

Abstract
The first total syntheses for the (Z)-15-methyl-10-hexadecenoic acid and the (Z)-13-methyl-8-tetradecenoic acid were accomplished in seven steps and in 31–32% overall yields. The (trimethylsilyl)acetylene was the key reagent in both syntheses. It is proposed that the best synthetic strategy towards monounsaturated iso methyl-branched fatty acids with double bonds close to the ω end of the acyl chain is first acetylide coupling of (trimethylsilyl)acetylene to a long-chain bifunctional bromoalkane followed by a second acetylide coupling to a short-chain iso bromoalkane, since higher yields are thus obtained. Spectral data is also presented for the first time for these two unusual fatty acids with potential as biomarkers and as topoisomerase I inhibitors.
Keywords: (Z)-15-methyl-10-hexadecenoic acid, (Z)-13-methyl-8-tetradecenoic acid, fatty acids, synthesis, biomarkers
1. Introduction
Methyl-branched fatty acids are of interest as biomarkers for alkane-utilizing bacteria in soils and as biomarkers for sulphate-reducing bacteria, such as Desulfovibrio species, also found in wetland sediments (Boon et al., 1977; Boon, et al., 1996). Particularly interesting fatty acids for the Desulfovibrio species are the iso methyl-branched fatty acids i-15:1 (n-7) and i-17:1 (n-7), which are characteristic for the Desulfovibrio and both fatty acids seem to be biosynthetically related by a two carbon extension of the shorter- chain C15 analog (Dowling et al., 1988; Edlund et al., 1985). While the iso C15–C17 (n-7) series has been well studied, little is known of another series of odd-chain fatty acids, namely the iso C15–C17 (n-6) series exemplified by the fatty acids (Z)-13-methyl-8- tetradecenoic acid (1a) and the (Z)-15-methyl-10-hexadecenoic acid (1b). Acid 1a has not been isolated from natural sources, but the (Z)-15-methyl-10-hexadecenoic acid (1b) has been identified before in several organisms, such as in the siphonarid limpet Siphonaria denticulata (Carballeira et al., 2001). Because of the nature of the (n-6) fatty acids 1a and 1b, i.e., C15–C17 chain lengths and iso ramification, there is a high probability for these compounds to be also of bacterial origin (Kaneda, 1991). In addition, these fatty acids remain under-explored as possible therapeutic agents despite the fact that iso-branched fatty acids with similar chain lengths have displayed considerable topoisomerase-I inhibition and also cytotoxicity (Lee et al., 1998; Reyes and Carballeira, 1996). Therefore, in order to provide spectral data so as to aid in their identification in nature and to supply enough material for further biological studies we have developed the first total syntheses for acids 1a and 1b following a similar synthetic sequence consisting of seven steps and using (trimethylsilyl)acetylene as the key reagent in the syntheses.
2. Materials and methods
2.1 Instrumentation
1H NMR (300 MHz) and 13C NMR (75 MHz) were recorded on a Bruker DPX-300 spectrometer. 1H NMR chemical shifts are reported with respect to internal (CH3)4Si, 13C NMR chemical shifts are reported in parts per million relative to CDCl3 (77.0 ppm). GC/MS analyses were recorded at 70 eV using a Hewlett Packard 5972A MS ChemStation equipped with a 30 m x 0.25 mm special performance capillary column (HP-5MS) of polymethyl siloxane crosslinked with 5% phenyl methylpolysiloxane. IR spectra were recorded on a Nicolet 600 FT-IR spectrophotometer (Thermo-Nicolet, Madison, WI, USA). Elemental analyses were performed by Galbraith Laboratories Inc., Knoxville, Tennessee. High resolution mass spectral data was performed at the Emory University Mass Spectrometry Center.
2.2 General procedure for the protection of the bromo-alcohols
To 4.00 g of either 7-bromo-1-heptanol or 9-bromo-1-nonanol in 12 mL of dichloromethane (CH2Cl2) was added dropwise 2,3-dihydro-2H-pyran (35.85-41.00 mmol) and catalytic amounts of p-toluene sulfonic acid (PTSA). The reaction mixture was stirred for 24 h at room temperature. The organic layer was washed with water (2 x 50 mL), NaHCO3 (3 x 50 mL), and dried over MgSO4, filtered, and evaporated in vacuo affording 5.55–5.59 g (97–100% yield) of either 3a or 3b. The products were purified using silica gel column chromatography eluting with hexane/ether (9:1).
2.2.1 7-Bromo-1-[(tetrahydropyran-2-yl)oxy]heptane (3a)
The 7-bromo-1-[(tetrahydropyran-2-yl)oxy]heptane (3a) was obtained as a colorless oil in a 97% yield from the reaction of 7-bromo-1-heptanol (4.00 g, 20.50 mmol) with 2,3-dihydro-2H-pyran (35.85 mmol) and catalytic amounts of PTSA in dichloromethane (12 mL) according to the general procedure described above. Compound 3a presented comparable spectral data to the one previously reported in the literature (Das et al., 2006).
2.2.2 9-Bromo-1-[(tetrahydropyran-2-yl)oxy]nonane (3b)
The 9-bromo-1-[(tetrahydropyran-2-yl)oxy]nonane (3b) was obtained as a colorless oil in a 100% yield from the reaction of 9-bromo-1-nonanol (4.06 g, 18.18 mmol) with 2,3-dihydro-2H-pyran (41.00 mmol) and catalytic amounts of PTSA in dichloromethane (12 mL) according to the general procedure described above. Compound 3b presented comparable spectral data to the one previously reported in the literature (Grube et al., 2006).
