

Abstract
Background & Aims
Neonatal intestinal obstruction (meconium ileus or MI) occurs in 15% of patients with cystic fibrosis (CF). Our aim was to determine the relative contribution of genetic and non-genetic modifiers to the development of this major complication of CF.
Methods
Using clinical data and DNA collected by the CF Twin and Sibling Study, 65 monozygous twin pairs, 23 dizygous twin/triplet sets, and 349 sets of siblings with CF were analyzed for MI status, significant covariates, and genome-wide linkage.
Results
Specific mutations in CFTR, the gene responsible for CF, correlated with MI indicating a role for CFTR genotype. Monozygous twins showed substantially greater concordance for MI than dizygous twins and siblings (p=1×10−5) demonstrating that modifier genes independent of CFTR contribute substantially to this trait. Regression analysis revealed that MI was correlated with distal intestinal obstruction syndrome (DIOS; p=8×10−4). Unlike MI, concordance analysis indicated that the risk for development of DIOS in CF patients is primarily due to non-genetic factors. Regions of suggestive linkage (logarithm of the odds of linkage >2.0) for modifier genes that cause MI (chromosomes 4q35.1, 8p23.1, and 11q25) or protect from MI (chromosomes 20p11.22 and 21q22.3) were identified by genome-wide analyses. These analyses did not support the existence of a major modifier gene within the CFM1 region on chromosome 19 that had previously been linked to MI.
Conclusions
The CFTR gene along with two or more modifier genes are the major determinants of intestinal obstruction in newborn CF patients, while intestinal obstruction in older CF patients is primarily due to non-genetic factors.
Keywords: Twins, siblings, linkage, association, intestinal obstruction, CFM-1
Introduction
Cystic fibrosis (CF [MIM 219700]) is an autosomal recessive disease caused by mutations in the CF transmembrane regulator (CFTR [MIM 602421]). Variation in the CFTR genotype has been associated with some aspects of the CF phenotype such as pancreatic status (1). On the other hand, many other variable features of CF are not correlated, indicating that factors independent of CFTR genotype play a major role in the complications and course of this “single gene” disorder (2; 3). Two such features are meconium ileus (MI), a form of intestinal obstruction observed in the neonatal period that occurs in 13–20% of CF patients (3–6) and distal intestinal obstruction syndrome (DIOS), a trait with clinical and pathologic similarities to MI that affects older CF patients (7; 8). Estimates of the prevalence of DIOS in CF patients vary considerably (2.4% to 41.3%) probably due to differences in the diagnostic criteria, length of follow-up and age range of the patients studied (7–10). Several reports indicate that the recurrence rate of MI in siblings is significantly higher than the prevalence of MI in unrelated patients suggesting that factors shared among siblings contribute to the development of MI (4; 11–13). However, it is not known whether familial concordance of MI is the result of shared genetic or environmental factors or a combination of both.
Evidence for genetic factors underlying intestinal obstruction has been derived from studies of the CF mouse model. The majority of homozygous CFTR null mice have a severe and usually fatal intestinal obstruction in early life (14). By capitalizing on strain-specific differences in survival among CFTR null mice, Rozmahel and colleagues were able to locate a modifier locus to the distal portion of mouse chromosome 7 (15). Several other loci for survival from intestinal obstruction in CF mice have since been identified (16). Zielenski and coworkers postulated that the locus on chromosome 7 responsible for murine intestinal obstruction may contain a gene that is responsible for MI in CF patients. This theory was tested by genotyping 185 North American CF sibling pairs and parents using 9 microsatellite markers that spanned 7.65 Mb on chromosome 19q13, a region of conserved synteny with the segment of mouse chromosome 7 that was linked to intestinal obstruction. Several markers from this region demonstrated significant linkage with the MI phenotype consistent with the presence of a MI modifier gene (CFM1 [MIM 603855]) within this region (17). However, the gene (or genes) within the CFM1 region that modifies MI has not been identified.
The CF Twin and Sibling Study in the United States is currently recruiting patients to assess genetic and non-genetic contribution to disease variation in CF. Monozygous (MZ) and dizygous (DZ) twins can differentiate the relative contribution of genetic factors since twins have a high degree of shared environment, but MZ and DZ twins differ in their degree of gene sharing (100% versus 50%) (18–21). On the other hand, siblings have the same degree of gene sharing as DZ twins (50%), but a lower degree of shared environment so that a comparison of DZ twins and siblings can estimate environmental contribution. Although recruitment for the CF Twin and Sibling study is not complete, MI and DIOS are sufficiently frequent to perform an analysis of concordance rates among the patients recruited to date. We also performed an initial genome-wide linkage analysis for MI including a re-evaluation of the CFM1 locus.
Materials and Methods
Family collection
The CF Twin and Sibling Study is a collaborative effort to collect detailed clinical information and DNA samples from affected siblings and their parents. Informed consent was obtained from all subjects prior to enrollment in the study. Enrollment was based on conclusive diagnosis of CF (22). The diagnosis of MI was based on the presence of the following features in the newborn period: lack of passage of stool within 24 hours after birth, evidence of obstruction on abdominal X-ray (ground glass appearance of intestine, air fluid levels and/or intra-abdominal calcifications), evidence of colonic abnormality (microcolon on X-ray) and treatment for obstruction (enema or surgery). Some cases were complicated by bilious vomiting, perforation and/or atresia of jejunum or ileum. The diagnosis of DIOS was based upon the clinical impression of the reporting CF center. A required feature was intestinal obstruction requiring treatment beyond laxatives (e.g., oral polyethylene glycol solution, enema or surgery); radiological documentation of DIOS was obtained for some, but not all patients. Pancreatic status was obtained from patient medical records. Pancreatic insufficient status was determined by a fecal fat test, a fecal elastase test, the appearance of the stool pattern, the presence of oil in the stool, the patient’s growth pattern, and/or the complaint of abdominal pains.
DNA isolation and genotyping methods
Genomic DNA was isolated from whole blood using the phenol/chloroform procedure (23). Zygosity status of twins was determined by AmpFISTR Profiler (Applied Biosystems, Foster City, CA). CFTR genotype was obtained from the patient’s medical record. In cases where CFTR genotyping had not been performed or was incomplete, DNA samples were typed for 58 CFTR alleles using the Roche PCR-based Line Probe Assay (Roche Molecular Systems, Alameda, CA) (24). DNA samples with one or more unidentified CFTR mutations following typing with the Line Probe Assay were subjected to DNA sequencing of all coding regions of CFTR and flanking introns (25). For genome-wide linkage studies, patients were genotyped by the Marshfield Genotyping Center for 402 polymorphic short tandem repeat markers with an average spacing of 10 cM, or 7.5 Mb. The average rate of genotypes called for the 1161 individuals was 95% (range 70–99.8%); identified mendelian errors numbered 642 (1.1%). Average heterozygosity in the study population was 75% (range 57–89%). SNP typing was performed using 100K SNP chips from Affymetrix (26); 90,689 markers met quality criteria for inclusion (minor allele frequency at least 1%; genotype obtained in at least 85% of samples) and yielded an average marker spacing of 23.6 kilobases. The average rate of genotypes called for the 86 individuals was 96% (range 80–100%); identified mendelian errors numbered 2115 (0.03%). Average heterozygosity was 30%. For the chromosome 19-specific map, patients were genotyped at 7 STR markers spanning 6.5 Mb of chr19q13: D19S211, D19S217, D19S219, D19S112, D19S412, D19S902, and D19S604 yielding an average marker spacing of 1.1 Mb. STR markers were sized using an ABI Prism 310 DNA Sequencer or an ABI 3100 DNA Sequencer (Applied Biosystems) and GENESCAN analysis software version 3.1.2. Genotyping was internally controlled by genotyping individuals of the same family on the same run. Any ambiguous genotypes and genotypes that were not consistent with mendelian inheritance were re-typed.
