

Abstract
Intensive efforts have been made to develop potent and selective ligands for certain human melanocortin receptors as possible treatments for obesity and sexual dysfunction due to the role of these receptors in feeding behavior, energy homeostasis, sexual function, etc. A number of novel α-MSH analogues were designed and synthesized primarily on the basis of our previous MTII NMR structure. In these peptide analogues, a disulfide or lactam bridge between residues at positions 5 and 8 was used as a conformational constraint to enhance the β-turn spanning His6 and d-Phe7, while the pharmacophore group in Arg8 was mimicked via Nα-alkylation of residues 8 or 9 with the guanidinylbutyl group. Biological assays for binding affinities and adenylate cyclase activities for the hMC1R, hMC3R, hMC4R, and hMC5R showed that three analogues have good binding affinity for the hMC4R (0.7–4.1 nM), but have no binding affinity up to 10 μM at the other three melanocortin receptors. Interestingly, the three hMC4R selective analogues display only 50% binding efficiency, suggesting there is allosteric modulation of the melanocortin-4 receptor. These analogues were found to act as antagonists of the hMC4R. This result represents a discovery of very selective peptide-based antagonists for the hMC4R. The high selectivity may be due to the strong conformational constraint via ring contraction as compared to MTII, and the rigid conformation preferred by these new ligands allows them to recognize only the hMC4R, but not to activate the second messenger. The MTII NMR structure-based design thus not only examined the structural model of melanocortin ligands, but also yielded new biologically unique α-MSH analogues.
Introduction
α-, β-, and γ-Melanocyte stimulating hormone (MSHa) and adrenocorticotropin (ACTH) are posttranslational products of the proopiomelanocortin (POMC) prohormone.1 As natural ligands interacting with five melanocortin receptors (MC1R-MC5R), these endogenous neuropeptides and hormones play an important role in regulating a large number of diverse physiological functions, including skin pigmentation,2,3 adrenal cortical function,3 energy homeostasis,4,5 feeding behavior,6,7 sexual function,8 exocrine gland function,9 pain,10 and many others. These physiological functions mediated in the melanocortin systems have made melanotropins useful lead peptides to develop drugs for treating obesity, sexual dysfunction, pigmentary problems, etc. To this end, earlier studies have focused on elucidation of the structure–activity relationships of α-MSH and identified His-Phe-Arg-Trp as the message sequence of melanotropin peptides.11-13 These studies also have led to early discoveries of several α-MSH analogues with biological profiles significantly different from those of α-MSH itself,14-17 thus providing diverse molecular tools for better understanding the biological roles of melanocortin pathways. More recently, much effort has been made to develop receptor selective analogues, peptidomimetics, and small molecule mimics of α-MSH.18-26 Selectivity for the MC4R is a particularly desirable goal due to the role of this receptor in feeding regulation and weight control.6,7,27 Suitable ligands highly selective for the human MC4R (hMC4R) may be developed as specific molecular probes to better study the receptor functions and as possible treatments for obesity.
One approach to development of receptor selective ligands is to apply conformational constraints28-36 to the existing lead peptides. In this regard, the structural insights obtained in our previous NMR study37 of MTII and other melanotropin analogues provide useful information on the design of potentially selective ligands with new conformational constraints. Despite the conformational differences observed, most of these α-MSH analogues were found to contain a type II β-turn structure spanning residues 6 and 7 distorted to different extents.37 In this study, we designed, synthesized, and biologically analyzed a number of new conformationally restricted analogues on the basis of the NMR-obtained conformation. The design was aimed to enhance the type II β-turn at positions 6 and 7 of α-MSH with a linker between residues 5 and 8. It turned out that several of these analogues selectively bind to the human MC4R only and act as antagonists of this receptor. The NMR structure-based design and synthesis of the new analogues thus not only served as a test of our previous structural model of melanocortin ligands, but also resulted in new biologically unique and receptor selective α-MSH analogues.
Design Considerations
As reported previously, MTII and several other analogues adopt a type II β-turn structure spanning His6 and d-Phe7.37 A more careful examination of our previous MTII NMR structure showed that the Hβ protons of Asp5 and Arg8 are in close proximity in space, as verified by observation of the weak interresidual ROESY cross-peaks between these protons. Such a short interproton distance is also consistent with the peptide chain reversal due to the β-turn structure at residues 6 and 7. It thus was reasonable to replace one of the two hydrogen atoms in each residue with a sulfur atom and then to use a disulfide bridge for the conformational restriction, in place of the original conformational constraint via the lactam bridge in MTII. As a result, Asp5 can be replaced by Cys5, while the Arg derivative (3R or 3S)-thio-Arg8 instead of Arg8 is introduced. The design led to the 5-residue 14-member cyclic peptides, Ac-c[Cys-His-d-Phe-(3R/S)-thio-Arg]-Trp-NH2, smaller cyclic peptides with a more rigid ring than MTII. The lowest energy structure of the peptide with (3R)-thio-Arg in the Monte Carlo conformational search was compared to the MTII NMR structure. The low backbone rmsd value for the superposition using the backbone atoms of the message sequence residues (0.76 Å) suggested a structural similarity between this peptide and MTII. However, thus far no practical synthetic methodology of β-thio-Arg with either configuration at Cβ has been developed.
An alternative design was to use Cys instead of β-thio-Arg at position 8 of the peptide for a disulfide bridge as the conformational constraint and to alkylate the Nα atom of Cys8 with the guanidinylbutyl group to mimic the Arg side chain group. This design led to a new peptide shown in Figure 1. Practically, peptide 1 may be synthesized on a solid phase support without resort to any unnatural amino acids which are difficult to obtain either commercially or synthetically.
Figure 1.
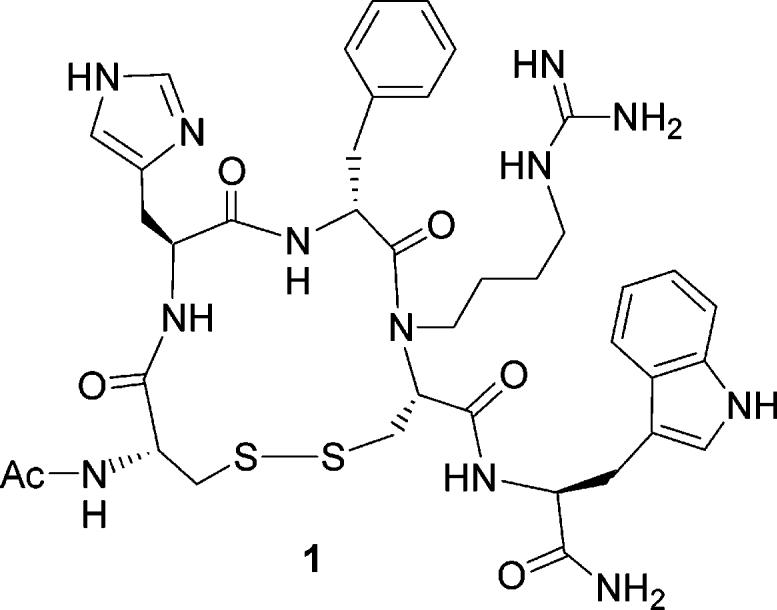
The structure of peptide 1, Ac-c[Cys-His-d-Phe-Nα-guanidinylbutyl-Cys]-Trp-NH2.
Guided by the MTII NMR structure, the design described above should reach the conformational space that is close to, but may be different from, that of MTII. The design of such a single peptide molecule may not be sufficient to pin down the structural requirements of the melanocortin receptors. It thus is necessary that the conformational space nearby be explored using a number of peptides with similar chemical structures. Fortunately, there were several ways to modify peptide 1 for such a purpose: altering the Nα-alkylation position (e.g. residue 8 or 9) or inverting the chirality of the Cα atom of the residue to be Nα-alkylated (e.g. use of l- or d-Cys at position 8). It is particularly noteworthy that, due to the Nα-alkylation of a residue, the backbone conformational and dynamics properties of that residue can be significantly different from the same residue without Nα-alkylation. This clearly justified the use of not only l-, but also d-amino acids for the Nα-alkylated residue. These considerations and modifications gave the following three new peptides with slightly different chemical structures from peptide 1: Ac-c[Cys-His-d-Phe-Nα-guanidinylbutyl-d-Cys]-Trp-NH2, 2; Ac-c[Cys-His-d-Phe-Cys]-Nα-guanidinylbutyl-Trp-NH2, 3; Ac-c[Cys-His-d-Phe-Cys]-Nα-guanidinylbutyl-d-Trp-NH2, 4.