2.3 General procedure for the first acetylide coupling reaction
To a stirred solution of (trimethylsilyl)acetylene (5.3–8.1 mL, 38.5–58.2 mmol) in dry THF (18.0 mL) and under argon was added n-BuLi (2.5M, 44.88–67.97 mmol) in hexane at −78º C. After 5 min HMPA (5.0–7.0 mL) (Caution! HMPA is toxic and safety precautions should be taken when handling this solvent) and either 3a or 3b (3.94–5.42 g) were added dropwise to the reaction mixture, maintaining the temperature at −78ºC. After 24h, the reaction mixture was quenched with water. The organic product was extracted with diethyl ether (2 x 15 mL), dried over MgSO4, filtered, and evaporated in vacuo, affording 4.02–4.74 g (87–98% yield) of either 9-(trimethylsilyl)-1-[(tetrahydropyran-2-yl)oxy]non-8-yne (4a) or 11-(trimethylsilyl)-1-[(tetrahydropyran-2- yl)oxy]undec-10-yne (4b). The products 4a and 4b were purified under vacuum distillation (Kugelrohr) at 100–115ºC/3 mm Hg for 1h.
2.3.1 9-(Trimethylsilyl)-1-[(tetrahydropyran-2-yl)oxy]non-8-yne (4a)
The 9-(trimethylsilyl)-1-[(tetrahydropyran-2-yl)oxy]non-8-yne (4a) was obtained as a colorless oil in a 87% yield from the reaction of 3a (5.42 g, 19.42 mmol) with n-BuLi in THF (18 mL) and (trimethylsilyl)acetylene (7 mL) according to the general procedure described above; IR (neat) νmax 2932, 2859, 2174, 1454, 1441, 1352, 1249, 1200, 1137, 1121, 1079, 1034, 843, 760 cm−1; 1H NMR (CDCl3) δ 4.57 (1H, m), 3.86–3.73 (2H, m), 3.50–3.35 (2H, m), 2.21 (2H, t, J = 7.1 Hz, H-7), 1.91–1.52 (10H, m, -CH2-), 1.33 (6H, m), 0.12 (9H, s, -Si(CH3)3); 13C NMR (CDCl3) δ 107.68 (s, C-2), 98.83 (d), 84.45 (s), 67.60 (t, C-1), 62.33 (t), 30.76 (t), 29.66 (t), 28.90 (t), 28.72 (t), 28.54 (t), 26.08 (t), 25.47 (t), 19.81 (t), 19.68 (t), 0.16 (q, -Si(CH3)3); GC-MS (70 eV) m/z (relative intensity) 295 (M+-1, 1), 223 (1), 183 (2), 173 (6), 123 (2), 111 (2), 109 (7), 103 (15), 101 (7), 85 (100), 75 (21), 73 (55), 69 (3), 67 (10), 59 (11), 57 (9), 55 (11). HRMS (APCI) Calcd for C17H33O2Si [M + H]+ 297.2250, found 225.2239.
2.3.2 11-(Trimethylsilyl)-1-[(tetrahydropyran-2-yl)oxy]undec-10-yne (4b)
The 11-(trimethylsilyl)-1-[(tetrahydropyran-2-yl)oxy]undec-10-yne (4b) was obtained as a colorless oil in a 98% yield from the reaction of 3b (3.94 g, 12.82 mmol) with n-BuLi in THF (18 mL) and (trimethylsilyl)acetylene (5 mL) according to the general procedure described above; IR (neat) νmax 2931, 2855, 2174, 1454, 1352, 1249, 1200, 1136, 1121, 1079, 1033, 843, 760 cm−1; 1H NMR (CDCl3) δ 4.57 (1H, m), 3.89–3.68 (2H, m), 3.51–3.35 (2H, m), 2.20 (2H, t, J = 7.1 Hz, H-9), 1.80–1.51 (10H, m, -CH2-), 1.31 (10H, m), 0.12 (9H, s, -Si(CH3)3); 13C NMR (CDCl3) δ 107.75 (s, C-10), 98.83 (d), 84.30 (s, C-11), 67.66 (t, C-1), 62.33 (t), 30.76 (t), 29.72 (t), 29.39 (t), 29.00 (t), 28.76 (t), 28.59 (t), 26.20 (t), 25.48 (t), 19.83 (t, C-9), 19.68 (t), 0.16 (q, -Si(CH3)3); GC-MS (70 eV) m/z (relative intensity) 323 (M+-1, 1), 309 (1), 183 (1), 173 (6), 159 (2), 149 (2), 137 (1), 119 (3), 103 (13), 101 (12), 97 (3), 93 (1), 91 (2), 86 (5), 85 (100), 83 (5), 81 (4), 75 (18), 73 (51), 69 (4), 67 (10), 61 (2), 59 (12), 57 (9), 56 (9), 55 (11). HRMS (APCI) Calcd for C19H37O2Si [M + H]+ 325.2552, found 325.2563.
