify PGC1α-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc. Natl. Acad. Sci. USA. 2004;101:6570–6575. doi: 10.1073/pnas.0401401101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, Neschen S, White MF, Bilz S, Sono S, Pypaert M, Shulman GI. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest. 2005;115:3587–3593. doi: 10.1172/JCI25151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neschen S, Morino K, Hammond LE, Zhang D, Liu ZX, Romanelli AJ, Cline GW, Pongratz RL, Zhang XM, Choi CS, Coleman RA, Shulman GI. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metabolism. 2005;2:55–65. doi: 10.1016/j.cmet.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Spiegelman BM. Peroxisome Proliferator-activated receptor γ coactivator 1α (PGC1α): Transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DK. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- Rustin P, Chretien D, Bourgeron T, Gérard B, Rötig A, Saudubray JM, Munnich A. Biochemical and molecular investigations in respiratory chain deficiencies. Clin. Chim. Acta. 1994;228:35–51. doi: 10.1016/0009-8981(94)90055-8. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- Savkur RS, Burris TP. The coactivator LXXLL nuclear receptor recognition motif. J Pept Res. 2004;63:207–212. doi: 10.1111/j.1399-3011.2004.00126.x. [DOI] [PubMed] [Google Scholar]
- Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, Oakeley EJ, Kralli A. The estrogen-related receptor α (ERRα) functions in PPARγ coactivator 1α (PGC-1α)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. USA. 2004;101:6472–6477. doi: 10.1073/pnas.0308686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre J, Lin J, Krauss S, Tarr PT, Yang R, Newgard CB, Spiegelman BM. Bioenergetic analysis of peroxisome proliferator-activated receptor-γ coactivators 1α and 1β (PGC-1α and PGC-1β) in muscle cells. J Biol Chem. 2003;278:26597–26603. doi: 10.1074/jbc.M301850200. [DOI] [PubMed] [Google Scholar]
- Warram JH, Martin BC, Krolewski AS, Soeldner JS, Kahn CR. Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offsprings of diabetic patients. Ann Intern Med. 1990;113:909–915. doi: 10.7326/0003-4819-113-12-909. [DOI] [PubMed] [Google Scholar]
- Wolfrum C, Stoffel M. Coactivation of Foxa2 through Pgc-1β promotes liver fatty acid oxidation and triglyceride/VLDL secretion. Cell Metabolism. 2006;3:99–110. doi: 10.1016/j.cmet.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang CY, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Youn JH, Buchanan TA. Fasting does not impair insulin-stimulated glucose uptake but alters intracellular glucose metabolism in conscious rats. Diabetes. 1993;42:757–763. doi: 10.2337/diab.42.5.757. [DOI] [PubMed] [Google Scholar]
- Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI. Mechanism by which fatty acids inhibit insulin activation of IRS-1 associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- Zimmet P, Alberti KGMM, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.