An isosteric analogue of peptide 1 would be obtained if we substituted Asp and Dapa (α,β-diaminopropionic acid) for the Cys residues at positions 5 and 8, respectively, followed by a cyclization using a lactam structure. The design yielded a peptide with the general sequence Ac-c[X-His-d-Phe-Nα-guanidinylbutyl-Y]-Trp-NH2, where X = Asp and Y = Dapa. In this alternative design, there were 16 possible combinations which would give a large number of peptides with a slightly different primary structure: X = Asp or Glu for position 5 and Y = Dab (α,γ-diaminobutyric acid), Dapa, Orn, or Lys for position 8, or X = Dab, Dapa, Orn, or Lys for position 5 and Y = Asp or Glu for position 8. In addition, it also was possible to apply the Nα-alkylation to Trp9 instead of residue 8 where the cyclization occurs, giving more possible peptides. Although these possible variations provided a good approach to extensively exploring the conformational space close to that of MTII, it would be time-consuming to synthesize all the peptides. Instead, a Monte Carlo conformational search followed by a structural comparison of the structures obtained with the MTII conformation allowed us to select the following two representative peptides which most closely mimicked the MTII NMR structure: Ac-c[Glu-His-d-Phe-Nα-guanidinylbutyl-Dab]-Trp-NH2, 5; Ac-c[Glu-His-d-Phe-Nα-guanidinylbutyl-Orn]-Trp-NH2, 6.
To investigate the effect of bulky aromatic residues at position 7, we also designed the following two peptides: Ac-c[Cys-His-d-Nal(2′)-Cys]-Nα-guanidinylbutyl-d-Trp-NH2, 7; Ac-c[Glu-His-d-Nal(2′)-Nα-guanidinylbutyl-Orn]-Trp-NH2, 8.
All of the eight analogues designed above were successfully synthesized using solid-phase peptide synthesis and purified using RP-HPLC. The syntheses and biological assay results are discussed below in detail.
Chemistry
A key design in the syntheses of all the above peptides was to utilize a Mitsunobu reaction38,39 to alkylate the α-amino group of a relevant residue using N,N′-diBoc-guanidinylbutanol40 for mimicking the pharmacophore in Arg8 of the melanotropin. Prior to the alkylation, the α-amino group was activated by the 2-nitrobenzenesulfonyl (oNBs) group (Scheme 1) which can be removed under mild conditions.41,42 Formation of such a sulfonamide made the amide proton acidic enough to be deprotonated easily by the azo compound. The nitrogen anion thus formed became a good nucleophile in the subsequent Mitsunobu reaction. The Mitsunobu alkylation is well suited for synthesis on a solid-phase support.41,43 It thus was possible to develop all the steps for synthesizing each peptide by solid phase, as exemplified by the synthesis of peptide 1 shown in Scheme 1.
A number of peptides were designed on the basis of the NMR structures of MTII, a lead α-MSH analogue. The solid-phase syntheses of these peptides were challenging for Nα-alkylation, secondary amine coupling, and macrocyclization.
Because of the mild conditions for removing the 2,4-dinitrobenzenesulfonyl (DNBs) group,44 sulfonylation of the free α-amino group of Cys8 with 2,4-dinitrobenzenesulfonyl chloride (3 equiv) and pyridine (3 equiv) in dry DCM (Scheme 2) was initially used for the synthesis of peptide 1, prior to the Nα-alkylation via a Mitsunobu reaction. However, an intramolecular nucleophilic aromatic substitution under the mild basic conditions for the sulfonylation was found to occur via a mechanism shown in Scheme 2.45 Such a substitution resulted in the migration of the aromatic ring from the sulfur atom to the nitrogen atom and the subsequent loss of SO2 (Scheme 2). The rearrangement falls into the group of reactions collectively known as Smiles rearrangements45 described originally by Smiles and co-workers46 in 1931, but reported herein for the first time to occur in a sulfonamide on a solid-phase support. It must be noted that the rearranged product in Scheme 2, a secondary amine, still was acidic enough to allow the Nα-alkylation to take place under Mitsunobu conditions to yield a tertiary amine, as suggested by the mass change after the Mitsunobu reaction. Presumably, the acidity of the secondary amine obtained after the rearrangement can be attributed to the electron-withdrawing effect of the two nitro groups.
Scheme 1.
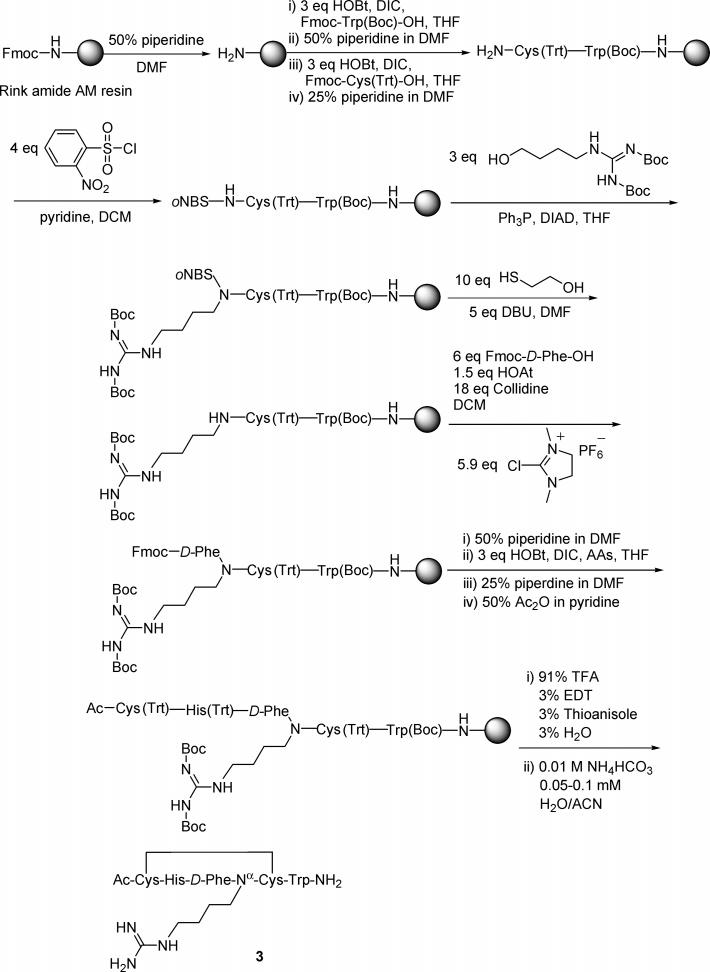
Synthesis of a Representative Peptide (peptide 1) on a Solid-Phase Support
Scheme 2.
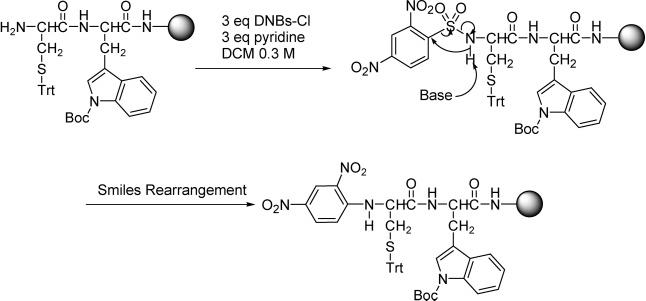
Smiles Rearrangement Occurred in the Initial Synthesis of Peptide 1 under a Mild Sulfonylation Condition
A consequence of the rearrangement was that a removable protecting group had been changed into one whose removal was impractical. In the Kaiser test after the sulfonylation, brown resin beads were observed, indicating the presence of a secondary amine. The rearrangement also was confirmed by the mass difference (−64) between the compound obtained and the target product after the sulfonylation step and the Mitsunobu alkylation reaction, respectively.
The rearrangement appeared to be sequence dependent. Under similar conditions, it was observed in the synthesis of DNBs-Nα-Dab(Alloc)-Trp(Boc)-Resin, DNBs-Nα-Orn(Alloc)-Trp(Boc)-Resin, and DNBs-Nα-Arg(Boc)2-Trp(Boc)-Resin, but not for DNBs-Nα-Trp(Boc)-Resin and DNBs-Nα-Arg(Pbf)-Trp(Boc)-Resin. It also was found that a change in the side chain protecting group of Cys8 in Scheme 2 from trityl to acetamidomethyl (Acm) completely avoided the rearrangement. In addition, we noted that no rearrangement was observed for DNBs-Nα-Cys(Trt)-OMe that was synthesized in solution phase under otherwise similar sulfonylation conditions. These observations implied that a steric effect rather than an electronic effect may have led to a conformation more favorable to the rearrangement which occurred in our study. To find a general solution to avoid the rearrangement problem, other synthetic strategies were thus sought.