2.4 General procedure for the removal of the silyl group
To a mixture of either 4a or 4b (2.18–3.67 g, 6.72–12.38 mmol) and dry THF (8.0 mL) was added dropwise tetrabutylammonium fluoride (1M, 6.72–12.38 mmol) at 0 ºC. After 48h at room temperature the reaction was quenched with HCl (2M), and the organic m layer was washed with a brine solution (1 x 50 mL), dried over MgSO4, filtered, and evaporated in vacuo, affording 1.48–2.67 g (5.89–11.91 mmol) of the desilylated products 5a and 5b in 88–96% isolated yields. The products were used in the next step without further purification.
2.4.1 1-[(Tetrahydropyran-2-yl)oxy]non-8-yne (5a)
The 1-[(tetrahydropyran-2-yl)oxy]non-8-yne (5a) was obtained as a colorless oil in a 96% yield in the reaction of 9-(trimethylsilyl)-1-[(tetrahydropyran-2-yl)oxy]non-8-yne (4a) (3.68 g, 12.38 mmol) with tetrabutylammonium fluoride (1 M) in THF (8 mL) following the general procedure described above. Compound 5a presented comparable spectral data to the one previously reported in the literature (Karpinska et al., 2004). HRMS (APCI) Calcd for C14H25O2 [M + H]+ 225.1855, found 225.1846.
2.4.2 1-[(Tetrahydropyran-2-yl)oxy]undec-10-yne (5b)
The 1-[(tetrahydropyran-2-yl)oxy]undec-10-yne (5b) was obtained as a colorless oil in a 88% yield in the reaction of 11-(trimethylsilyl)-1-[(tetrahydropyran-2-yl)oxy]undec-10-yne (4b) (2.18 g, 6.72 mmol) with tetrabutylammonium fluoride (1 M) in THF (8 mL) following the general procedure described above. Compound 5b presented comparable spectral data to the one previously reported in the literature (Cryle et al., 2005). HRMS (APCI) Calcd for C16H29O2 [M + H]+ 253.2159, found 253.2168.
2.5 General procedure for the second acetylide coupling reaction
To a solution of either 5a or 5b (1.42–2.57 g, 5.64–11.45 mmol) and dry THF (12.0 mL) at 0ºC n-BuLi (2.5M, 19.73–40.01 mmol) in hexane was added while stirring under an argon atmosphere. After 45 min 1-bromo-4-methylpentane (16.91–34.34 mmol) in HMPA (2.5–4.5 mL) (Caution! HMPA is toxic and safety precautions should be taken when handling this solvent) was added to the reaction mixture and the reaction was left stirring for 24h at room temperature. After this time the reaction mixture was quenched with water and the organic products were extracted with hexane (1 x 50 mL), dried over MgSO4, filtered and evaporated in vacuo, affording 1.29–2.76 g (3.84–8.94 mmol) of either 13-methyl-1-[(tetrahydropyran-2-yl)oxy]tetradec-8-yne (6a) or 15-methyl-1-[(tetrahydropyran-2-yl)oxy]hexadec-10-yne (6b) in 68–78% isolated yields. The products were purified by distilling the impurities under vacuum distillation (Kugelrohr) at 110–120 ºC/3 mm Hg.
2.5.1 13-Methyl-1-[(tetrahydropyran-2-yl)oxy]tetradec-8-yne (6a)
The 13-methyl-1-[(tetrahydropyran-2-yl)oxy]tetradec-8-yne (6a) was obtained as a colorless oil in a 78% yield from the reaction of 1-[(tetrahydropyran-2-yl)oxy]non-8-yne (5a) (2.57 g, 11.45 mmol) with 1-bromo-4-methylpentane (34.34 mmol) in HMPA (4.5 mL) and n-BuLi (40.01 mmol) according to the general procedure described above; IR (neat) νmax 2932, 2857, 1466, 1441, 1384, 1366, 1352, 1261, 1201, 1137, 1121, 1078, 1034, 905, 869, 815 cm−1; 1H NMR (CDCl3) δ 4.57 (1H, m), 3.86–3.70 (2H, m), 3.51–3.35 (2H, m), 2.14 (4H, m), 1.84–1.45 (13H, m, -CH2-), 1.34 (8H, m, -CH2-), 0.87 (6H, d, J = 6.6 Hz, -CH(CH3)2); 13C NMR (CDCl3) δ 98.83 (d), 80.31 (s), 80.16 (s), 67.63 (t, C-1), 62.32 (t), 38.20 (t, C-12), 30.77 (t), 29.71 (t, C-2), 29.09 (t), 29.01 (t), 28.79 (t), 27.64 (d, C-13), 27.06 (t), 26.16 (t, C-3), 25.49 (t), 22.57 (q, C-14, C-15), 19.68 (t), 18.72 (t, C-10), 18.36 (t, C-7); GC-MS (70 eV) m/z (relative intensity) 308 (M+, 1), 223 (1), 195 (1), 193 (1), 179 (1), 123 (2), 121 (3), 111 (1), 109 (7), 107 (3), 101 (14), 97 (2), 95 (12), 93 (5), 91 (3), 85 (100), 81 (15), 79 (9), 77 (3), 69 (18), 67 (21), 57 (10), 55 (22).