Concordance Analysis
Concordance for disease (MI or DIOS) was calculated by the number of pairs concordant for disease divided by the total number of pairs in which at least one member had disease. Concordance for lack of disease was calculated by the number of pairs in which neither member had disease divided by the total number of pairs in which at least one member did not have disease. Heritability estimates were estimated as described by Falconer (27; 28). For DIOS concordance calculations, siblings were used as a proxy for DZ twins by correcting the DIOS status for age difference, by using the DIOS status of the older sibling when he/she was the age of the younger sibling. Power calculation was done using DSTPLAN (M. D. Anderson Cancer Center). One set of nonidentical triplets in which 1 member had MI and none had DIOS was counted as a single MI-discordant pair and as a single pair without DIOS. Three families had a set of MZ twins and a sibling all with CF and contributed to both the MZ and sibling pair concordance calculations. With one exception, the MZ twin set was concordant for disease (MI or DIOS) or lack of disease, and it was counted as a single individual within a sibling pair. The exception was an MZ twin pair discordant for DIOS with a sibling without DIOS; this set was counted as a single discordant sibling pair.
Statistical Analysis
Statistical calculations were performed using STATA (StataCorp, College Station, TX). For all tests, a p value <0.05 was considered statistically significant. The Fisher exact test was used when one or more cells had 5 or fewer observations (http://www.unc.edu/~preacher/fisher/fisher.htm). Student’s t-test was used to evaluate means derived from normally distributed continuous data. Multiple logistic regression was performed with STATA using clinical data from the CF Twin and Sibling Study. Each clinical variable was tested for correlation with MI along with age and gender in a three-variable logistic model. Variance was estimated using the robust Huber/White/sandwich estimator, and observations within each family were not considered to be independent. Factors tested individually for correlation with MI were: age, gender, DIOS, surgery for DIOS, pancreatic insufficiency, number of ΔF508 alleles, sweat chloride, positive cultures for P.aeruginosa, mucoid pseudomonas, antibiotic resistant pseudomonas, B.cepacia, atypical mycobacteria, aspergillus, S.aureus, methicillin-resistant S.aureus, S.maltophilia, A.xylosoxidans, K.pneumoniae, E.coli, H.influenzae, streptococcus, sinus disease, surgery for sinus disease, nasal polyps, surgery for nasal polyps, elevated transaminases, pancreatitis, surgery for pancreatitis, diabetes, cholelithiasis, cholecystectomy, gastroesophageal reflux, Nissen fundoplication, gastrostomy tube placement, rectal prolapse, appendiceal disease, failure to thrive, steatorrhea, neonatal jaundice, average FEV1 (CF-specific percentile score (29)), average BMI Z-score, age at diagnosis of CF, lung transplantation, number of pulmonary exacerbations in the last 1 or last 5 years, and medication compliance. Factors significantly correlated with MI in the three-variable analysis were tested for inclusion in a multivariate logistic regression model. Factors were removed if they did not contribute significantly to the risk of MI and their inclusion did not improve the model (likelihood ratio test), or if they were strongly correlated with another covariate. In the final multivariate model, the 3 parameters regarding number of ΔF508 alleles were correlated (as expected), but no other pair of factors had a correlation coefficient of greater than 0.25.
Genetic Analysis
The genome-wide and chromosome 19-specific STR marker data were screened for mendelian inconsistencies by PedCheck (30) and SIB-PAIR 0.99.9 (31); inconsistent markers were retyped or eliminated. Single- and multipoint, parametric and nonparametric linkage analyses were performed using GENEHUNTER (32) and MERLIN (33). Parametric analyses were performed with dominant, additive, and recessive models of inheritance with penetrance set at 0.80 and phenocopy rate set at 0.10 (both derived from observed rates of MI; see below), and with alternative penetrance values of 0.60 and 1.00 and phenocopy values of 0.00 and 0.20. Analysis using MERLIN included error detection through identification of improbable recombination events. For analysis of high-density SNP data, MERLIN was again used; for this analysis, linkage disequilibrium between markers was modeled by clustering correlated markers (34). For the 7 STR map of chromosome 19, SIB-PAIR 0.99.9 (31) was used to perform identical-by-descent (IBD) analyses. If parental genotype data was not available, then IBD was estimated from identical-by-state (IBS). ASP 26.07.2001 (35) was used to simulate (n=1000) the statistical power of our dataset under a dominant (disease allele frequency = 0.10), recessive (disease allele frequency = 0.43), or additive model (disease allele frequency = 0.10) with the disease gene located at a recombination fraction θ = 0.01 from the simulated marker and a phenocopy rate of 0.05. The above disease allele frequencies were calculated assuming Hardy-Weinberg equilibrium, a trait prevalence of 15% (derived from the observed rate of MI), and a trait penetrance of 80% (derived from the concordance rate in MZ twins). With disease allele frequency defined as q, calculations were as follows: q2 × 0.8 = 0.15 giving q = 0.43 for recessive; (1 − p2) × 0.8 = 0.15 giving p = 0.90 and q = 0.10 for dominant; (2pq + 2q2) × 0.8 = 0.15 giving q = 0.10 for additive. For all three models, the number of alleles and the allele frequencies calculated by Genehunter for marker D19S112 were used (13 alleles: 0.0029, 0.1556, 0.0039, 0.0616, 0.0352, 0.2076, 0.2074, 0.0020, 0.1301, 0.1223, 0.0577, 0.0108, 0.0029). The total power of the study was calculated using restricted model estimates and the equation: [1 − (1 − concordant estimated power)(1 − discordant estimated power)] × 100% = % power.
Results
Clinical information collected from 65 pairs of monozygous (MZ) twins, 22 pairs of dizygous (DZ) twins, one set of non-identical triplets, 14 single siblings, 365 sibling pairs (includes 4 sibling-MZ twin pairs), 29 sets of 3 siblings, and one set of 5 siblings (1009 patients total) was obtained from the CF Twin and Sibling Study. All participants meet diagnostic criteria for CF. These patients account for approximately 85% of all twins affected with CF and 35% of all families with two or more children affected with CF in the United States according to the CF Patient Registry maintained by the U.S. CF Foundation. Of the 940 patients with information about MI, 160 had MI (17%); treatment was known in 159 of these: 108 patients had surgical treatment, 49 patients were treated with an enema, 1 patient had an enema and surgery, and 1 resolved spontaneously.
Pancreatic insufficiency and CFTR genotype are correlated with MI
Approximately 95% of CF patients have pancreatic insufficiency (PI), and prior studies of MI have shown a close correlation between pancreatic status and MI (1). Indeed, 159 of the 160 MI patients in this study are diagnosed as PI. As expected, pancreatic sufficiency (PS) was present at a higher rate in patients without MI (91 of 762; 12%; p=3×10−7). Since MI correlates with pancreatic status, and pancreatic status correlates with CFTR genotype, we expected that MI should also correlate with CFTR genotype. As predicted, the common CF mutation ΔF508, which is highly associated with PI, was found at increased frequency in MI vs. non-MI patients (79% vs. 68%; p=9×10−5), and homozygosity for ΔF508 was also increased in MI vs. non-MI patients (64% vs. 47%; p=2×10−4). Furthermore, of the 39 CFTR genotypes found in PS patients in this study, only 1 genotype (ΔF508/2184insA) also occurred in a patient with MI: the single MI patient who remains PS (complete list of CFTR mutations available from corresponding author). Intriguingly, 31 patients with PI carried one mutation observed in PS patients, and none of these patients had MI, whereas among 784 PI patients with no PS mutations, 153 had MI (p=0.002). The latter observation suggests that CFTR genotype is more predictive of MI than pancreatic status. To test this concept, we selected only PI patients and recalculated the frequency of the ΔF508 mutation and the frequency of homozygosity for ΔF508 in MI vs non-MI patients. Both ΔF508 and homozygosity for ΔF508 were found at greater frequency in MI patients than in non-MI patients (ΔF508 allele frequency: 79% vs. 73%, p= 0.02; ΔF508 homozygote frequency: 64% vs. 54%, p= 0.01). Finally, we performed a multivariate regression analysis in which PI was subdivided based on the number of ΔF508 alleles. As shown in Table 1, PI with ΔF508 homozygosity was an independent risk factor for MI (OR 13.0). PI in the context of other CFTR genotypes had a large effect size (OR 7.8–8.4) but was of borderline significance. Thus, while PI is highly correlated with MI, CFTR genotype can decrease the association with MI (e.g. PS mutations) or increase association with MI (e.g. ΔF508 homozygosity) in PI patients.
Table 1.