During sulfonylation of the α-amino groups with 2-nitrobenzenesulfonyl (oNBs) chloride and pyridine in DCM,41 no Smiles rearrangement was found in our study regardless of the peptide sequence. This may be attributed to the fact that removal of the para-nitro group reduced the electrophilicity of the aromatic carbon at position 1′, making the nucleophilic attack of the α-nitrogen atom toward the aromatic carbon significantly unfavored. To remove the 2-nitrobenzenesulfonyl group,41 however, conditions (DBU and 2-mercaptoethanol, Scheme 1) which were harsher than those needed to remove the 2,4-dinitrobenzenesulfonyl group44 were required. Fortunately, no detectable racemization was observed in the analytical HPLC of the dipeptide samples cleaved from the resin after the deprotection step in the syntheses of these peptides. In addition, the 2-nitrobenzenesulfonamides were as easily alkylated as the rearranged products of the 2,4-dinitrobenzenesulfonamides. Thus, the 2-nitrobenzenesulfonyl group instead of the 2,4-dinitrobenzenesulfonyl group was used throughout the final syntheses of all the target peptides in our study and should be generally applicable to other solid-phase syntheses whenever the rearrangement described above is a concern.
The alkylation of the sulfonamides with N,N′-diBoc-guanidinylbutanol for the syntheses of all the peptides was readily achieved using the Mitsunobu reaction (diisopropyl azodicarboxylate (DIAD), Ph3P, THF, 0 °C, 10 min, and then room temperature, 15 h). The reaction was monitored by analytical HPLC and mass spectrometry after periodic resin cleavage and was complete in 15 h. We also tried to use diethyl azodicarboxylate (DEAD) for the Mitsunobu alkylation in the synthesis of peptide 1 and found that the reaction was not clean. Rew and Goodman43 reported that use of diethyl azodicarboxylate (DEAD) instead of DIAD in the Mitsunobu reaction always gave the ethylated byproduct in high yield (> 40%), in addition to the target alkylation product. Thus, DIAD is superior to DEAD in the solid-phase Mitsunobu reaction and was used for the Mitsunobu alkylation throughout this study.
It was noted that the coupling between the secondary amines and the subsequent amino acid (particularly Fmoc-d-Phe-OH) was very difficult. However, the coupling was finally achieved using Fmoc-d-Phe-OH (6 equiv), 2-chloro-1,3-dimethylimidazolidium hexafluorophospate (CIP, 5.9 equiv), 2,4,6-collidine (18 equiv), and HOAt (1.5 equiv) in DCM at room temperature.47-49 To minimize the formation of side products, couplings under the above condition were conducted for a short time (1–2 h) and repeated several times until the total coupling time was ca. 10 h. It was found using analytical HPLC that the coupling was clean and about 90% of the secondary amine, depending on the peptide sequence, was successfully coupled. No detectable racemization was observed during the repetitive coupling.
Fmoc-Cys(Trt)-OH was coupled to the secondary amines in the syntheses of peptides 3, 4, and 7 using the HBTU/DIEA procedure (Experimental Section), followed by another coupling using the symmetric anhydride of Fmoc-Cys(Trt)-OH in THF (Experimental Section). It was found that this protocol yielded a cleaner reaction for coupling Fmoc-Cys(Trt)-OH to the secondary amines than the CIP approach.
The syntheses of these peptides hereafter were relatively straightforward, although the cyclization of these peptides via a disulfide or lactam bridge was slow (Experimental Section). Gel filtration through a Sephadex G-15 column using 1.0 M acetic acid aqueous solution was used for all the samples, and the peptides were finally purified using preparative HPLC. The analytical data of these peptides are shown in Table 1. The purity of all the peptides was found to be >98%.
Table 1.
Analytical Data for the New Melanotropin Peptides
| TLC
Rfa |
HPLC |
HR-MSc |
|||||
|---|---|---|---|---|---|---|---|
| peptide sequence | A | B | C | kb | obsd | calcd | |
| 1 | Ac-c[Cys-His-d-Phe-Nα-guanidinylbutyl-Cys]-Trp-NH2 | 0.64 | 0.43 | 0.18 | 9.98 | 847.3517 | 847.3496 |
| 2 | Ac-c[Cys-His-d-Phe-Nα-guanidinylbutyl-d-Cys]-Trp-NH2 | 0.66 | 0.47 | 0.21 | 10.41 | 847.3510 | 847.3496 |
| 3 | Ac-c[Cys-His-d-Phe-Cys]-Nα-guanidinylbutyl-Trp-NH2 | 0.63 | 0.43 | 0.18 | 10.66 | 847.3502 | 847.3496 |
| 4 | Ac-c[Cys-His-d-Phe-Cys]-Nα-guanidinylbutyl-d-Trp-NH2 | 0.64 | 0.44 | 0.20 | 9.58 | 847.3510 | 847.3496 |
| 5 | Ac-c[Glu-His-d-Phe-Nα-guanidinylbutyl-Dab]-Trp-NH2 | 0.37 | 0.18 | 0.07 | 8.79 | 854.4442 | 854.4426 |
| 6 | Ac-c[Glu-His-d-Phe-Nα-guanidinylbutyl-Orn]-Trp-NH2 | 0.38 | 0.19 | 0.07 | 8.58 | 868.4592 | 868.4582 |
| 7 | Ac-c[Cys-His-d-Nal(2)′-Cys]-Nα-guanidinylbutyl-d-Trp-NH2 | 0.71 | 0.54 | 0.26 | 13.09 | 897.3669 | 897.3652 |
| 8 | Ac-c[Glu-His-d-Nal(2)′-Nα-guanidinylbutyl-Orn]-Trp-NH2 | 0.48 | 0.28 | 0.11 | 12.24 | 918.4725 | 918.4739 |
Rf values were obtained using thin-layer silica gel chromatography in the following three solvent systems: (A) water/HOAc/tert-butyl alcohol/EtOAc (1:1:1:1); (B) water/HOAc/tert-butyl alcohol/EtOAc (1:1:1:2); and (C) water/HOAc/tert-butyl alcohol/EtOAc (1:1:2:2).
HPLC k′ = [(peptide retention time – solvent retention time)/solvent retention time] in the solvent system (A: 0.1% aqueous TFA; B: acetonitrile). A linear gradient from 10% to 40% of solvent B over 30 min was applied. An analytical Waters C18 column (3.5 μm, 4.6 × 75 mm) was used with a flow rate of 1 mL/min. c High-resolution MS.
High-resolution MS.
It is noteworthy that our 1D 1H NMR study of all the peptides in D2O suggested the presence of conformers with a different ratio only in peptides 1, 3, 5, 6, and 8, all of which contain an l-amino acid residue at the Nα-alkylation site, but not in peptides 2, 4, and 7 with a d-amino acid at the alkylation position. The occurrence of conformers was further confirmed by a 2D ROESY50,51 experiment on peptide 5 as an example (see the ROESY spectrum in the Supporting Information). The same sign between the ROESY cross and diagonal peaks in the region of 8.3–8.6 ppm indicates that these cross-peaks arise from the conformational exchange between each resonance instead of a direct cross-relaxation effect of two nearby protons or impurities. Presumably, these conformers in peptides 1, 3, 5, 6, and 8 are due to the cis–trans isomerization of the Nα-alkylated peptide bond.
Results and Discussion
The binding affinity and adenylate cyclase activity of the peptides developed in this study were evaluated for various human melanocortin receptors (hMC1R, hMC3R, hMC4R, and hMC5R) using MTII as a standard control, and the results of these assays are summarized in Table 2. Peptide 1 was found to have good binding affinity toward the hMC4R (IC50 = 1.8 nM) but exhibited no receptor–ligand binding for the other melanocortin receptor subtypes up to 10−5 M. Interestingly, this analogue was an antagonist with a binding efficiency of about 50% (Figure 2). This may suggest the existence of an allosteric function for 1 on the hMC4R. Such an allosterically linked binding site prohibits the binding of a ligand to the other site of the receptor protein. Furthermore, the allosteric modulator changes the shape of the receptor, probably through conformational selection and stabilization of one or some of the receptor's ensemble of states. This results in a completely new set of reactivity toward the ligand and the cellular components.52-54 To verify the existence of allosteric function of the melanocortin receptors, we did the experiments where dose–response curves of MTII were obtained in the presence of antagonists. No significant shift in dose–response curves were observed (data not shown) which supports the existence of an allosteric binding site for antagonist peptide 1. In contrast to peptide 1, peptide 2 was obtained by using the d-enantiomer of Cys in position 8. The resulting peptide was found to be entirely inactive at all the melanocortin receptor subtypes, which suggests that such substitution drastically affects the secondary structure of the corresponding peptide. A shift of the Nα-guanidylbutyl moiety to the Trp9 position resulted in peptide 3, which demonstrated a potent binding affinity (IC50 = 4.1 nM), and weak partial agonist activity (18% maximal stimulation) at the hMC4R. Peptide 4 was obtained by substituting Trp9 in the peptide 3 sequence with d-Trp9, which led to a further improvement in binding potency to hMC4R (IC50 = 0.7 nM). In the cAMP accumulation assay, this compound was determined to be a weak partial agonist (19% maximal stimulation) at the hMC4R, similar to peptide 3. As was observed for peptide 1, peptides 3 and 4 also exhibited remarkable receptor selectivity for the hMC4R and once again the binding efficiency could reach to the level of about 50% only. This evidence provides further support for the existence of an allosteric binding site at the hMC4R. These results also suggest that the Cα stereochemistry of Trp9 does not appear to be as critical to biological activity as that of the residue at position 8. Presumably, this is because Trp9 is located outside the ring structure, which affords much more flexibility of its side chain group for a favorable orientation for interaction with the receptor.