2.5.2 15-Methyl-1-[(tetrahydropyran-2-yl)oxy]hexadec-10-yne (6b)
The 15-methyl-1-[(tetrahydropyran-2-yl)oxy]hexadec-10-yne (6b) was obtained as a colorless oil in a 68% yield from the reaction of 1-[(tetrahydropyran-2-yl)oxy]undec-10-yne (5b) (1.29 g, 3.84 mmol) with 1-bromo-4-methylpentane (16.91 mmol) in HMPA (2.5 mL) and n-BuLi (19.73 mmol) according to the general procedure described above; IR (neat) νmax 2937, 2856, 1467, 1366, 1255, 1201, 1140, 1121, 1080, 1033, 904, 869, 815 cm−1; 1H NMR (CDCl3) δ 4.57 (1H, m), 3.86–3.70 (2H, m), 3.50–3.35 (2H, m), 2.12 (4H, m), 1.76–1.48 (13H, m), 1.29 (12H, m, -CH2-), 0.87 (6H, d, J = 6.6 Hz, -CH(CH3)2); 13C NMR (CDCl3) δ 98.83 (d), 80.10 (s), 80.11 (s), 67.67 (t, C-1), 62.33 (t), 38.19 (t, C-14), 30.77 (t), 29.73 (t, C-2), 29.43 (t), 29.14 (t), 28.82 (t), 27.63 (t), 27.06 (d, C-15), 26.21 (t), 25.48 (t), 22.57 (q, C-16, C-17), 19.68 (t), 18.99 (t, C-12), 18.73 (t, C-9); GC-MS (70 eV) m/z (relative intensity) 336 (M+, 1), 251 (1), 149 (1), 137 (2), 123 (2), 121 (2), 111 (1), 109 (8), 107 (3), 105 (1), 101 (22), 96 (4), 95 (13), 93 (4), 91 (3), 85 (100), 81 (15), 79 (8), 77 (3), 71 (2), 69 (20), 67 (22), 65 (2), 57 (11), 56 (10), 55 (23); analysis calculated for C22H40O2: C, 78.51; H, 11.98; found: C, 79.12; H, 12.67.
2.6 General procedure for the removal of the tetrahydropyranyl protecting group
To a mixture of methanol (10.0 mL) and either 6a or 6b (1.23–2.69 g, 3.67–8.73 mmol) was added catalytic amounts of PTSA and the reaction mixture was stirred at 35 ºC for 24h. After this time the organic extract was washed with a saturated solution of sodium bicarbonate (3 x 50 mL), dried over MgSO4, filtered, and evaporated in vacuo, affording 0.82–1.79 g (3.24–7.96 mmol) of the alkynols 7a and 7b for 88–91% yields. The product was used in the next step without further purification.
2.6.1 13-Methyl-8-tetradecyn-1-ol (7a)
The 13-methyl-8-tetradecyn-1-ol (7a) was obtained as a colorless oil in a 91% yield from the reaction of 13-methyl-1-[(tetrahydropyran-2-yl)oxy]tetradec-8-yne (2.69 g, 8.73 mmol) with catalytic amounts of PTSA in methanol (10 mL) according to the general procedure described above; IR (neat) νmax 3360 (OH, broad), 2927, 2856, 1466, 1434, 1384, 1367, 1333, 1117, 1058, 725 cm−1; 1H NMR (CDCl3) δ 3.63 (2H, t, J = 6.6 Hz, H-1), 2.12 (4H, m, H-7, H-10), 1.60–1.45 (7H, m, -CH2-), 1.35 (8H, m, -CH2-), 0.86 (6H, d, J = 6.6 Hz, -CH(CH3)2); 13C NMR (CDCl3) δ 80.34 (s), 80.10 (s), 62.99 (t, C-1), 38.19 (t, C-12), 32.71 (t, C-2), 29.01 (t), 28.91 (t), 28.74 (t), 27.60 (d, C-13), 27.02 (t, C-11), 25.61 (t), 22.56 (q, C-14, C-15), 18.98 (t, C-10),18.70 (t, C-7); GC-MS (70 eV) m/z (relative intensity) 224 (M+, 1), 209 (1), 150 (5), 135 (6), 125 (2), 124 (8), 123 (8), 122 (4), 121 (20), 119 (1), 111 (6), 110 (10), 109 (54), 108 (9), 107 (19), 105 (4), 98 (11), 97 (12), 96 (24), 95 (66), 94 (16), 93 (34), 91 (17), 85 (5), 84 (6), 83 (17), 82 (57), 81 (85), 80 (31), 79 (59), 78 (6), 77 (21), 71 (8), 70 (14), 69 (100), 68 (42), 67 (90), 66 (11), 65 (14), 63 (2), 59 (1), 57 (19), 56 (12), 55 (81), 54 (36). HRMS (APCI) Calcd for C15H29O [M + H]+ 225.2213, found 225.2213.
2.6.2 15-Methyl-10-hexadecyn-1-ol (7b)
The 15-methyl-10-hexadecyn-1-ol (7b) was obtained as a colorless oil in an 88% yield from the reaction of 15-methyl-1-[(tetrahydropyran-2-yl)oxy]hexadec-10-yne (1.24 g, 3.67 mmol) with catalytic amounts of PTSA in methanol (10 mL) according to the general procedure described above; IR (neat) νmax 3360 (OH, broad), 2932, 2856, 1467, 1386, 1366, 1320, 1058 cm−1; 1H NMR (CDCl3) δ 3.63 (2H, t, J = 6.6 Hz, H-1), 2.12 (4H, m, H-9, H-12), 1.55–1.45 (7H, m, -CH2-), 1.29 (12H, m, -CH2-), 0.87 (6H, d, J = 6.6 Hz, -CH(CH3)2); 13C NMR (CDCl3) δ 80.29 (s), 80.20 (s), 63.05 (t, C-1), 38.19 (t, C-14), 32.77 (t, C-2), 29.48 (t), 29.36 (t), 29.12 (t), 29.08 (t), 28.81 (t), 27.63 (d, C-15), 27.05 (t), 25.70 (t), 22.56 (q, C-16, C-17), 18.99 (t, C-12), 18.72 (t, C-9); GC-MS (70 eV) m/z (relative intensity) 252 (M+, 1), 237 (1), 196 (1), 178 (1), 163 (1), 153 (1), 151 (1), 149 (3), 137 (2), 136 (3), 135 (7), 121 (11), 111 (5), 110 (12), 109 (59), 107 (12), 97 (10), 96 (29), 95 (62), 94 (10), 93 (23), 91 (11), 83 (17), 82 (54), 81 (77), 80 (20), 79 (40), 77 (14), 70 (11), 69 (100), 68 (37), 67 (81), 65 (10), 57 (16), 55 (75), 54 (31). HRMS (APCI) Calcd for C17H33O [M + H]+ 253.2526, found 253.2525.