Multivariate regression analysis of MI covariates
| Odds Ratio | 95% Conf. Interval | P value | |
|---|---|---|---|
| Pancreatic insufficiency with no ΔF508 alleles | 8.4 | (0.9, 77.2) | 0.061 |
| Pancreatic insufficiency with one ΔF508 allele | 7.8 | (1.0, 61.2) | 0.050 |
| Pancreatic insufficiency with two ΔF508 alleles | 13.0 | (1.7, 100) | 0.014 |
| DIOSa treated nonsurgically | 2.52 | (1.47, 4.32) | 0.001 |
| DIOS treated with surgery | 24.6 | (8.34, 72.6) | 7 × 10−9 |
| Elevated ALT/ASTb | 4.15 | (2.25, 7.67) | 5 × 10−6 |
| Current Agec | 0.93 | (0.89, 0.97) | 0.001 |
| BMI Z-scored | 1.34 | (1.03, 1.75) | 0.032 |
| B. cepacia positive culture | 3.33 | (1.48, 7.47) | 0.004 |
Distal intestinal obstruction syndrome
Alanine aminotransferase (ALT) or aspartate aminotransferase (AST) levels greater than twice normal upper limit at least twice
Odds ratio reflects decreased odds of MI for each year of age.
Odds ratio reflects increased odds of MI for each unit decrease in BMI Z-score.
Complications correlated with MI and mode of treatment of MI
To determine if other features of CF were correlated with MI, a series of disease characteristics (listed in Methods) was tested for association with MI by single and multiple logistic regression analysis. In addition to pancreatic status and CFTR genotype (shown above), MI was also correlated with distal intestinal obstruction syndrome (DIOS), DIOS treated surgically, elevated transaminases, age, decreased BMI Z-score and B.cepacia positive culture (Table 1). We then considered treatment modality (surgical vs. nonsurgical) for MI under the hypothesis that surgical treatment is a marker for more severe intestinal disease. By multiple regression analysis, the only variable significantly correlated with treatment mode for MI was DIOS (odds ratio of having had surgery=2.6; 95% confidence interval 1.1–5.9; p=0.03). Further examination of DIOS and MI revealed that the rate of DIOS in patients without MI (96 of 733, 13%) was lower than patients with MI treated surgically (52 of 155, 34%; p=9×10−9) but about the same in patients with MI treated nonsurgically (10 of 51, 20%; p=0.2). Of note, CFTR genotype was not correlated with MI treatment modality. No significant correlation between MI or MI treatment modality was observed for gender or other markers for CF disease severity.
Modifier genes contribute substantially to MI
To determine if genetic or nongenetic factors cause MI, we compared the rates of concordance for MI among affected twin pairs and sibling sets, who share both copies of CFTR but otherwise have different degrees of gene sharing (Table 2). Among families where at least one member had MI, MZ twins show significantly greater concordance than DZ twins and siblings. Concordance rates for MI did not differ between DZ twins and siblings. In the subjects homozygous for ΔF508, concordance rates were similarly greater among MZ twins (11 of 13 MZ twin pairs; 1 of 5 DZ twin pairs, p=0.02; 12 of 44 sibling pairs, p=0.0003; 2 of 6 sibling trios, p=0.05). It is possible that intestinal obstruction due to MI may affect all CF patients but common protective factors (genetic and otherwise) prevent the development of this complication in most patients. A protective role for modifying factors of MI is suggested by the observation that intestinal obstruction is a consistent rather than uncommon complication in the murine model of CF (15). The concordance rates for the absence of MI are significantly greater in MZ twins than in DZ twins or sibling sets (Table 2). Concordance for lack of MI did not differ between DZ twins and siblings, and results were essentially identical when restricting analysis to homozygous ΔF508 subjects (not shown). Finally, we investigated whether sharing the mode of MI treatment correlated with degree of gene sharing. However, MZ twins did not differ from DZ twins or 2 affected siblings when distributed according to treatment modality (both treated with surgery; one treated with surgery and the other with enema; both treated with enema; data not shown). These data show that MZ twins demonstrate greater concordance in both causative and protective models for MI, suggesting predominance of genetic factors in each model.
Table 2.
MI Status of Twins and Siblings
| MZ twins | DZ twins | 2 affected siblings | 3 affected siblings | |
|---|---|---|---|---|
| Total # of Families | 65 | 23 | 326 | 27 |
| # Sets MI Concordant | 14 | 2 | 19 | 2 |
| # Sets MI Discordant | 3 | 7 | 61 | 8a |
| # Sets without MI | 48 | 14 | 246 | 17 |
| Concordance for MI | 82% | 22%b | 24%c | 20%d |
| Concordance for no MI | 94% | 67%e | 80%f | 68%g |
Two families had 2 affected children with MI; 5 families had one affected child with MI.
p=0.009 vs. MZ twins
p= 1 × 10−5 vs. MZ twins
p=0.003 vs. MZ twins
p=0.005 vs. MZ twins
p=0.02 vs. MZ twins
p=0.04 vs. MZ twins
Modifier genes do not contribute substantially to DIOS
Since DIOS was highly correlated with surgically treated MI and DIOS, and MI have similar clinical manifestations (8; 36), we explored the possibility that they have the same genetic etiology. As compared to MI, the ΔF508 allele of CFTR is less well correlated with DIOS. Although the frequency of the ΔF508 allele is greater (76% vs. 70%; p=0.02) in subjects with DIOS, the frequency of ΔF508 homozygosity is similar (58% vs. 50%; p=0.1). Furthermore, DIOS occurred at similar rates (20%) in subjects homozygous for ΔF508 and in those with other CFTR genotypes (15%; p=0.08). In striking contrast to MI, the concordance rates for DIOS (Table 3) are low in MZ twins, and MZ twins and sibling pairs have similar concordance rates (p=0.5; Fisher exact) indicating that genetic factors do not play a major role in DIOS. We estimated that these patients provided 80% power for detecting a rate difference greater than 0.25. Concordance rates could not be estimated precisely in DZ twins and 3 affected sibling sets due to small numbers in these groups. Concordance rates for absence of DIOS did not differ among the 2-member groups; the decrease in concordance for lack of DIOS among sets of 3 siblings is no longer present when siblings in each set of three are taken pairwise (concordance rate 75%). DIOS concordance rates for ΔF508 homozygotes were similarly low in all twin and sibling groups (1 of 11 MZ twin pairs; 0 of 3 DZ twin pairs, p=1; 7 of 34 sibling pairs, p=0.7; 0 of 4 sibling trios, p=1). No significant differences were seen in the concordance for lack of DIOS in ΔF508 homozygote twin and sibling groups (not shown). The frequency of DIOS is not significantly different among MZ twins and siblings, while DZ twins have a trend toward a lower rate that likely reflects their younger median age. To assess whether the study population adequately captured the range of ages at which DIOS is usually diagnosed, we compared the mean age at diagnosis of DIOS to mean age of the entire twin or sibling groups. For the MZ twins and the siblings, the mean age at diagnosis of DIOS is lower (12.0 years and 9.0 years, respectively) than the mean age of the MZ twins and siblings (15.5 years and 13.4 years, respectively). Only 5 of 47 DZ twins/triplets were diagnosed with DIOS (average age of 6.4 years at diagnosis), likely reflecting the younger mean age of this population (8.4 years), as previously noted. Unlike MI, these results indicate that modifier genes do not contribute substantially to DIOS, suggesting that DIOS and MI have different etiologies.
Table 3.
DIOS status of twins and siblings
| MZ twins | DZ twins | 2 affected siblingsa | 3 affected siblingsa | |
|---|---|---|---|---|
| Total # of families | 60 | 23 | 312 | 25 |
| # sets DIOS concordant | 5 | 0 | 16 | 0 |
| # sets DIOS discordant | 13 | 5 | 56 | 12b |
| # sets without DIOS | 42 | 18 | 240 | 13 |
| Concordance for DIOS | 28% | 0% | 22% | 0% |
| Concordance for no DIOS | 76% | 78% | 81% | 52% |
Siblings were corrected for age difference by determining the DIOS status of each sibling at the age of the youngest sibling in the sibship.
Includes 9 sets of three siblings in which one member had DIOS and 3 sets in which two members had DIOS.