Table 2.
Binding Affinity (IC50, nM) and cAMP Activity (EC50, nM) of Cyclic Melanotropin Peptides at Various Human Melanocortin Receptors
|
hMC1R |
hMC3R |
hMC4R |
hMC5R |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| peptide | IC50a (nM) | binding eff. (%)c | EC50b (nM) | act. % | IC50a (nM) | binding eff. (%)c | EC50b (nM) | act. % | IC50a (nM) | binding eff. (%)c | EC50b (nM) | act. % | IC50a (nM) | binding eff. (%)c | EC50b (nM) | act. % |
| MTII | 0.20±0.01 | 100 | 0.30±0. 04 | 100 | 2±0.2 | 100 | 1.9 ±0.2 | 100 | 1.1±0.3 | 100 | 2.9±0. 52 | 100 | 7.5±0.2 | 100 | 3.3±0.7 | 100 |
| 1 | NB | 0 | NA | 0 | NB | 0 | NA | 0 | 1.8±0.4 | 50 | NA | 0 | NB | 0 | NA | 0 |
| 2 | 1000 | <10 | NA | 0 | NB | 0 | NA | 0 | NB | 0 | NA | 0 | NB | 0 | NA | 0 |
| 3 | >1000 | <10 | NA | 0 | >1000 | 50 | NA | 0 | 4.1±1 | 50 | >1000 | 18 | NB | 0 | NA | 0 |
| 4 | NB | 0 | NA | 0 | >1000 | <10 | NA | 0 | 0.7±0.1 | 50 | >1000 | 19 | NB | 0 | NA | 0 |
| 5 | >1000 | <10 | NA | 0 | >1000 | 50 | NA | 0 | NB | 0 | NA | 0 | NB | 0 | NA | 0 |
| 6 | 750±100 | <10 | NA | 0 | >1000 | 50 | NA | 0 | 101±20 | 50 | >1000 | <10 | NB | 0 | NA | 0 |
| 7 | >1000 | <10 | NA | 0 | NB | 0 | NA | 0 | NB | 0 | NA | 0 | 209±30 | <10 | NA | 0 |
| 8 | >10000 | <10 | NA | 0 | >1000 | 50 | NA | 0 | NB | 0 | NA | 0 | NB | 0 | NA | 0 |
IC50 = Concentration of peptide at 50% specific binding (N = 4–6). The peptides were tested in a range of concentrations (10−10 to 10−5 M).
EC50 = Concentration of peptide at 50% maximal cAMP generation (n = 4). The peptides were tested in a range of concentrations (10−10 to 10−5 M).
Binding efficiency: maximal percentage binding of test compounds vs MTII agonist binding when competed with [125I]NDP-α-MSH. NB = No binding; NA = no cAMP activity. Three experiments were done in duplicate (n = 6) for binding as well as cAMP assays.
Figure 2.

Competitive binding assay of MTII and peptide 1 with [125I]-NDP-α-MSH at hMC4R. Note that peptide 1 does not displace the [125I]NDP-α-MSH to the extent MTII does.
Furthermore, employment of cyclic lactam templates (rather than disulfides as above) in peptides 5 (Ac-c[Glu-His-d-Phe-Nα-guanidylbutyl-Dab]-Trp-NH2) and 6 (Ac-c[Glu-His-d-Phe-Nα-guanidylbutyl-Orn]-Trp-NH2) led to a loss of binding affinity and biological activities at all the receptor subtypes for peptide 5, whereas peptide 6 showed modest binding affinity to the hMC4R (IC50 = 100 nM), with a binding efficiency of 50%. Thus, the cyclic disulfide scaffold (peptides 1–4) turned out to be superior to the cyclic lactams (peptides 5 and 6), possibly due to the greater flexibility of the disulfide bridge. Peptide 7 was produced by replacing d-Phe7 in peptide 4 with d-Nal(2′),7 which resulted in an hMC5R-selective weak binder (IC50) 210 nM). Similar substitution in the peptide 6 sequence led to peptide 8 (Ac-c[Glu-His-d-Nal(2′)-Nα-guanidylbutyl-Orn]-Trp-NH2) which showed total loss of binding affinity and biological activity at all of the hMCRs.
Conclusion
A number of new cyclic α-MSH analogues was designed on the basis of the MTII NMR structure. In these peptide analogues, novel disulfide or lactam bridges with macrocyclization were used as conformational constraints, while the pharmacophore group in Arg8 was mimicked via Nα-alkylation of certain amino acid residues with the guanidinylbutyl group. All eight cyclic peptides were successfully synthesized on a solid-phase support. The Mitsunobu reaction was used to achieve the Nα-alkylation for mimicking the Arg8 pharmacophore group in these syntheses. The binding affinity and adenylate cyclase activity assays of these peptides at human melanocortin receptors showed that three of the new α-MSH analogues act as antagonists and exhibited high selectivity toward the human melanocortin-4 receptor. The selectivity may be due to the strong conformational constraint via ring contraction as compared to MTII. The MTII NMR structure-based design thus not only examined the structural model of melanocortin ligands, but also yielded new biologically unique α-MSH analogues. As observed in this study, allosterically modulated GPCRs have been recently recognized as an alternative approach to gain selectivity in drug action. It is highly important to further modify the majority of assays currently being used to study such novel compounds that could be a great lead in the drug discovery process. A number of peptide analogues are in the works in our laboratory that are based on this unique design concept.
Experimental Section
General Procedures for Solid-Phase Peptide Synthesis
All the peptides were manually synthesized based on the Nα-Fmoc chemistry solid-phase method. A specific amount of Rink amide AM resin (0.63 mmol/g) was swollen in DMF for 2 h. After carefully washing the resin with DMF, the Fmoc protecting group of the resin or Nα-Fmoc peptidyl resin was removed using 50% (or 25% when a sensitive residue such as Cys was involved) piperidine in DMF (2 min × 1 and 20 min × 1). The resin was washed with DMF, 1.0 M HOBt in DMF, 1.0 M HOBt in DMF with 1–2 drops of 1.0 mM bromophenol blue in DMF as an indicator, and then DMF. For coupling between a primary amine and an amino acid, the Nα-Fmoc protected amino acid (3 equiv) with appropriate side chain protection was first activated by the formation of HOBt ester (0.25 M) using HOBt (3 equiv) and DIC (3 equiv) in THF and then mixed with the Fmoc-deprotected resin or peptidyl resin. For amino acids such as Fmoc-Nα-His(Trt)-OH that are not very soluble in THF, a mixture of DMF and THF (1:1), instead of THF, was used. The blue resin slurry was gently stirred on a shaker until the slurry became yellow or until a negative Kaiser test was obtained (generally 1–3 h). When the coupling was achieved, the resin was washed with DMF. The same procedure for deprotection and coupling was repeated for the next amino acid until all amino acids in the sequence were attached. The experimental procedures for the Nα-alkylation and the coupling of a secondary amine with an amino acid are discussed below in detail. The α-amino group of the last residue in the N-terminus of a peptide, after removal of its Fmoc group, was acetylated using 50% acetic anhydride in pyridine (4 min × 1 and 20 min × 1).
The peptidyl resin was thoroughly washed with DCM before the peptide was cleaved. Two different cleavage solutions were chosen depending on the sequence of the peptide. For peptides containing a trityl protecting group, a mixture (10 mL/g resin) of trifluoroacetic acid (91%), water (3%), 1,2-ethanedithiol (3%), and thioanisole (3%) was added to the resin. For peptides with no trityl protecting group, the cleavage mixture consisted of trifluoroacetic acid (95%), water (2.5%), and triisopropylsilane (2.5%). In either case, the cleavage cocktail was stirred for 3 h at room temperature. After precipitation of the peptide with cooled ethyl ether, followed by washing and centrifuging three times, ethyl ether was decanted and the peptide was further dried. To remove the residual Boc group from the Trp side chain, the peptide was dissolved in a 10% acetic acid aqueous solution for 3–5 h and then lyophilized. The sample was then purified using HPLC.