2.7 General procedure for the catalytic hydrogenation of the alkynes
Into a 25-mL two-necked round-bottomed flask were placed dry hexane, the alkyne, quinoline (1 mL), and palladium in activated carbon (Lindlar’s catalyst). One of the necks was capped with a rubber septum and the other was connected via tygon tubing to a 25-mL graduated pipet ending in a 150-mL beaker with distilled water. While stirring at room temperature a 20-mL syringe with needle was used to withdraw air from the system and to draw water up into the graduated pipet to the 0.0-mL mark. Hydrogen was then introduced into the system using a balloon filled with hydrogen attached to a hose barb-to-luer lock adapter with a stopcock and a needle. The reaction mixture consumed 51.0–54.3 mL of hydrogen during 1 h. The mixture was filtered and the solvent removed in vacuo obtaining 0.47–0.51 g (1.99–2.07 mmol) of the desired alkenols 8a and 8b accounting for 94–96% yields. The alkenols were purified under vacuum distillation (Kugelrohr) by removing impurities at 110 ºC/3 mm Hg.
2.7.1 (Z)-13-Methyl-8-tetradecen-1-ol (8a)
The (Z)-13-methyl-8-tetradecen-1-ol (8a) was obtained as a colorless oil in a 94% yield from the catalytic hydrogenation, using Lindlar’s catalyst, of 13-methyl-8-tetradecyn-1-ol (0.50 g, 2.22 mmol) according to the general procedure described above; IR (neat) νmax 3360 (OH, broad), 3005, 2927, 2856, 1660, 1465, 1384, 1366, 1117, 1058, 724 cm−1; 1H NMR (CDCl3) δ 5.34 (2H, m, H-8,H-9), 3.63 (2H, t, J = 6.6 Hz, H-1), 2.00 (5H, m, H-7, H-10, H-13), 1.70 (1H, brs, -OH), 1.68–1.55 (2H, m, H-2), 1.31 (12H, m, -CH2-), 0.86 (6H, d, J = 6.6 Hz, -CH(CH3)2); 13C NMR (CDCl3) δ 129.99 (d), 129.75 (d), 63.03 (t, C-1), 38.62 (t, C-12), 32.74 (t, C-2), 29.65 (t), 29.29 (t), 29.21 (t), 27.86 (d, C-13), 27.52 (t, C-10), 27.41 (t, C-7), 27.14 (t), 25.68 (t), 22.60 (q, C-14, C-15); GC-MS (70 eV) m/z (relative intensity) 226 (M+, 1), 208 (3), 180 (1), 152 (2), 137 (4), 124 (7), 123 (10), 111 (5), 110 (15), 109 (23), 97 (16), 96 (45), 95 (53), 83 (32), 82 (80), 81 (61), 79 (10), 69 (74), 68 (40), 67 (73), 57 (40), 56 (53), 55 (100), 54 (39).
2.7.2 (Z)-15-Methyl-10-hexadecen-1-ol (8b)
The (Z)-15-methyl-10-hexadecen-1-ol (8b) was obtained as a colorless oil in a 96% yield from the catalytic hydrogenation, using Lindlar’s catalyst, of 15-methyl-10-hexadecyn-1-ol (0.51 g, 1.99 mmol) according to the general procedure described above; IR (neat) νmax 3360 (OH, broad), 3005, 2926, 2854, 1622, 1504, 1467, 1384, 1366, 1120, 1058, 805, 722 cm−1; 1H NMR (CDCl3) δ 5.34 (2H, m, H-10, H-11), 3.63 (2H, t, J = 6.6 Hz, H-1), 2.00 (5H, m, H-9, H-12, H-15), 1.70 (1H, brs, -OH), 1.58–1.50 (2H, m, H-2), 1.28 (16H, m, -CH2-), 0.86 (6H, d, J = 6.6 Hz, -CH(CH3)2); 13C NMR (CDCl3) δ129.93 (d), 129.85 (d), 63.03 (t, C-1), 38.63 (t, C-14), 32.79 (t, C-2), 29.73 (t), 29.56 (t), 29.46 (t), 29.41 (t), 29.26 (t), 27.87 (d, C-15), 27.54 (t, C-12), 27.42 (t, C-9), 27.18 (t), 25.73 (t, C-3), 22.61 (q, C-16, C-17); GC-MS (70 eV) m/z (relative intensity) 254 (M+, 1), 236 (3), 208 (1), 180 (1), 165 (1), 151 (2), 137 (5), 123 (11), 111 (8), 110 (15), 109 (22), 97 (21), 96 (45), 95 (46), 83 (36), 82 (75), 81 (50), 71 (10), 70 (18), 69 (80), 68 (34), 67 (55), 57 (42), 56 (56), 55 (100), 54 (31); analysis calculated for C17H34O: C, 80.24; H, 13.47; found: C, 79.63; H, 12.37.