Identification of regions that may contain MI modifier genes
The high rate of concordance for MI among MZ twins compared to DZ twins and siblings (all of whom share CFTR alleles) indicates that one or more modifier genes exist that cause or protect from MI. To identify regions that may contain MI modifier genes, we performed a genome-wide linkage analysis using short tandem repeat (STR) polymorphisms. Since CFTR genotype contributes to MI risk, linkage analysis was also performed on the subset of subjects who were homozygous for the ΔF508 mutation. Forty-six patients with MI who had at least one sibling with MI (2 DZ twin pairs, 18 sibling pairs, and 2 sibling trios; total 26 pairs) and all available parents were genotyped for 402 STR markers with an average spacing of 10 cM. Of these patients, 34 were homozygous for ΔF508 (14 pairs and 2 trios; total 20 pairs). Identity-by-descent (IBD) allele sharing methods were used to identify regions linked to MI. The only region exhibiting significant linkage (logarithm of odds of linkage or LOD >3.0) encompassed the CFTR gene on chromosome 7 (multipoint nonparametric LOD score 4.17; see Figure 1A, open circles). Peaks suggestive of linkage (LOD >2.0) were detected on chromosomes 8p23.1 (LOD 2.14) and 11q25 (LOD 2.18) (Figure 1B–C, open circles). When analysis was restricted to ΔF508 homozygotes concordant for MI, the odds of linkage diminished for all three peaks (Table 4). To search for genes that protect CF patients from developing MI, we performed a linkage analysis using 282 pairs (224 nuclear families, 16 trios, and 1 set of 5) concordant for the absence of MI. No regions of linkage with a LOD score greater than 2.0 were found when all CFTR genotypes were included (not shown). Because this group without MI includes individuals who may not be susceptible to MI (such as pancreatic sufficient patients), a second analysis was restricted to 128 pairs of ΔF508 homozygotes, all with pancreatic insufficiency (97 nuclear families, 7 trios, and 1 set of 5). Suggestive LOD scores were found on chromosomes 20p11.22 (2.20) and 21q22.3 (2.38); the LOD score for the STR marker nearest to CFTR was 11.35 (Figure 2). Thus, multiple loci of suggestive linkage for causative or protective MI modifier genes were found.
Figure 1. Linkage peaks identified in 26 CF sibling pairs concordant for presence of MI.
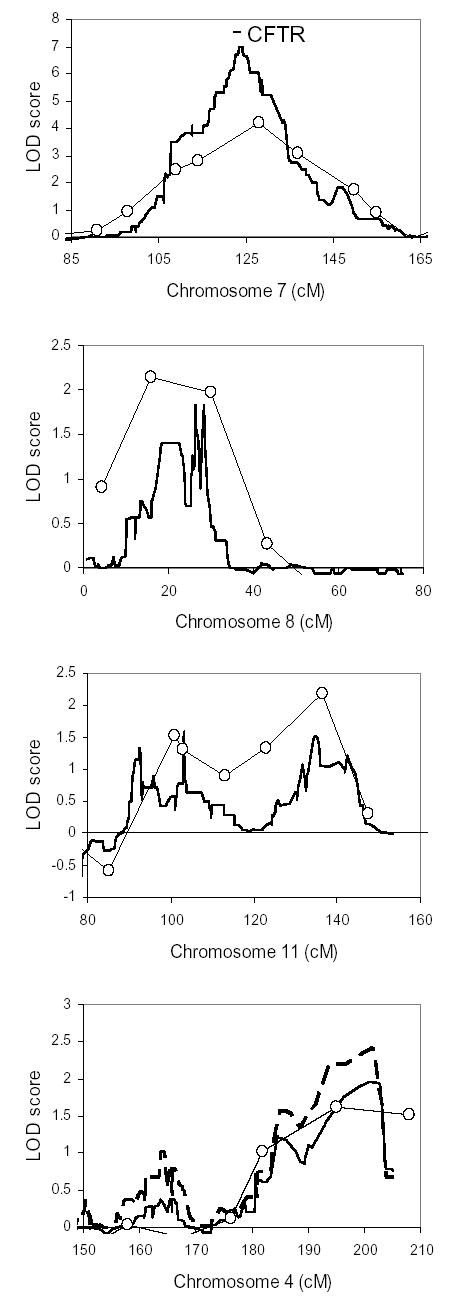
Shown are multipoint nonparametric LOD scores for linkage using the STR map (open circles) and SNP map (solid line) in 26 pairs with all CFTR genotypes included. The CFTR gene location on chromosome 7 is marked by the bar. For chromosome 4, also shown are linkage results from SNP genotyping in the subset of 20 MI-concordant pairs homozygous for ΔF508 (dashed line). For comparison of peak widths, 80 cM width is shown for each chromosome.
Table 4A.
Multipoint nonparametric LOD scores exceeding 2.0 from genome-wide STR and SNP linkage analysis of patients concordant for presence of MI.
| STR markers | SNP markers | |||||
|---|---|---|---|---|---|---|
| Chromosome | cM (Marshfield) | All (n=26) | ΔF508/ΔF508 (n=20) | cM (deCODE) | All (n=26) | ΔF508/ΔF508 (n=20) |
| 7 (CFTR) | 128 | 4.17 | 3.42 | 123.559–124.152 | 6.96 | 5.09 |
| 8p23.1 | 16 | 2.14 | 2.07 | 18.065–22.332 | 1.40 | 0.90 |
| 11q25 | 136.5 | 2.18 | 1.94 | 134.172–135.623 | 1.52 | 1.76 |
| 4q35.1 | 195.06 | 1.62 | 1.71 | 193.7–201.4 | 1.96 | 2.42 |
NOTE: Analyses are shown from all CFTR genotypes (All) and ΔF508 homozygotes (ΔF508/ΔF508); n refers to the number of pairs.
Figure 2. Linkage peaks identified in patients concordant for the absence of MI.
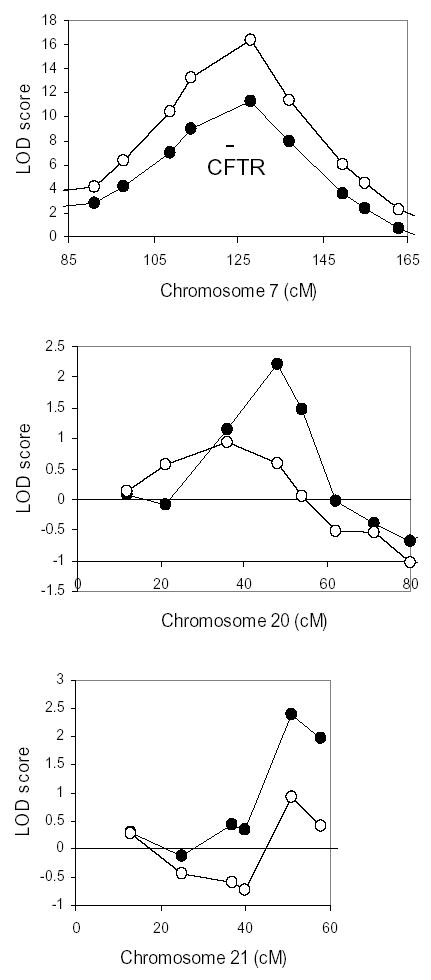
Shown are multipoint nonparametric LOD scores for linkage using the STR map in 282 pairs with all CFTR genotypes included (open circles) and in the subset of 128 pairs without MI and homozygous for ΔF508 (filled circles). The CFTR gene location on chromosome 7 is marked by the bar. For comparison of peak widths, 80 cM width is shown for each chromosome.
To evaluate the regions of possible linkage identified with STR markers and to search for additional regions of linkage, we performed a high-density linkage analysis on the same group of families concordant for the presence of MI (26 affected sibling pairs) using the Affymetrix Centurion single nucleotide polymorphism (SNP) array. 90,689 SNPs with an average spacing of 34 Kb were genotyped. As expected for a denser marker map, the peak LOD score for the region encompassing the CFTR gene was increased (multipoint nonparametric LOD 6.96), and the region of maximal linkage was narrower (0.6 cM) than in the STR analysis (Figure 1A, solid line). Linkage peaks were again found on chromosomes 8 and 11, though the LOD scores were lower (Figure 1B–C, solid lines; Table 4). The only region with LOD near 2.0 in the SNP analysis was on chromosome 4q35.1 (nonparametric LOD 1.96; Figure 1D, solid line), a region whose nearest marker in the previous 10 cM STR map had a LOD score of 1.62. In analysis restricted to the 20 pairs of MI-concordant ΔF508 homozygotes, LOD scores for CFTR on chromosome 7 and for loci on chromosome 8 and 11 decreased (Table 4), while the peak LOD on chromosome 4 increased to 2.42 (Figure 1D, dashed line). Thus, the SNP map improved sensitivity and resolution for detecting CFTR linkage on chromosome 7 and provided evidence of linkage to MI, in ΔF508 homozygotes, on chromosome 4q35.1.