Rink amide AM resin (200–400 mesh, 0.6–0.7 mmol/g), Nα-Fmoc-d-Trp(Boc)-OH, Nα-Fmoc-d-Phe-OH, and Nα-Fmoc-His(Trt)-OH were purchased from Novabiochem; Nα-Fmoc-Dab(Alloc)-OH, Nα-Fmoc-Orn(Alloc)-OH, and Nα-Fmoc-Glu(Allyl)-OH from Neo-System; Nα-Fmoc-Trp(Boc)-OH and Nα-Fmoc-Arg(Pbf)-OH from SynPep; Nα-Fmoc-Cys(Trt)-OH from American Peptide Company; Nα-Fmoc-Cys(Acm)-OH from Bachem; Nα-Fmoc-d-Cys(Trt)-OH and HBTU from Advanced ChemTech; Nα-Fmoc-Arg(Boc)2-OH and piperidine from Chem-Impex International; pyridine from Mallinckrodt; 2-chloro-1,3-dimethylimidazolidium hexafluorophosphate and sodium diethyl dithiocarbamate trihydrate from Fluka; HOAt from AnaSpec Inc.; DMF, DCM, ethyl ether, and acetonitrile from EMD Chemicals Inc.; THF from DriSolv; bromophenol blue from Sigma; acetic acid from EM Science; tetrakis(triphenylphosphine)palladium(0) from Strem Chemicals Inc.; DIC from Acros; and trifluoroacetic acid from Du Pont. All the other reagents were purchased from Aldrich. Reagents and solvents were used as packaged, and no further purification was performed. The structures of the pure peptides were confirmed by ESI-MS (Finnigan, Thermoelectron, LCQ classic) and high-resolution FAB-MS (JEOL HX-110A sector instrument) at the University of Arizona Mass Spectrometry and Protein Sequencing Facility. Thin-layer chromatography (TLC) was performed with Merck silica gel 60 F254 (0.25 mm layer thickness). Flash liquid chromatography was performed with 230–400 mesh size silica gel which was purchased from Aldrich.
Preparation of 2-Nitrobenzenesulfonamide
After the Nα-Fmoc group was removed, the resin was washed with DMF (10 mL/g resin × 3) and DCM (10 mL/g resin × 3). A solution of 2-nitrobenzenesulfonyl chloride (4 equiv) and pyridine (4 equiv) in dry DCM (0.3 M) was added. The resin slurry was stirred for 3 h at room temperature. The reaction was repeated until a Kaiser test became negative, which usually took about 10–15 h. The resin was finally washed with DCM (10 mL/g resin × 3) and THF (10 mL/g resin × 3) for the next step.
Preparation of N,N′-DiBoc-guanidinylbutanol
To the solution of 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (5.0 mmol) in THF (10 mL) was added 4-amino-1-butanol (1.5 equiv) at room temperature. The reaction was then heated to 50 °C, and thin-layer chromatography (TLC) was used to monitor the completion of the reaction in about 2 h. The solvent was first removed via evaporation in vacuo, and the product was redissolved in DCM. The HCl aqueous solution (1.0 M) was used to extract the residual starting material 4-amino-1-butanol (10 mL × 4). The organic phase was finally washed with the brine solution and dried over MgSO4 prior to evaporation in vacuo. Instead of this workup procedure, flash column chromatography sometimes was carried out to improve the product purity and yield using the solvent mixture (1:1 hexane/ethyl acetate) as indicated by TLC.
Nα-Alkylation via a Mitsunobu Reaction
To the 2-nitrobenzenesulfonamide peptidyl resin pre-washed with THF were added N,N′-diBoc-guanidinylbutanol (3 equiv) and triphenylphosphine (3 equiv) in dry THF. The resin slurry was cooled to 0 °C and diisopropyl azodicarboxylate (DIAD, 3 equiv, 15% in dry THF, also cooled to 0 °C) was injected into the resin slurry. The amount of solvent was used such that the reactants had concentrations of 0.23 M. The reaction was stirred at room temperature for 15 h.
Removal of a 2-Nitrobenzenesulfonyl Group
After the Nα-alkylation described above, the resin was washed with THF (10 mL/g resin × 3) and DMF (10 mL/g resin × 3), to which was added 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 5 equiv) and 2-mercaptoethanol (10 equiv) in DMF (10 mL/g resin). The reaction was stirred at room temperature for 2 min and repeated for 20 min. The resin was finally washed with DMF (10 mL/g resin × 3) and DCM (10 mL/g resin × 3) before the next coupling reaction sequence was begun.
Coupling of a Secondary Amine with Nα-Fmoc-d-Phe-OH
To the peptidyl resin with a free secondary amine were added Nα-Fmoc-d-Phe-OH (6 equiv), 2-chloro-1,3-dimethylimidazolidium hexafluorophosphate (CIP, 5.9 equiv), HOAt (1.5 equiv), and 2,4,6-collidine (18 equiv) in dry DCM with drops of DMF for better solubility of the reagents (amino acid concentration: 0.3 M). The reaction was stirred for 1.5-2 h at room temperature and repeated until the total coupling time was 10 h. The chloranil test was performed after each coupling period to qualitatively monitor the amount of the residual secondary amine. The resin was washed with DCM (10 mL/g resin × 3) and DMF (10 mL/g resin × 3) for the further deprotection and coupling cycle described in the general procedures.
Coupling of a Secondary Amine withNα-Fmoc-Cys(Trt)-OH
To the peptidyl resin with a free secondary amine were added Nα-Fmoc-Cys(Trt)-OH (4 equiv), HBTU (3.8 equiv), and DIEA (4 equiv) in DMF (0.4 M). The resin slurry was stirred for 45 min, and the coupling was repeated one more time at room temperature. Another coupling using the symmetrical anhydride of Nα-Fmoc-Cys(Trt)-OH (0.2 M, 4 equiv of amino acid activated using 2 equiv of DIC) in THF was carried out at room temperature. The chloranil test was performed after each coupling period to qualitatively monitor the amount of residual secondary amine. The resin was washed with THF (10 mL/g resin × 3) and DMF (10 mL/g resin × 3) for the further deprotection and coupling cycle described in the general procedures.
Cyclization via a Disulfide Bridge55
After the cleavage from the resin, the peptide was dissolved in ammonium bicarbonate buffer (0.01 M) with a peptide concentration of about 0.08 M. A small amount of acetonitrile (0.3%) was added to the solution. The mixture was then vigorously stirred at room temperature in a flask open to the air until the cyclization was completed as periodically analyzed using HPLC. To the solution was added 10% acetic acid (in excess to the amount of NH4HCO3 used), and the reaction was then concentrated by removing water via evaporation in vacuo at 50 °C, followed by lyophilization.
Deprotection of Allyl and Alloc Groups
After all amino acids were coupled, the N-terminus was modified via acetylation, and the peptidyl resin was washed with DCM (10 mL/g resin × 3), to which were added tetrakis(triphenylphosphine)palladium(0) (0.2 equiv) and phenylsilane (25 equiv) in dry DCM (10 mL/g resin).56,57 The reaction was stirred for 30 min at room temperature, and the deprotection was repeated one more time. The resin was then washed with DCM (10 mL/g resin × 3), sodium diethyldithiocarbamate trihydrate (0.5%) in DMF (10 mL/g resin × 6), DMF (10 mL/g resin × 3), and THF (10 mL/g resin × 3).
Cyclization via a Lactam Bridge
To the resin-bound peptide with free side chain amino and carboxylic groups were added HOBt (3 equiv) and DIC (3 equiv) in THF (0.28 M). The reaction was stirred for 16 h at room temperature. After washing with DMF (10 mL/g resin × 3), HOAt (1.2 equiv) and DIC (1.2 equiv) in DMF/THF (1:1, 0.13 M) were added for the cyclization reaction at room temperature for 24 h, followed by another cyclization reaction using HOAt (1 equiv), DMAP (2 equiv), and PyBrop (1 equiv) in DMF (0.13 M) at room temperature for 20 h. Finally, HOAt (1 equiv) and DIC (1 equiv) in DMF (0.13 M) were used for the cyclization at room temperature for 28 h. A Kaiser test was performed after each step to estimate the completion of the cyclization.
Purification of Peptides by RP-HPLC
The crude peptides were first dissolved in water (with an addition of <15% acetonitrile in case of poor solubility) to give a concentration of 5 mg/mL. The peptide solutions were then filtered through a 0.45 μ cellulose acetate filter (Aerodisk) to remove insoluble impurities. The crude peptides were then purified using a Hewlett-Packard 1100 series HPLC instrument with a semipreparative Vydac reverse-phase C18-bonded column (10 × 220 mm, 10 μm, 300 Å, Vydac, Hesperia, CA, Catalog No. 218TP1010). Peptides were eluted with a linear acetonitrile/0.1% TFA aqueous solution gradient at a flow rate of 3.0 mL/min. The separation was monitored at 220, 254, and 280 nm with a Hewlett-Packard 1100 series using UV detection. The injection volume and the gradient depended on the resolution of the peptide peak from impurities and the retention time of the target peptide.