2.8 General procedure for the oxidation of the alcohols
To a solution of pyridinium dichromate (8.7–10.1 mmol) and 2 mL of dimethylformamide (DMF) was added under argon (or nitrogen) a solution of the alcohol (1.7–2.0 mmol) in 12 ml of DMF and the reaction mixture was left stirring at room temperature for 24 h. After this time, the mixture was washed with water (3 x 15 mL), hexane (1 x 15 mL), dried over MgSO4, filtered, and evaporated in vacuo affording 1.1–1.2 mmol of the corresponding carboxylic acids in 63% yields.
2.8.1 (Z)-13-methyltetradec-8-enoic acid (1a )
The (Z)-13-methyltetradec-8-enoic acid (1a) was obtained as a colorless oil in a 63% yield from the reaction of (Z)-13-methyltetradec-8-en-1-ol (0.46 g, 2.0 mmol) and pyridinium dichromate (3.8 g, 10.1 mmol) in DMF according to the general procedure described above; IR (neat) νmax 3500–2500, 3004, 2925, 2854, 1708 (C=O), 1462, 1412, 1384, 1366, 1169, 1116, 909, 732 cm−1; 1H NMR (CDCl3) δ 5.34 (2H, m, H-8, H-9), 2.34 (2H, t, J = 7.5 Hz, H-2), 2.00 (5H, m, H-7, H-10, H-13), 1.56 (2H, m, H-3), 1.35 (10 H, m, -CH2-), 0.86 (6H, d, J = 6.6 Hz, -CH(CH3)2); 13C NMR (CDCl3) δ 179.38 (s, C-1), 130.11 (d), 129.61 (d), 38.63 (t, C-12), 33.93 (t, C-2), 29.49 (t, C-6), 28.94 (t, C-5), 28.85 (t, C-4), 27.88 (d, C-13), 27.53 (t, C-10), 27.43 (t, C-7), 27.09 (t), 24.63 (t), 22.61 (q, C-14, C-15); GC-MS (70 eV) m/z (relative intensity) 240 (M+, 8), 222 (3), 207 (1), 185 (2), 179 (3), 167 (9), 161 (2), 149 (6), 143 (1), 137 (4), 129 (2), 125 (5), 123 (7), 115 (3), 111 (11), 109 (10), 101 (5), 98 (12), 97 (21), 87 (4), 85 (8), 84 (25), 83 (33), 81 (23), 73 (14), 71 (13), 70 (31), 69 (97), 67 (34), 60 (15), 57 (52), 55 (100). HRMS (ESI) Calcd for C15H29O2 [M + H]+ 241.2168, found 241.2163.
2.8.2 (Z)-15-methylhexadec-10-enoic acid (1b )
The (Z)-15-methylhexadec-10-enoic acid (1b) was also obtained as a colorless oil in a 63% yield from the reaction of (Z)-15-methylhexadec-10-en-1-ol (0.44 g, 1.7 mmol) and pyridinium dichromate (2.28 g, 8.7 mmol) in DMF according to the general procedure described above; IR (neat) νmax 3500–2500, 3003, 2924, 2853, 1708 (C=O), 1463, 1412, 1384, 1366, 1117, 1089, 1027, 807, 784, 732 cm−1; 1H NMR (CDCl3) δ 5.34 (2H, m, H-10, H-11), 2.34 (2H, t, J = 7.5 Hz, H-2), 2.01 (5H, m, H-9, H-12, H-15), 1.61 (2H, m, H-3), 1.29 (14H, m, -CH2-), 0.86 (6H, d, J = 6.6 Hz, -CH(CH3)2); 13C NMR (CDCl3) δ 179.43 (s, C-1), 129.97 (d), 129.81 (d), 38.63 (t, C-14), 33.96 (t, C-2), 29.70 (t), 29.30 (t), 29.20 (t), 29.04 (t), 27.87 (d, C-15), 27.54 (t), 27.42 (t), 27.16 (t), 24.68 (t), 22.61 (q, C-16, C-17); GC-MS (70 eV) m/z (relative intensity) 268 (M+, 6), 250 (4), 235 (1), 213 (2), 207 (2), 195 (5), 177 (3), 165 (2), 161 (1), 151 (3), 149 (1), 143 (1), 137 (4), 129 (3), 125 (6), 123 (5), 115 (3), 111 (13), 109 (7), 101 (4), 98 (12), 97 (24), 95 (14), 87 (4), 85 (8), 84 (22), 83 (36), 81 (19), 73 (12), 71 (11), 70 (29), 69 (85), 67 (28), 60 (18), 57 (51), 55 (100); analysis calculated for C17H32O2: C, 76.06; H, 12.02; O, 11.92; found: C, 76.46; H, 12.05; O, 12.25.