MI is not linked to the CFM-1 locus on chromosome 19q13
Meconium ileus had been previously linked to the CFM1 region on chromosome 19q13, but our genome-wide analyses with low- and high-density marker maps did not reveal a linkage peak on this chromosome (Figure 3A). To provide a direct comparison with previously used STR markers, we typed 7 STR markers spanning 6.5 Mb of the CFM1 region in a subset of the above group, including 24 pairs concordant for the presence of MI, 65 discordant pairs, and 130 pairs without MI. Analyses were performed on the entire group and on those concordant for MI or lack of MI. Five of the 7 markers were the same as those used in the study by Zielenski et al. (D19S219, D19S112, D9S412, D19S902, and D19S604; Figure 3, solid diamonds). The highest nonparametric LOD score was 0.14 for all pairs (0.42 in MI concordant pairs). Under recessive, dominant and additive models of inheritance, LOD scores never exceeded 0.00 (0.06 in the concordant pairs) and the heterogeneous LOD scores never exceeded 0.25 (0.08 in the concordant pairs). Analysis to evaluate linkage to a protective allele on chromosome 19q13 produced a LOD score of 0.42 (0.67 in pairs concordant for the absence of MI and 1.18 when restricted to ΔF508 homozygotes). As a second means of assessing linkage, haplotypes were manually constructed based on identity-by-state from markers D19S217, D19S219, and D19S112 for the siblings concordant for MI, discordant for MI, or unaffected with MI. The proportion of siblings sharing 0, 1, or 2 haplotypes across the region did not deviate from the expected distribution of 25%, 50% and 25%, respectively (p=1, χ2). Finally, to evaluate for genetic heterogeneity, 5 sets of concordant siblings that did not share at least one haplotype and 14 sets of discordant siblings sharing both haplotypes were excluded, and the linkage analysis was repeated. Without these families, the highest LOD score was 0.40 (1.74 in the concordant pairs). Thus, we found no evidence for linkage of MI to the CFM1 locus in this sample of CF twins and siblings.
Figure 3. Linkage analysis of the chromosome 19q13 region encompassing the CFM1 locus.
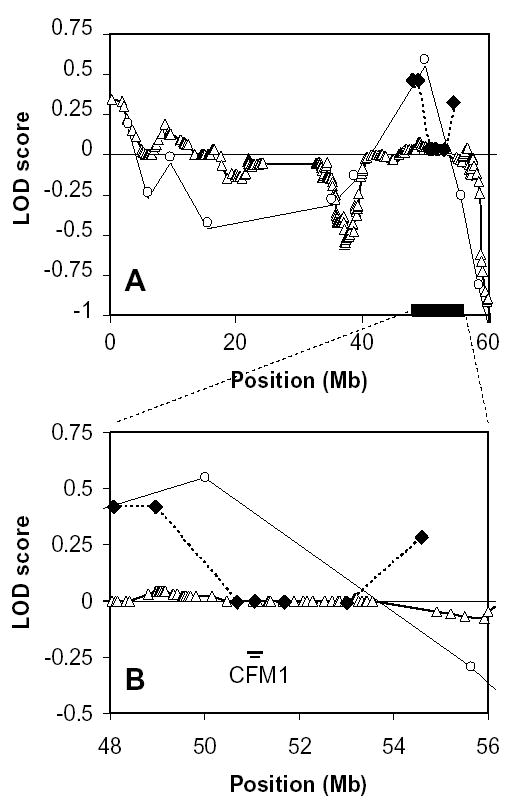
(A) Entire chromosome 19, (B) the CFM1 region at chromosome 19q13. Shown are linkage results from the genome-wide STR map (open circles), the genome-wide SNP map (open triangles), and the chromosome 19-specific map of 7 STR markers (diamonds) which includes 5 STR markers used in a prior study of MI (17) and 2 additional STR markers (diamonds with asterisks). The region from 48–56 Mb encompasses 2 of the Marshfield STR markers, the 7 STR marker map, and 165 of the SNP markers. The location of CFM1 is indicated by the bar.
Discussion
Cystic fibrosis (CF), like many other “single gene” disorders, displays considerable variability that cannot be attributed to allelic differences in the disease-causing gene (3). Meconium ileus is a well-recognized complication of CF that has been a paradigm for the elucidation of modifying factors (37). Prior studies of CF siblings documented that familial recurrence of MI exceeded the incidence of MI in unrelated patients (4; 11–13; 17). However, siblings not only share 50% of their genes but they also have similar environmental exposures (e.g. parents, home, clinic). The significantly higher concordance rate of MI in affected MZ twins that share 100% of their genes, compared to affected DZ twins who share 50% of their genes, indicates that genetic factors are likely to play a substantial role in MI. While acknowledging that the number of study subjects is small, we can estimate that the contribution of genetic variation (heritability or h2) to developing MI approaches 1.0 (h2 = 2(MZ concordance − DZ concordance) (28)). A more robust estimate which considers the disease prevalence (27) is required to estimate heritability for absence of MI (which “occurs” in ~85% of the CF population). Heritability estimates for lack of MI also approach 1.0 (0.80 to 1.4 as estimated by DZ and sibling pairs, respectively). Thus, there is evidence for the presence of both protective and susceptibility modifier genes for MI.
Comparison of the recurrence rates of MI among twins and siblings facilitates predictions of the penetrance, number and inheritance pattern of the modifier gene(s). The high rate of concordance in the MZ twins suggests that penetrance of genetic factors is high (e.g. >80%). In the 2 affected sibling sets, 19 pairs were concordant for MI and 61 pairs were discordant indicating a sibling recurrence rate of 0.24. Within the ten sets of 3 affected siblings with MI (2 concordant sets and 8 discordant sets in Table 3), there are 30 possible pairwise combinations; nine pairs are concordant and 16 pairs are discordant for MI giving a sibling recurrence rate of 0.36. These estimates compare favorably with recurrence rates that can be derived from families with 2 or more siblings affected with CF provided by Donnison et al (Concordant 16/Discordant 54; 0.23; (11)), Allan et al (Concordant 7/Discordant 16; 0.3;(12)), Kerem et al (Concordant 5/Discordant 15; 0.25;(4)), Zielenski et al (Concordant 7/Discordant 33; 0.18; (17)) and Picard et al (Concordant 10/Discordant 40; 0.2;(13)). A recurrence risk of 0.24 among siblings is similar to that observed for single gene recessive disorders such as CF. The results of the genome-wide linkage analyses indicate that a single modifier gene is unlikely to account for MI. Thus, we postulate that two or more modifier genes of relatively high penetrance are primarily responsible for MI.
While the classic twin study has proven to be a reliable means of distinguishing the effects of shared environment from shared genes, potential limitations have been discussed (38). For example, although it is commonly agreed that MZ and DZ twins have similar degrees of shared environment, there have been examples where MZ twins have a unique shared environment (e.g. placenta) causing elevated concordance rates in MZ twins leading to an inflated estimate of heritability (38). At this point, we cannot exclude this possibility. Furthermore, the collection of DZ twins in this study has an excess of males, indicating an incomplete ascertainment of this group, so the concordance rates in DZ twins may be different than calculated. This could alter our heritability estimates, possibly increasing the predicted role for non-genetic factors. The presence of three sets of MZ twins discordant for MI in this study and the published report of an MZ pair discordant for MI (39) indicate that non-heritable factor(s) play a role in MI. Nevertheless, the above findings, particularly the high rate of concordance in MZ twins, support the conclusion that the risk for MI is primarily inherited.
Although MI has been reported in a few patients that do not manifest CF (40; 41), MI in CF patients has almost always been reported in the context of pancreatic insufficiency (PI) (1). Exocrine pancreatic function in CF patients depends on the underlying mutations in CFTR, with mutations resulting in loss of CFTR function being almost invariably associated with PI. While most of the patients with MI in this study had PI, we discovered that the frequency of MI in PI patients differed depending upon CFTR genotype. These results suggest that the CFTR genotype affects the risk for MI beyond its role in determining pancreatic status. This conclusion is consistent with prior studies of unrelated CF patients. CF patients bearing the G551D mutation have been shown to have a lower rate of MI than those homozygous for ΔF508 even though both mutations are almost invariably associated with PI (42; 43). Thus, MI appears to be the consequence of interaction between the CFTR gene and MI modifier genes.