Receptor Binding Assay
Competition binding experiments were performed on whole cells.58 Transfected HEK293 cell lines with the various hMCRs were seeded on 96-well plates, 48 h before the assay was done, 100 000 cells/well. For the assay, the cell culture medium was expirated and cells were washed twice with a freshly prepared binding buffer containing 100% minimum essential medium with Earle's salt (MEM; GIBCO, Carlsbad, CA), 25 mM HEPES (pH 7.4), 0.2% bovine serum albumin, 1 mM 1,10-phenanthrolone, 0.5 mg/L leupeptin, and 200 mg/L bacitracin. Cells were then incubated with different concentrations of an unlabeled peptide and labeled [125I]-[Nle4,d-Phe7]-α-MSH (20 000 cpm/well, 0.1386 nm; Perkin-Elmer Life Science, Freemont, CA) for 30 min at 37 °C. The assay medium was subsequently removed and each well was washed twice with the binding buffer. The cells were lysed by the addition of 250 μL of 0.1 M NaOH and 250 μL of 1% Triton X-100. The lysed cells were transferred to the 12 × 75 mm glass tubes, and the radioactivity was measured by Wallac 1470 WIZARD Gamma Counter (Wallac, Jefferson, NY).
Adenylate Cyclase Assay
HEK 293 cells transfected with human melanocortin receptors58 were grown to confluence in MEM medium (GIBCO) containing 10% fetal bovine serum, 100 units/mL penicillin and streptomycin, and 1 mM sodium pyruvate. The cells were seeded on 96-well plates 48 h before assay (50 000 cells/well). For the assay, the cell culture medium was removed and the cells were rinsed with 1 mL of MEM buffer (GIBCO) or with Earle's balanced salt solution (EBSS, GIBCO). An aliquot (0.4 mL) of the Earle's balanced salt solution was placed in each well along with 5 μL 0.5 mM isobutylmethylxanthine (IBMX) for 1 min at 37 °C. Next, varying concentration aliquots of melanotropin peptides (0.1 mL) were added, and the cells were incubated for 3 min at 37 °C. The reaction was stopped by aspirating the assay buffer and adding 0.15 mL of ice-cold Tris/EDTA buffer to each well. After dislodging the cells with the help of trypsin, the cells were transferred to polypropylene microcentrifuge tubes and placed in a boiling water bath for 15 min. The cell lysate was then centrifuged for 2 min at 6500 rpm, and 50 μL of the supernatant was aliquoted into an Eppendorf tube. The total cAMP content was measured by competitive binding assay according to the TRK 432 assay kit instructions (Amersham Corp., Piscataway, NJ). The antagonist properties of the lead compounds were evaluated by its ability to competitively displace the MT-II agonist in a dose-dependent manner, at up to 10 μM.
Data Analysis
IC50 and EC50 values represent the mean of two experiments performed in triplicate. IC50 and EC50 estimates and their associated standard errors were determined by fitting the data using a nonlinear least-squares analysis, with the help of GraphPad Prism 4 (GraphPad Software, San Diego, CA).
General Procedure for the Conformational Study
Molecular dynamics simulated annealing and Monte Carlo multiple-minimum (MCMM)59 conformational search methods were used to explore the low energy conformations of the designed peptides using the MacroModel 8.1 software package within Maestro 5.1 (Schrödinger, Portland, OR). The OPLS-AA force field, no cutoff for nonbonded interactions, constant dielectric constant (ε = 1.0) for the electrostatic treatment, and generalized-Born/surface-area solvation model for water were applied in all the calculations.60
Starting from the MTII NMR structure described previously,37 the target peptides were built in Maestro 5.1 by modifying the covalent structure of MTII. A 2000 step Polak-Ribier conjugate gradient minimization was performed to optimize the structures. In the simulated annealing run, the starting structure was fully minimized in 5000 steps. The temperature was increased from 50 to 1000 K over 30 ps, followed by a 20 ps equilibrium run at 1000 K. The molecule was then cooled to 500 K in 50 ps and finally to 50 K over 500 ps. The SHAKE protocol was used to constrain all the bonds to a hydrogen atom to their natural values. The time step was set to 1.0 fs for the heating, equilibrating, and the first cooling runs, but to 2.0 fs for the final cooling run. In all the heating and cooling steps, the system was coupled to a thermal bath of the target temperature with a bath time constant of 5 ps.
The lowest energy conformations obtained in the simulated annealing calculations were compared to the MTII NMR structure. If the backbone structures were close to each other, the conformations of the designed peptides were further studied using the MCMM search. In each MCMM conformational search, torsional variations were automatically set up in MacroModel. The conformational search was broken up into four blocks. In each block, a 5000-step Monte Carlo conformational search was conducted with an energy window value of 25 kJ/mol. Each new conformation generated in the search was minimized using 2000-step Polak-Ribier conjugate gradient minimization with a gradient convergence threshold of 0.05. After the search in each block was completed, conformations found in the search were further minimized using the same minimization method for 5000 steps to ensure that all conformations were converged. The searches in each block were seeded with the conformations from the previous block, but with a different starting value for the random number generator.
All results were analyzed within the Maestro graphical user interface. All calculations were performed on a Beowulf Linux cluster of personal computer workstations.
Supplementary Material
Acknowledgment
We thank Professor Richard Glass for his helpful discussion. This work was supported by a grant from U.S. Public Health Service, National Institutes of Health, Grant No. DK17420, and by a fellowship to J.Y. from Merck Research Laboratories.
Footnotes
Abbreviations: NMR, nuclear magnetic resonance; ROESY, rotating frame Overhauser effect spectroscopy; MCMM, Monte Carlo multiple-minimum; MSH, melanocyte stimulating hormone; POMC, proopiomelanocortin; MCR, melanocortin receptor; hMCR, human melanocortin receptor; Dapa, R,α,β-diaminopropionic acid; Dab, R,α,β-diaminobutyric acid; DEAD, diethyl azodicarboxylate; DIAD, diisopropyl azodicarboxylate; oNBs, 2-nitrobenzenesulfonyl; DNBs, 2,4-dinitrobenzenesulfonyl; ACM, acetamidomethyl; CIP, 2-chloro-1,3-dimethylimidazolidium hexafluorophosphate; DIC, N,N-diisopropylcarbodiimide; HOBt, 1-hydroxybenzotriazole; DMF, N,N-dimethylformamide; THF, tetrahydrofuran; HOAt, 1-hydroxy-7-azabenzotriazole; DMAP, 4-(dimethylamino)pyridine; DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene; HBTU, 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate; DIEA, N,N-diisopropylethylamine; DCM, dichloromethane.