3. Results and discussion
Several synthetic approaches have been employed in the synthesis of monounsaturated iso methyl-branched fatty acids with double bonds close to the ω end of the acyl chain (Reyes and Carballeira, 1996; Carballeira et al., 2004). A reported methodology employs the Wittig strategy where the best combination for generating the cis double bond was using the phosphonium salt of a short-chain iso methyl-branched bromoalkane and a long-chain aldehyde bearing a functional group for further transformation into a carboxylic acid. This approach was used in the synthesis of the (Z)-15-methylhexadec-11-enoic acid (Reyes and Carballeira, 1996). The synthetic limitations to this approach were the low yields (50%) in the Wittig coupling and obtaining Z/E mixtures, which can be a serious limitation when only the Z isomer is desired.
A more recent approach aiming at a 100% Z selectivity involves the use of the versatile reagent (trimethylsilyl)acetylene, as exemplified in the synthesis of the (Z)-14-methyl-9-pentadecenoic acid (Carballeira et al., 2004). In this particular synthesis the synthetic approach involved the initial coupling of the (trimethylsilyl)acetylene to a short-chain iso bromoalkane resulting in a volatile iso-alkyne followed, after removal of the silyl protecting group, by a second acetylide coupling to a longer chain bromoalkane bearing a functional group (e.g., a protected alcohol) that could later be transformed into a carboxylic acid. The advantage of this strategy is that a 100% Z stereoselectivity was obtained for the double bond, i.e., after Lindlar hydrogenation of the resulting internal alkyne. However, still a synthetic limitation to this approach is that the initial short-chain iso-alkane (in the example giving above the 6-methyl-1-heptyne) is quite volatile, and the yields for the second alkyne-bromide coupling reaction were rather low (33% yields). Therefore, we envisioned that by reversing the coupling order, i.e., initial coupling of the (trimethylsilyl)acetylene to a long-chain bifunctional bromoalkane followed by a second acetylide coupling to a short-chain iso bromoalkane would result in higher yields for the second acetylide coupling reaction. This is the strategy we are reporting herein for the seven-step syntheses of the fatty acids (Z)-13-methyl-8-tetradecenoic acid (1a) and the (Z)-15-methyl-10-hexadecenoic acid (1b), as outlined in Scheme 1.
Scheme 1.
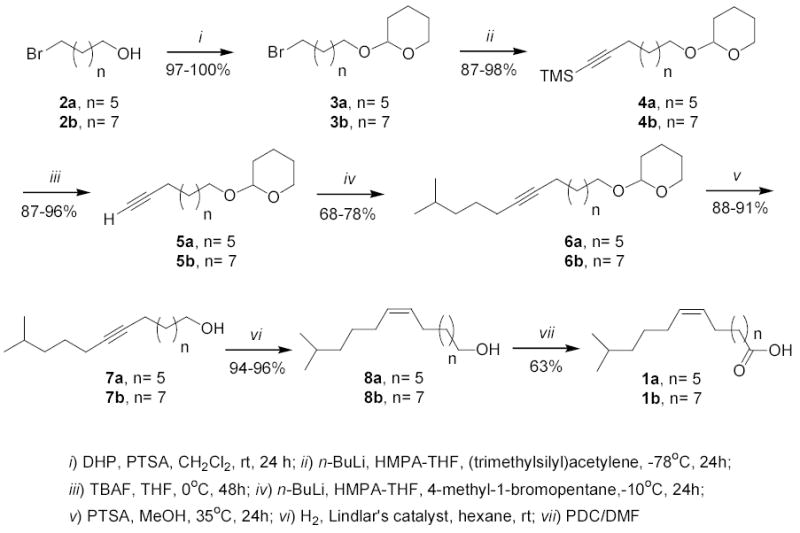
Synthesis of (Z)-13-methyltetradec-8-enoic acid (1a) and (Z)-15-methylhexadec-10-enoic acid (1b).
Both syntheses started with commercially available (Aldrich) bromo-alcohols, i.e., the 7-bromo-1-heptanol (2a) and the 9-bromo-1-nonanol (2b), which were protected with 3,4-dihydro-2H-pyran (DHP) in the presence of p-toluenesulfonic acid (PTSA) affording the corresponding dihydropyranyl protected alcohols 3a and 3b, respectively, in 97–100% isolated yields. The bromo dihydropyranyl protected alcohols 3a and 3b were then submitted to the first acetylide coupling reaction with the versatile reagent (trimethylsilyl)acetylene using n-BuLi in THF-HMPA affording the trimethylsilyl acetylenic derivatives 4a and 4b in 87–98% yields, respectively. Deprotection of the terminal trimethylsilyl group with tetrabutylammonium fluoride (TBAF) afforded the terminal alkynes 5a and 5b in 87–96% yields. A second acetylide coupling with 4-methyl-1-bromopentane using n-BuLi, THF-HMPA at −10° C resulted in the iso-branched alkynes 6a and 6b in 68–78% isolated yields (Scheme 1). Deprotection of the dihydropyranyl group with PTSA in methanol at 35° C for 24 h afforded the 13-methyl-8-tetradecyn-1-ol (7a) and the 15-methyl-10-hexadecyn-1-ol (7b) in 88–91% isolated yields. Catalytic hydrogenation, under Lindlar’s conditions of 7a and 7b, afforded the cis-alkenols (Z)-13-methyl-8-tetradecen-1-ol (8a) and (Z)-15-methyl-10-hexadecen-1-ol (8b) in 94–96% yields. Final oxidation of the alcohols with pyridinium dichromate in dimethylformamide (DMF) resulted in the formation of the desired acids 1a and 1b, both in 63% yields (Scheme 1). The overall total yields for the seven steps, in both syntheses, were 31–32%.