Cross-sectional and longitudinal studies indicate that CF patients with MI follow a different course than those without MI (6; 44; 45). Some have suggested that complications arising from surgical treatment of MI are the primary reason for this difference (46). To determine if the effect of MI modifier genes extends to other manifestations of CF, we performed multivariate regression analysis to identify significant covariates of MI. In addition to pancreatic status and CFTR genotype, we found that MI was highly correlated with several other gastrointestinal manifestations of CF including elevated liver enzyme levels, DIOS, and reduced BMI. An inverse correlation with increasing age suggests that the strict criteria for diagnosis of MI in the current study are more difficult to fulfill in older patients (for whom neonatal records are less accessible, for instance); alternatively, this could reflect a shorter life expectancy for MI patients as has been reported (6). Although MI was associated with B. cepacia infection, we were unable to correlate MI with any other markers of pulmonary disease severity. MI has been previously associated with liver disease (47), poorer nutritional outcomes (48), and DIOS (46) though correlation with mode of MI treatment was not specifically addressed in these studies. The clinical and pathological similarities between DIOS and MI have suggested a common etiology (8; 9; 46). However, our concordance analysis suggests that MI and DIOS are caused by different modifying factors. At present, the complexity of covariate analysis is limited by the number of patients in the study; more exhaustive evaluation of possible covariates of MI and of DIOS will be performed when recruitment for the CF Twin and Sibling Study is completed.
Genome-wide linkage analyses did not identify a single major locus for MI, although we had ample power as indicated by the significant LOD scores obtained for the region encompassing CFTR. Instead, multiple loci with LOD scores suggestive of linkage were identified, in agreement with expectations from sibling recurrence risks (above). While it is clear that the data support a polygenic origin for MI, further studies are required to determine whether the identified loci contain modifier genes for MI. Of note, the peak on chromosome 11q25 is in a region of conserved synteny with a segment of mouse chromosome 9 associated with intestinal obstruction in a CF mouse model (49). Two additional points are worth noting. First, the high density map composed of SNP markers localized CFTR on chromosome 7 with a substantially higher LOD score and better defined peak of linkage than the coarse map composed of STR markers. This pattern is expected for a linkage region containing a bona fide trait causing gene. Although the high density map showed lower odds of linkage than the STR map for chromosomes 8p23.1 and 11q25, it did increase for a region on chromosome 4 that had a LOD score in the coarse STR map that did not exceed the threshold of suggestive linkage (LOD≥ 2.0) (50). The LOD score for chromosome 4q35.1 was further enhanced when linkage analysis was restricted to ΔF508 homozygotes despite a reduction in the number of pairs analyzed from 26 to 20. The latter behavior is consistent with evidence that CFTR genotype influences risk of developing MI. Under this circumstance, one would predict that restricting linkage analysis to patients bearing high-risk CFTR genotypes should optimize the power to detect MI modifier genes. Second, we were unable to detect the previously reported linkage of MI to a locus on chromosome 19q13 (17). The power to detect linkage to a single locus in the previous study at an alpha value of 0.05 was 68%, 35%, and 58% under a recessive, additive, or dominant model, respectively. The power of this study to detect linkage at the same alpha value was 92% under a recessive model; 56% under an additive model and 80% under a dominant model. Furthermore, removal of unambiguously unlinked sibling pairs from the analysis did not expose linkage disguised by locus heterogeneity. There are several possible reasons for the difference between our result and that of Zielenski and colleagues. Meconium ileus may be the consequence of multiple modifier genes as suggested by studies of the CF mouse (16; 49) and the results of the linkage scans presented here. Differences in the stratification of the sibling populations including differences in CFTR genotype prevalence may have exposed a modifier gene of minor effect on chromosome 19 in the initial study but not in the current study. Finally, the diagnosis of MI involves subjective criteria that may have led to differences in the phenotyping of the MI populations in each study.
In summary, we have used the classic twin design to estimate the contribution of genetic and non-genetic modifiers to a well-known recognized complication of CF. Demonstration that genetic modifiers contribute substantially to MI justifies efforts to identify MI modifier genes by virtue of their location. Evidence that CFTR genotype contributes to MI risk emphasizes the importance of considering this variable in future modifier studies of MI. Our results indicating that MI is polygenic will inform the design of future searches for modifier genes responsible for this intestinal complication.
Table 4B.
Multipoint nonparametric LOD scores exceeding 2.0 from genome-wide STR linkage analysis of patients concordant for absence of MI.
| STR markers | |||
|---|---|---|---|
| Chromosome | cM (Marshfield) | All (n=282) | ΔF508/ΔF508 (n=128) |
| 7 (CFTR) | 128 | 16.4 | 11.4 |
| 20p11.22 | 48 | 0.58 | 2.20 |
| 21q22.3 | 51 | 0.93 | 2.38 |
Acknowledgments
The authors would like to thank Shin Lin, PhD for helpful discussions and Nulang Wang for DNA extraction and CFTR genotyping. Genotyping of the 402 marker STR map was done by the Mammalian Genotyping Service in the Center for Medical Genetics at the Marshfield Clinic (Marshfield, WI). This study would not be possible without CF patients and their families, Research Coordinators and CF Center Directors and Physicians participating in the CF Twin and Sibling Study. The following Centers are participants in the CF Twin and Sibling Study: Advocate Hope Children’s Hospital, Oak Lawn, IL; Children’s Hospital and Medical Center of Akron, Akron, OH; Children’s Asthma Respiratory & Exercise Specialists, Glenview, IL; Children’s Hospital and Medical Center of Cincinnati, Cincinnati, OH; Children’s Hospital and Medical Center, Seattle, WA; The Children’s Hospital of Boston, Boston, MA; Children’s Hospital of Buffalo, Buffalo, NY; Children’s Hospital of Denver, Denver, CO; Children’s Hospital of Los Angeles, Los Angeles, CA; Children’s Hospital of Oakland, CA; Children’s Hospital of Orange County, Orange, CA; Children’s Hospital of Philadelphia, Philadelphia, PA; Children’s Hospital of Pittsburgh, Pittsburgh, PA; Children’s Hospital of Wisconsin, Milwaukee, WI; Children’s Medical Center of Dallas, Dallas, TX; Children’s Memorial Medical Center, Chicago, IL; Children’s Mercy Hospital Kansas City, KS; Columbus Children’s Hospital, Columbus, OH; Connecticut Children’s Medical Center, Hartford, CT; Cook Children’s Medical Center, Fort Worth, TX; Dartmouth-Hitchcock Medical Center, Lebanon, NH; Eastern Maine Medical Center, Bangor, ME; Emory University School of Medicine, Atlanta, GA; Fletcher Allen Health Care, Burlington, VT; Georgia Pediatric Pulmonology Associates, Atlanta, GA; Great Falls Clinic, Great Falls, MT; Hasbro Children’s Hospital, Providence, RI; Hershey Medical Center, Hershey, PA; Hospital of the University of Pennsylvania, Philadelphia, PA; Joe DiMaggio Pulmonary Center, Hollywood, FL; Johns Hopkins Hospital, Baltimore, MD; Kaiser Permanente Medical Center, Oakland, CA; Kaiser Permanente, Portland, OR; Kaiser-Permanente, Panorama City, CA; Massachusetts General Hospital, Boston, MA; Medical College of Georgia, Augusta, GA; Medical University of South Carolina, Charleston, SC; Memphis Lung Physicians, South Haven, MS; Methodist Children’s Hospital, San Antonio, TX; Methodist Hospital, Houston, TX; Michigan State University, Lansing, MI; Monmouth Medical Center, Long Branch, NJ; Morristown Memorial Hospital, NJ; Mountain State CF Center, Morgantown, WV; National Naval Medical Center, Bethesda, MD; Naval Medical Center San Diego, San Diego, CA; New York Medical College, Valhalla, NY; Oklahoma Health Science Center, Oklahoma City, OK; Pediatric Diagnostic Center, Ventura, CA; Pediatric Pulmonary Associates, Columbia, SC; The Prince Charles Hospital, Australia; Rainbow Babies and Children’s Hospital, Cleveland, OH; Riley Children’s Hospital, Indianapolis, IN; Royal Hospital for Sick Children, Yorkhill, Scotland; Rush Presbyterian St Luke’s Medical Center, Chicago, IL; Saint Vincent’s Hospital & Medical Center, New York, NY; Schneider Children’s Hospital, New Hyde Park, NY; Sparks Regional Medical Center, Fort Smith, AK; St Christopher’s Hospital for Children, Philadelphia, PA; St Mary’s Medical Center, West Palm Beach, FL; St. Louis Children’s Hospital, St. Louis, MO; Stanford University Medical Center, Palo Alto, CA; SUNY Pediatric Pulmonary & CF Center, Syracuse, NY; Sutter Medical Center, Sacramento, CA; TC Thompson Children’s Hospital, Chattanooga, TN; Texas Children’s Hospital, Houston TX; University of Alabama CF Center, Birmingham, AL; University of Arizona, Tucson, AZ; University of California at San Francisco; University of Iowa Hospitals and Clinics, Iowa City, IA; University of Kentucky, Lexington, KY; University of Massachusetts Medical School, Worcester, MA; University of Michigan, Ann Arbor, MI; University of Minnesota, Minneapolis, MN; University of Mississippi Medical Center, Jackson, MS; University of Nebraska, Omaha, NE; University of New Mexico Health Sciences Center, Albuquerque, NM; University of North Carolina, Chapel Hill, NC; University of Tennessee, Memphis, TN; University of Texas Health Center, Tyler TX; University of Utah, Salt Lake City, UT; University of Virginia Health System, Charlottesville, VA; University of Wisconsin Hospital, Madison, WI; Vanderbilt University Medical Center, Nashville, TN; Wake Forest University, Baptist Medical Center, Winston-Salem, NC; Wayne State University School of Medicine, Detroit, MI; Western Carolina CF Center, Charlotte, NC; Wichita Clinic, PA, Wichita, KS; Wilford Hall Medical Center, San Antonio, TX; Yale University, New Haven, CT.