Supporting Information Available: Proton NMR spectra in D2O for peptides 1–8; list of chemical shifts and coupling constants for peptides 2, 4, and 7; 2D ROESY spectrum for peptide 5. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hruby VJ, Han G. The Melanocortin Receptors. Humana Press; Totowa, NJ: 2000. The Molecular Pharmacology of α-MSH–Structure Activity Relationships for Melanotropins at the Melanocortin Receptors; pp. 239–261. [Google Scholar]
- 2.Hruby VJ, Wilkes BC, Cody WL, Sawyer TK, Hadley ME. Melanotropins: Structural, Conformational and Biological Considerations in the Development of Superpotent and Superprolonged Analogs. Pept. Protein Rev. 1984;3:1–64. [Google Scholar]
- 3.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The Cloning of a Family of Genes that Encode the Melanocortin Receptors. Science. 1992;257:1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 4.Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van der Ploeg LHT. Inactivation of the Mouse Melanocortin-3 Receptor Results in Increased Fat Mass and Reduced Lean Body Mass. Nat. Genet. 2000;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- 5.Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelley-mounter MA, Dekoning J, Baetscher M, Cone RD. A Unique Metabolic Syndrome Causes Obesity in the Melanocortin-3 Receptor-Deficient Mouse. Endocrinology. 2000;141:3518–3521. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- 6.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of Melanocortinergic Neurons in Feeding and the Agouti Obesity Syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 7.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeir LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted Disruption of the Melanocortin-4 Receptor Results in Obesity in Mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 8.Wessells H, Hruby VJ, Hackett J, Han G, Balse-Srinivasan P, Vanderah TW. Ac–Nle-c[Asp-His-DPhe-Arg-Trp-Lys]-NH2 Induces Penile Erection via Brain and Spinal Melanocortin Receptors. Neuroscience. 2003;118:755–762. doi: 10.1016/s0306-4522(02)00866-7. [DOI] [PubMed] [Google Scholar]
- 9.Chen WB, Kelly MA, Opitz-Araya X, Thomas RE, Low MJ, Cone RD. Exocrine Gland Dysfunction in MC5–R-Deficient Mice: Evidence for Coordinated Regulation of Exocrine Gland Function by Melanocortin Peptides. Cell. 1997;91:789–798. doi: 10.1016/s0092-8674(00)80467-5. [DOI] [PubMed] [Google Scholar]
- 10.Mogil JS, Wilson SG, Chesler EJ, Rankin AL, Nemmani VS, Lariviere WR, Groce MK, Wallace MR, Kaplan L, Staud R, Ness TJ, Glover TL, Stankova M, Mayorov AV, Hruby VJ, Grisel JE, Fillingim RB. The Melanocortin-1 Receptor Gene Mediates Female-Specific Mechanisms of Analgesis in Mice and Humans. Proc. Natl. Acad. Sci. U.S.A. 2003;100:4867–4872. doi: 10.1073/pnas.0730053100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hruby VJ, Wilkes BC, Hadley ME, Al-Obeidi F, Sawyer TK, Staples DJ, deVaux AE, Dym O, de Lauro Castrucci AM, Hintz MF, Riehm JP, Rao KR. α-Melanotropin: The Minimal Active Sequence in the Frog Skin Bioassay. J. Med. Chem. 1987;30:2126–2130. doi: 10.1021/jm00394a033. [DOI] [PubMed] [Google Scholar]
- 12.de Lauro Castrucci AM, Hadley ME, Sawyer TK, Wilkes BC, Al-Obeidi F, Staples DJ, deVaux AE, Dym O, Hintz MF, Riehm JP, Rao KR, Hruby VJ. α-Melanotropin: The Minimal Active Sequence in the Lizard Skin Bioassay. Gen. Comput. Endocrinol. 1989;73:157–163. doi: 10.1016/0016-6480(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 13.Eberle AN. The Melanotropins. Chemistry, Physiology and Mechanisms of Action. Karger; Basel, Switzerland: 1988. [Google Scholar]
- 14.Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. 4-Norleucine, 7-d-Phenylalanine-α-Melanocyte-Stimulating Hormone: A Highly Potent α-Melanotropin with Ultralong Biological Activity. Proc. Natl. Acad. Sci. U.S.A. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sawyer TK, Hruby VJ, Darman PS, Hadley ME. [half-Cys4, half-Cys10]-α-Melanocyte-Stimulating Hormone: A Cyclic α-Melanotropin Exhibiting Superagonist Biological Activity. Proc. Natl. Acad. Sci. U.S.A. 1982;79:1751–1755. doi: 10.1073/pnas.79.6.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Al-Obeidi F, Hadley ME, Pettitt BM, Hruby VJ. Design of a New Class of Superpotent Cyclic α-Melanotropins Based on Quenched Dynamic Simulations. J. Am. Chem. Soc. 1989;111:3413–3416. [Google Scholar]
- 17.Hruby VJ, Lu D, Sharma SD, Castrucci AL, Kesterson RA, Al-Obeidi FA, Hadley ME, Cone RD. Cyclic Lactam α-Melanotropin Analogues of Ac-Nle4-c[Asp5,d-Phe7,Lys10]α-MSH(4–10)-NH2 with Bulky Aromatic Amino Acids at Position 7 Show High Antagonist Potency and Selectivity at Specific Melanocortin Receptors. J. Med. Chem. 1995;38:3454–3461. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- 18.Kask A, Mutulis F, Muceniece R, Pahkla R, Mutule I, Wikberg JES, Rago L, Schioth HB. Discovery of a Novel Superpotent and Selective Melanocortin-4 Receptor Antagonist (HS024): Evaluation in Vitro and in Vivo. Endocrinology. 1998;139:5006–5014. doi: 10.1210/endo.139.12.6352. [DOI] [PubMed] [Google Scholar]
- 19.Bednarek MA, Macneil T, Kalyani RN, Tang R, Van der Ploeg LHT, Weinberg DH. Analogs of MTII, Lactam Derivatives of α-Melanotropin, Modified at the N–Terminus, and Their Selectivity at Human Melanocortin Receptors 3, 4, and 5. Biochem. Biophys. Res. Commun. 1999;261:209–213. doi: 10.1006/bbrc.1999.0981. [DOI] [PubMed] [Google Scholar]
- 20.Kavarana MJ, Trivedi D, Cai M, Ying J, Hammer M, Cabello C, Grieco P, Han G, Hruby VJ. Novel Cyclic Templates of α-MSH Give Highly Selective and Potent Antagonists/Agonists for Human Melanocortin-3/4 Receptors. J. Med. Chem. 2002;45:2644–2650. doi: 10.1021/jm020021z. [DOI] [PubMed] [Google Scholar]
- 21.Grieco P, Lavecchia A, Cai M, Trivedi D, Weinberg D, MacNeil T, Van der Ploeg LHT, Hruby VJ. Structure–Activity Studies of the Melanocortin Peptides: Discovery of Potent and Selective Affinity Antagonists for the hMC3 and hMC4 Receptors. J. Med. Chem. 2002;45:5287–5294. doi: 10.1021/jm0202526. [DOI] [PubMed] [Google Scholar]
- 22.Sebhat IK, Martin WJ, Ye Z, Barakat K, Mosley RT, Johnston DBR, Bakshi R, Palucki B, Weinberg DH, MacNeil T, Kalyani RN, Tang R, Stearns RA, Miller RR, Tamvakopoulos C, Strack AM, McGowan E, Cashen DE, Drisko JE, Hom GJ, Howard AD, MacIntyre DE, van der Ploeg LHT, Patchett AA, Nargund RP. Design and Pharmacology of N-[(3R)-1,2,3,4-Tetrahydroisoquinolinium-3-ylcarbonyl]-(1R)-1-(4-chlorobenzyl)-2-[4-cyclohexyl-4-(1H-1,2,4-triazol-1-ylmethyl)piperidin-1-yl]-2-oxoethylamine, a Potent, Selective, Melanocortin Subtype-4 Receptor Agonist. J. Med. Chem. 2002;45:4589–4593. doi: 10.1021/jm025539h. [DOI] [PubMed] [Google Scholar]
- 23.Fotsch C, Smith DM, Adams JA, Cheetham J, Croghan M, Doherty EM, Hale C, Jarosinski MA, Kelly MG, Norman MH, Tamayo NA, Xi N, Baumgartner JW. Design of a New Peptidomimetic Agonist for the Melanocortin Receptors Based on the Solution Structure of the Peptide Ligand, Ac-Nle-cyclo[Asp-Pro-DPhe-Arg-Trp-Lys]-NH2. Bioorg. Med. Chem. Lett. 2003;13:2337–2340. doi: 10.1016/s0960-894x(03)00412-8. [DOI] [PubMed] [Google Scholar]
- 24.Richardson TI, Ornstein PL, Briner K, Fisher MJ, Backer RT, Biggers CK, Clay MP, Emmerson PJ, Hertel LW, Hsiung HM, Husain S, Kahl SD, Lee JA, Lindstrom TD, Martinelli MJ, Mayer JP, Mullaney JT, O'Brien TP, Pawlak JM, Revell KD, Shah J, Zgombick JM, Herr RJ, Melekhov A, Sampson PB, King CHR. Synthesis and Structure–Activity Relationships of Novel Arylpiperazines as Potent and Selective Agonists of the Melanocortin Subtype-4 Receptor. J. Med. Chem. 2004;47:744–755. doi: 10.1021/jm0304109. [DOI] [PubMed] [Google Scholar]