Acids 1a and 1b presented comparable spectral data that confirmed their structures. The iso methyl branching was easily identified by both 1H NMR and 13C NMR spectrometry as well as by infrared spectroscopy (IR). In the 1H NMR spectra both terminal isopropyl methyl groups appeared as a doublet at δ 0.86 ppm, while in 13C NMR both methyl groups resonated at δ 22.6 ppm. The IR was more characteristic since the terminal isopropyl group was observed as a doublet at 1384 and 1366 cm−1. The presence of the olefinic hydrogens was easily detected in the 1H NMR spectra by the olefinic signals at δ 5.34 ppm. The 13C NMR spectra were most informative since both allylic methylene carbons resonated at δ 27.4 and 27.5 ppm, confirming the presence of the cis double bond. The cis double bond stereochemistry was also further confirmed by IR since a strong absorption at 732 cm−1 was observed for both acids. The carboxylic acid carbonyl was observed in 13C NMR at δ 179.4 ppm and in IR at 1,708 cm−1. The mass spectra of both acids 1a and 1b presented a base peak at m/z 55 and the typical McLafferty rearrangement at m/z 60.
In summary, we have shown that higher yields are indeed obtained in the synthesis of monounsaturated iso methyl-branched fatty acids with double bonds close to the ω end of the acyl chain when (trimethylsilyl)acetylene is first coupled to a long-chain bifunctional bromoalkane (87–98% yields) followed by a second acetylide coupling to a short-chain iso-bromoalkane (68–78% yields). Complete spectral data is also provided for the first time for the acids 1a and 1b. The synthetic methodology presented herein will also certainly furnish enough material for systematic bioassay studies.
Acknowledgments
This work was supported by a grant from the SCORE program of the National Institutes of Health (Grant No. S06GM08102). L.F. Padilla thanks the NIH-MARC program for an undergraduate fellowship. We thank Fred Strobel (Emory University) for the high resolution mass spectral data.
References
- Boon JJ, de Leeuw JW, vd Hoek GJ, Vosjan JH. Significance and taxonomic value of iso and anteiso monoenoic fatty acids and branched β-hydroxy acids in Desulfovibrio desulfuricans. J Bacteriol. 1977;129:1183–1191. doi: 10.1128/jb.129.3.1183-1191.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon PI, Virtue P, Nichols PD. Microbial consortia in wetland sediments: a biomarker analysis of the effects of hydrological regime, vegetation and season on benthic microbes. Marine and Freshwater Research. 1996;47:27–41. [Google Scholar]
- Carballeira NM, Cruz H, Hill CA, De Voss JJ, Garson M. Identification and total synthesis of novel fatty acids from the siphonarid limpet Siphonaria denticulata. J Nat Prod. 2001;64:1426–1429. doi: 10.1021/np010307r. [DOI] [PubMed] [Google Scholar]
- Carballeira NM, Sanabria D, Ayala NL, Cruz C. A stereoselective synthesis for the (5Z,9Z)-14-methyl-5,9-pentadecadienoic acid and its monounsaturated analog (Z)-14-methyl-9-pentadecenoic acid. Tetrahedron Lett. 2004;45:3761–3763. [Google Scholar]
- Cryle MJ, Ortiz de Montellano PR, De Voss JJ. Cyclopropyl containing fatty acids as mechanistic probes for cytochromes P450. J Org Chem. 2005;70:2455–2469. doi: 10.1021/jo047985d. [DOI] [PubMed] [Google Scholar]
- Das B, Reddy MR, Reddy KR, Ramu R, Thirupathi P. Studies on novel synthetic methodologies. Part 73 An efficient direct conversion of THP ethers into acetates using Amberlyst-15. J Mol Catal A: Chem. 2006;248:185–188. [Google Scholar]
- Dowling NJE, Nichols PD, White DC. Phospholipid fatty acid and infrared spectroscopic analysis of a sulfate-reducing consortium. FEMS Microbiol Ecol. 1988;53:325–333. [Google Scholar]
- Edlund A, Nichols PD, Roffey R, White DC. Extractable and lipopolysaccharide fatty acid and hydroxy acid profiles from Desulfovibrio species. J Lipid Res. 1985;26:982–988. [PubMed] [Google Scholar]
- Grube A, Timm C, Koeck M. Synthesis and mass spectrometric analysis of cyclostellettamines H, I, K and L. Eur J Org Chem. 2006:1285–1295. [Google Scholar]
- Kaneda T. Iso- and anteiso-fatty acids in bacteria: biosynthesis, function, and taxonomic significance. Microbiol Rev. 1991;55:288–302. doi: 10.1128/mr.55.2.288-302.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpinska M, Lewandowska M, Grodner J. Ethynylation of ether derivatives of -haloalkanols with lithium acetylide-ethylenediamine complex. Pol J Chem. 2004;78:937–942. [Google Scholar]
- Lee HK, Lee DS, Lim J, Kim JS, Im KS, Jung JH. Topoisomerase I inhibitors from the Streptomyces sp strain KM86-9B isolated from a marine sponge. Arch Pharm Res. 1998;21:729–733. doi: 10.1007/BF02976766. [DOI] [PubMed] [Google Scholar]
- Reyes ED, Carballeira NM. A short synthesis of (Z)-15-methylhexadec-11-enoic acid. Synthesis. 1996:693–694. [Google Scholar]