This work was funded by grant HL 68927 from the National Institute of Heart, Lung and Blood and by grant CUTTIN00A0 from the U.S. Cystic Fibrosis Foundation.
Footnotes
Grant support: HL 68927 from the National Institute of Heart, Lung and Blood and CUTTIN00A0 from the U.S. Cystic Fibrosis Foundation. S. Blackman is supported by T32 DK07751 from the National Institute of Digestive and Diabetes and Kidney Disease. R. Deering-Brose is supported by T32 GM007814 from the National Institute of General Medical Sciences.
References
- 1.Kristidis P, Bozon D, Corey M, Markiewicz D, Rommens J, Tsui LC, Durie P. Genetic determination of exocrine pancreatic function in cystic fibrosis. Am J Hum Genet. 1992;50:1178–1184. [PMC free article] [PubMed] [Google Scholar]
- 2.Kerem E, Corey M, Kerem B-S, Rommens J, Markiewicz D, Levison H, Tsui LC, Durie P. The relation between genotype and phenotype in cystic fibrosis--analysis of the most common mutation (deltaF508) N Engl J Med. 1990;323:1517–1522. doi: 10.1056/NEJM199011293232203. [DOI] [PubMed] [Google Scholar]
- 3.Correlation between genotype and phenotype in patients with cystic fibrosis. The Cystic Fibrosis Genotype-Phenotype Consortium. N Engl J Med. 1993;329:1308–1313. doi: 10.1056/NEJM199310283291804. [DOI] [PubMed] [Google Scholar]
- 4.Kerem E, Corey M, Kerem B-S, Durie P, Tsui LC, Levison H. Clinical and genetic comparisons of patients with cystic fibrosis, with or without meconium ileus. J Pediatr. 1989;114:767–773. doi: 10.1016/s0022-3476(89)80134-9. [DOI] [PubMed] [Google Scholar]
- 5.Cystic Fibrosis Foundation. Cystic Fibrosis Foundation Patient Registry Annual Data Report 2000. 2001. [Google Scholar]
- 6.Lai HJ, Cheng Y, Cho H, Kosorok MR, Farrell PM. Association between initial disease presentation, lung disease outcomes, and survival in patients with cystic fibrosis. Am J Epidemiol. 2004;159:537–546. doi: 10.1093/aje/kwh083. [DOI] [PubMed] [Google Scholar]
- 7.Rosenstein BJ, Langbaum TS. Incidence of distal intestinal obstruction syndrome in cystic fibrosis. J Pediatr Gastroenterol Nutr. 1983;2:299–301. [PubMed] [Google Scholar]
- 8.Dray X, Bienvenu T, Desmazes-Dufeu N, Dusser D, Marteau P, Hubert D. Distal intestinal obstruction syndrome in adults with cystic fibrosis. Clin Gastroenterol Hepatol. 2004;2:498–503. doi: 10.1016/s1542-3565(04)00169-7. [DOI] [PubMed] [Google Scholar]
- 9.Jaffe BF, Graham WP, III, Goldman L. Postinfancy intestinal obstruction in children with cystic fibrosis. Arch Surg. 1966;92:337–343. doi: 10.1001/archsurg.1966.01320210017003. [DOI] [PubMed] [Google Scholar]
- 10.O’Halloran SM, Gilbert J, McKendrick OM, Carty HM, Heaf DP. Gastrografin in acute meconium ileus equivalent. Arch Dis Child. 1986;61:1128–1130. doi: 10.1136/adc.61.11.1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donnison AB, Shwachman H, Gross RE. A review of 164 children with meconium ileus seen at the Children’s Hospital Medical Center, Boston. Pediatrics. 1966;37:833–850. [PubMed] [Google Scholar]
- 12.Allan JR, Robbie M, Phelan PD, Danks DM. Familial occurence of meconium ileus. Eur J Ped. 1981;135:291–292. doi: 10.1007/BF00442105. [DOI] [PubMed] [Google Scholar]
- 13.Picard E, Aviram M, Yahav Y, Rivlin J, Blau H, Bentur L, Avital A, Villa Y, Schwartz S, Kerem B, Kerem E. Familial concordance of phenotype and microbial variation among siblings with CF. Pediatr Pulmonol. 2004;38:292–297. doi: 10.1002/ppul.20111. [DOI] [PubMed] [Google Scholar]
- 14.Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, Koller BH. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257:1083–1088. doi: 10.1126/science.257.5073.1083. [DOI] [PubMed] [Google Scholar]
- 15.Rozmahel R, Wilschanski M, Matin A, Plyte S, Oliver M, Auerbach W, Moore A, Forstner J, Durie P, Nadeau J, Bear C, Tsui LC. Modulation of disease severity in cystic fibrosis transmembrane conductance regulator deficient mice by a secondary genetic factor. Nature Genet. 1996;12:280–287. doi: 10.1038/ng0396-280. [DOI] [PubMed] [Google Scholar]
- 16.Haston CK, Tsui LC. Loci of intestinal distress in cystic fibrosis knockout mice. Physiol Genomics. 2003;12:79–84. doi: 10.1152/physiolgenomics.00114.2002. [DOI] [PubMed] [Google Scholar]
- 17.Zielenski J, Corey M, Rozmahel R, Markiewicz D, Aznarez I, Casals T, Larriba S, Mercier B, Cutting GR, Krebsova A, Macek M, Jr, Langfelder-Schwind E, Marshall BC, DeCelie-Germana J, Claustres M, Palacio A, Bal J, Nowakowska A, Ferec C, Estivill X, Durie P, Tsui LC. Detection of a cystic fibrosis modifier locus for meconium ileus on human chromosome 19q13. Nature Genet. 1999;22:128–129. doi: 10.1038/9635. [DOI] [PubMed] [Google Scholar]
- 18.MacGregor AJ, Snieder H, Schork NJ, Spector TD. Twins. Novel uses to study complex traits and genetic diseases. Trends Genet. 2000;16:131–134. doi: 10.1016/s0168-9525(99)01946-0. [DOI] [PubMed] [Google Scholar]
- 19.Hammond CJ, Snieder H, Spector TD, Gilbert CE. Genetic and environmental factors in age-related nuclear cataracts in monozygotic and dizygotic twins. N Engl J Med. 2000;342:1786–1790. doi: 10.1056/NEJM200006153422404. [DOI] [PubMed] [Google Scholar]
- 20.Bronsveld I, Mekus F, Bijman J, Ballmann M, Greipel J, Hundrieser J, Halley DJ, Laabs U, Busche R, De Jonge HR, Tummler B, Veeze HJ. Residual chloride secretion in intestinal tissue of deltaF508 homozygous twins and siblings with cystic fibrosis. The European CF Twin and Sibling Study Consortium. Gastroenterology. 2000;119:32–40. doi: 10.1053/gast.2000.8524. [DOI] [PubMed] [Google Scholar]
- 21.Selmi C, Mayo MJ, Bach N, Ishibashi H, Invernizzi P, Gish RG, Gordon SC, Wright HI, Zweiban B, Podda M, Gershwin ME. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology. 2004;127:485–492. doi: 10.1053/j.gastro.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 22.Rosenstein BJ, Cutting GR. The diagnosis of cystic fibrosis: A consensus statement. J Pediatr. 1998;132:589–595. doi: 10.1016/s0022-3476(98)70344-0. [DOI] [PubMed] [Google Scholar]