- 25.Cai M, Mayorov AV, Ying J, Stankova M, Trivedi D, Cabello C, Hruby VJ. Design of Novel Melanotropin Agonists and Antagonists with High Potency and Selectivity for Human Melanocortin Receptors. Peptides. 2005;26:1481–1485. doi: 10.1016/j.peptides.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 26.Kruijtzer JAW, Nijenhuis WAJ, Wanders N, Gispen WH, Liskamp RMJ, Adan RAH. Peptoid-Peptide Hybrids as Potent Novel Melanocortin Receptor Ligands. J. Med. Chem. 2005;48:4224–4230. doi: 10.1021/jm0490033. [DOI] [PubMed] [Google Scholar]
- 27.Kask A, Rago L, Mutulis F, Pahkla R, Wikberg JES, Schioth HB. Selective Antagonist for the Melanocortin 4 receptor (HS014) Increases Food Intake in Free-Feeding Rats. Biochem. Biophys. Res Commun. 1998;245:90–93. doi: 10.1006/bbrc.1998.8389. [DOI] [PubMed] [Google Scholar]
- 28.Veber DF, Holly FW, Paleveda WJ, Nutt RF, Bergstrand SJ, Torchiana M, Glitzer MS, Saperstein R, Hirschmann R. Conformationally Restricted Bicyclic Analogs of Somatostatin. Proc. Natl. Acad. Sci. U.S.A. 1978;75:2636–2640. doi: 10.1073/pnas.75.6.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Veber DF, Holly FW, Nutt RF, Bergstrand SJ, Brady SF, Hirschmann R, Glitzer MS, Saperstein R. Highly Active Cyclic and Bicyclic Somatostatin Analogues of Reduced Ring Size. Nature. 1979;280:512–514. doi: 10.1038/280512a0. [DOI] [PubMed] [Google Scholar]
- 30.DiMaio J, Schiller PW. A Cyclic Enkephalin Analog with High in vitro Opiate Activity. Proc. Natl. Acad. Sci. U.S.A. 1980;77:7162–7166. doi: 10.1073/pnas.77.12.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hruby VJ. Conformational Restrictions of Biologically Active Peptides via Amino Acid Side Chain Groups. Life Sci. 1982;31:189–199. doi: 10.1016/0024-3205(82)90578-1. [DOI] [PubMed] [Google Scholar]
- 32.Hruby VJ, Mosberg HI. Conformational and Dynamic Considerations in Peptide Structure–Function Studies. Peptides. 1982;3:329–336. doi: 10.1016/0196-9781(82)90095-x. [DOI] [PubMed] [Google Scholar]
- 33.DiMaio J, Nguyen TMD, Lemieux C, Schiller PW. Synthesis and Pharmacological Characterization in Vitro of Cyclic Enkephalin Analogues: Effect of Conformational Constraints on Opiate Receptor Selectivity. J. Med. Chem. 1982;25:1432–1438. doi: 10.1021/jm00354a008. [DOI] [PubMed] [Google Scholar]
- 34.Hruby VJ, Mosberg HI, Sawyer TK, Knittel JJ, Rockway TW, Ormberg J, Darman P. Conformational and Dynamic Considerations in the Design of Peptide Hormone Analogs. Biopolymers. 1983;22:517–530. doi: 10.1002/bip.360220165. [DOI] [PubMed] [Google Scholar]
- 35.Gilon C, Halle D, Chorev M, Selinger Z, Byk G. Backbone Cyclization – A New Method for Conferring Conformational Constraint on Peptides. Biopolymers. 1991;31:745–750. doi: 10.1002/bip.360310619. [DOI] [PubMed] [Google Scholar]
- 36.Chalmers DK, Marshall GR. Pro-D-NMe-Amino Acid and D-Pro-NMe-Amino Acid: Simple, Efficient Reverse-Turn Constraints. J. Am. Chem. Soc. 1995;117:5927–5937. [Google Scholar]
- 37.Ying J, Kövér KE, Gu X, Han G, Trivedi D, Kavarana MJ, Hruby VJ. Solution Structures of Cyclic Melanocortin Agonists and Antagonists by NMR. Biopolymers (Pept. Sci.) 2003;71:696–716. doi: 10.1002/bip.10596. [DOI] [PubMed] [Google Scholar]
- 38.Mitsunobu O. The Use of Diethyl Azodicarboxylate and Triphenylphosphine in Synthesis and Transformation of Natural Products. Synthesis. 1981:1–28. [Google Scholar]
- 39.Hughes DL. The Mitsunobu Reaction. Org. React. 1992;42:335–656. [Google Scholar]
- 40.Botta M, Corelli F, Maga G, Manetti F, Renzulli M, Spadari S. Research on Anti-HIV-1 Agents. Part 2: Solid-Phase Synthesis, Biological Evaluation and Molecular Modeling Studies of 2,5,6-Trisubstituted-4(3H)-Pyrimidinones Targeting HIV-1 Reverse Transcriptase. Tetrahedron. 2001;57:8357–8367. [Google Scholar]
- 41.Miller SC, Scanlan TS. Site-Selective N-Methylation of Peptides on Solid Support. J. Am. Chem. Soc. 1997;119:2301–2302. [Google Scholar]
- 42.Miller SC, Scanlan TS. oNBS-SPPS: A New Method for Solid-Phase Peptide Synthesis. J. Am. Chem. Soc. 1998;120:2690–2691. [Google Scholar]
- 43.Rew Y, Goodman M. Solid-Phase Synthesis of Amine-Bridged Cyclic Enkephalin Analogues via On-Resin Cyclization Utilizing the Fukuyama-Mitsunobu Reaction. J. Org. Chem. 2002;67:8820–8826. doi: 10.1021/jo020447l. [DOI] [PubMed] [Google Scholar]
- 44.Fukuyama T, Cheung M, Jow C-K, Hidai Y, Kan T. 2,4-Dinitrobenzenesulfonamides: A Simple and Practical Method for the Preparation of a Variety of Secondary Amines and Diamines. Tetrahedron Lett. 1997;38:5831–5834. [Google Scholar]
- 45.Bunnett JF, Zahler RE. Aromatic Nucleophilic Substitution Reactions. Chem. Rev. 1951;49:273–412. [Google Scholar]
- 46.Levy AA, Rains HC, Smiles S. Rearrangement of Hydroxy Sulfones. I. J. Chem. Soc. 1931:3264–3269. [Google Scholar]
- 47.Akaji K, Kuriyama N, Kimura T, Fujiwara Y, Kiso Y. Anchoring of Fmoc Amino Acid to 4-Alkoxybenzyl alcohol Resin using a New Esterification Reagent. Tetrahedron Lett. 1992;33:3177–3180. [Google Scholar]
- 48.Akaji K, Kuriyama N, Kiso Y. Efficient Coupling of α,α-Dimethyl Amino Acid using a New Chloro Imidazolidium Reagent, CIP. Tetrahedron Lett. 1994;35:3315–3318. [Google Scholar]
- 49.Han S-Y, Kim Y-A. Recent Development of Peptide Coupling Reagents in Organic Synthesis. Tetrahedron. 2004;60:2447–2467. [Google Scholar]
- 50.Bothner-By AA, Stephens RL, Lee J, Warren CD, Jeanloz RW. Structure Determination of a Tetrasaccharide: Transient Nuclear Overhauser Effects in the Rotating Frame. J. Am. Chem. Soc. 1984;106:811–813. [Google Scholar]
- 51.Bax A, Davis DG. Practical Aspects of Two-Dimensional Transverse NOE Spectrospy. J. Magn. Reson. 1985;63:207–213. [Google Scholar]
- 52.Soudijn W, van Wijngaarden I, Ijzerman AP. Allosteric Modulation of G Protein-Coupled Receptors: Perspectives and Recent Developments. Drug Discovery Today. 2004;9:752–758. doi: 10.1016/S1359-6446(04)03220-9. [DOI] [PubMed] [Google Scholar]
- 53.Kenakin T. New Concepts in Drug Discovery: Collateral Efficacy and Permissive Antagonism. Nature Rev. Drug Discuss. 2005;4:919–927. doi: 10.1038/nrd1875. [DOI] [PubMed] [Google Scholar]
- 54.Hoare SRJ. Mechanisms of Peptide and Nonpeptide Ligand Binding to Class B G-Protein-Coupled Receptors. Drug Discovery Today. 2005;10:417–427. doi: 10.1016/S1359-6446(05)03370-2. [DOI] [PubMed] [Google Scholar]
- 55.Chan WC, White PD. Fmoc Solid-Phase Peptide Synthesis. Oxford University Press Inc.; New York: 2000. [Google Scholar]
- 56.Thieriet N, Alsina J, Giralt E, Guibé F, Albericio F. Use of Alloc-Amino Acids in Solid-Phase Peptide Synthesis. Tandem Deprotection-Coupling Reactions using Neutral Conditions. Tetrahedron Lett. 1997;38:7275–7278. [Google Scholar]
- 57.Grieco P, Gitu PM, Hruby VJ. Preparation of ′Side-Chain-to-Side-Chain′ Cyclic Peptides by Allyl and Alloc Strategy: Potential for Library Synthesis. J. Pept. Res. 2001;57:250–256. doi: 10.1111/j.1399-3011.2001.00816.x. [DOI] [PubMed] [Google Scholar]
- 58.Cai M, Mayorov AV, Cabello C, Stankova M, Trivedi D, Hruby VJ. Novel 3D Pharmacophore of α-MSH/γ-MSH Hybrids Leads to Selective Human MC1R and MC3R Analogues. J. Med. Chem. 2005;48:1839–1848. doi: 10.1021/jm049579s. [DOI] [PubMed] [Google Scholar]
- 59.Chang G, Guida WC, Still WC. An Internal Coordinate Monte Carlo Method for Searching Conformational Space. J. Am. Chem. Soc. 1989;111:4379–4386. [Google Scholar]
- 60.Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical Treatment of Solvation for Molecular Mechanics and Dynamics. J. Am. Chem. Soc. 1990;112:6127–6129. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.