- 23.Cutting GR, Antonarakis SE, Buetow KH, Kasch LM, Rosenstein BJ, Kazazian HH., Jr Analysis of DNA polymorphism haplotypes linked to the cystic fibrosis locus in North American Black and Caucasian families supports the existence of multiple mutations of the cystic fibrosis gene. Am J Med Genet. 1989;44:307–318. [PMC free article] [PubMed] [Google Scholar]
- 24.Wang X, Myers A, Saiki RK, Cutting GR. Development and Evaluation of a PCR-based, Line Probe Assay for the Detection of 58 Alleles in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Gene. Clin Chem. 2002;48:1121–1123. [PubMed] [Google Scholar]
- 25.Groman JD, Meyer ME, Wilmott RW, Zeitlin PL, Cutting GR. Variant cystic fibrosis phenotypes in the absence of CFTR mutations. N Engl J Med. 2002;347:401–407. doi: 10.1056/NEJMoa011899. [DOI] [PubMed] [Google Scholar]
- 26.Matsuzaki H, Dong S, Loi H, Di X, Liu G, Hubbell E, Law J, Berntsen T, Chadha M, Hui H, Yang G, Kennedy GC, Webster TA, Cawley S, Walsh PS, Jones KW, Fodor SP, Mei R. Genotyping over 100,000 SNPs on a pair of oligonucleotide arrays. Nat Methods. 2004;1:109–111. doi: 10.1038/nmeth718. [DOI] [PubMed] [Google Scholar]
- 27.Falconer DS. Inheritance of Liability to Certain Diseases Estimated from Incidence Among Relatives. Ann Hum Genet. 1965;29:51. [Google Scholar]
- 28.Falconer DS, Mackay TFC. Introduction to quantitative genetics. 4th ed. Essex: Pearson Education Limited; 1996. Heritability; pp. 160–183. [Google Scholar]
- 29.Kulich M, Rosenfeld M, Campbell J, Kronmal R, Gibson RL, Goss CH, Ramsey B. Disease-specific Reference Equations for Lung Function in Patients with Cystic Fibrosis. Am J Respir Crit Care Med. 2005;172:885–891. doi: 10.1164/rccm.200410-1335OC. [DOI] [PubMed] [Google Scholar]
- 30.O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duffy DL. SIB-PAIR 0.99.9: A program for elementary genetic analysis. Queensland, Australia: Queensland University of Medical Research; 2004. [Google Scholar]
- 32.Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 33.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 34.Abecasis GR, Wigginton JE. Handling marker-marker linkage disequilibrium: pedigree analysis with clustered markers. Am J Hum Genet. 2005;77:754–767. doi: 10.1086/497345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krawczak M. ASP--a simulation-based power calculator for genetic linkage studies of qualitative traits, using sib-pairs. Hum Genet. 2001;109:675–677. doi: 10.1007/s00439-001-0634-x. [DOI] [PubMed] [Google Scholar]
- 36.JENSEN KG. Meconium-ileus equivalent in a 15-year-old patient with mucoviscidosis. Acta Paediatr. 1962;51:344–348. doi: 10.1111/j.1651-2227.1962.tb06550.x. [DOI] [PubMed] [Google Scholar]
- 37.Nadeau JH. Modifier genes in mice and humans. Nat Rev Genet. 2001;2:165–174. doi: 10.1038/35056009. [DOI] [PubMed] [Google Scholar]
- 38.Martin N, Boomsma D, Machin G. A twin-pronged attack on complex traits. Nat Genet. 1997;17:387–392. doi: 10.1038/ng1297-387. [DOI] [PubMed] [Google Scholar]
- 39.Santis G, Osborne L, Knight R, Smith M, Davison T, Hodson M. Genotype-phenotype relationship in cystic fibrosis: Results from the study of monozygotic and dizygotic twins with cystic fibrosis. Ped Pulm. 1992;(Suppl 8):239–240. [Google Scholar]
- 40.Dolan TF, Jr, Touloukian RJ. Familial meconium ileus not associated with cystic fibrosis. J Pediatr Surg. 1974;9:821–824. doi: 10.1016/s0022-3468(74)80215-0. [DOI] [PubMed] [Google Scholar]
- 41.Tal A, Carmi R, Chai-Am E, Zirkin H, Bar-Ziv J, Freud E. Familial meconium ileus with normal sweat electrolytes. Clin Pediatr (Phila) 1985;24:460–462. doi: 10.1177/000992288502400809. [DOI] [PubMed] [Google Scholar]
- 42.Hamosh A, King TM, Rosenstein BJ, Corey M, Levison H, Durie P, Tsui LC, McIntosh I, Keston M, Brock DJH, Macek M, Jr, Zemkova D, Krasnicanova H, Vavrovä V, Macek M, Sr, Golder N, Schwarz MJ, Super M, Watson EK, Williams C, Bush A, O’Mahoney SM, Humphries P, DeArce MA, Reis A, Burger J, Stuhrmann M, Schmidtke J, Wulbrand U, Dörk T, Tümmler B, Cutting GR. Cystic fibrosis patients bearing the common missense mutation Gly->Asp at codon 551 and the deltaF508 are indistinguishable from deltaF508 homozygotes except for decreased risk of meconium ileus . Am J Hum Genet. 1992;51:245–250. [PMC free article] [PubMed] [Google Scholar]
- 43.Feingold J, Guilloud-Bataille M. Genetic comparisons of patients with cystic fibrosis with or without meconium ileus. Clinical Centers of the French CF Registry. Ann Genet. 1999;42:147–150. [PubMed] [Google Scholar]
- 44.Li Z, Lai HJ, Kosorok MR, Laxova A, Rock MJ, Splaingard ML, Farrell PM. Longitudinal pulmonary status of cystic fibrosis children with meconium ileus. Pediatr Pulmonol. 2004;38:277–284. doi: 10.1002/ppul.20092. [DOI] [PubMed] [Google Scholar]
- 45.Evans AK, Fitzgerald DA, McKay KO. The impact of meconium ileus on the clinical course of children with cystic fibrosis. Eur Respir J. 2001;18:784–789. doi: 10.1183/09031936.01.00053701. [DOI] [PubMed] [Google Scholar]
- 46.Fuchs JR, Langer JC. Long-term outcome after neonatal meconium obstruction. Pediatrics. 1998;101:E7. doi: 10.1542/peds.101.4.e7. [DOI] [PubMed] [Google Scholar]
- 47.Maurage C, Lenaerts C, Weber A, Brochu P, Yousef I, Roy CC. Meconium ileus and its equivalent as a risk factor for the development of cirrhosis: an autopsy study in cystic fibrosis. J Pediatr Gastroenterol Nutr. 1989;9:17–20. [PubMed] [Google Scholar]
- 48.Lai HC, Kosorok MR, Laxova A, Davis LA, FitzSimmon SC, Farrell PM. Nutritional status of patients with cystic fibrosis with meconium ileus: a comparison with patients without meconium ileus and diagnosed early through neonatal screening. Pediatrics. 2000;105:53–61. doi: 10.1542/peds.105.1.53. [DOI] [PubMed] [Google Scholar]
- 49.Norkina O, De Lisle RC. Potential genetic modifiers of the cystic fibrosis intestinal inflammatory phenotype on mouse chromosomes 1, 9, and 10. BMC Genet. 2005;6:29. doi: 10.1186/1471-2156-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sawcer S, Jones HB, Judge D, Visser F, Compston A, Goodfellow PN, Clayton D. Empirical genomewide significance levels established by whole genome simulations. Genet Epidemiol. 1997;14:223–229. doi: 10.1002/(SICI)1098-2272(1997)14:3<223::AID-GEPI1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]