

Abstract
The molecular mechanisms underlying hypoxic responses in pulmonary and systemic arteries remain obscure. Here we for the first time report that acute hypoxia significantly increased total PKC and PKCɛ activity in pulmonary, but not mesenteric arteries, while these two tissues showed comparable PKCɛ protein expression and activation by the PKC activator phorbol 12-myristate 13-acetate. Hypoxia induced an increase in intracellular reactive oxygen species (ROS) generation in isolated pulmonary artery smooth muscle cells (PASMCs), but not in mesenteric artery SMCs. Inhibition of mitochondrial ROS generation with rotenone, myxothiazol, or glutathione peroxidase-1 overexpression, prevented hypoxia-induced increases in total PKC and PKCɛ activity in pulmonary arteries. The inhibitory effects of rotenone were reversed by exogenous hydrogen peroxide. A PKCɛ translocation peptide inhibitor or PKCɛ gene deletion decreased hypoxic increase in [Ca2+]i in PASMCs, whereas the conventional PKC inhibitor GÖ6976 had no effect. These data suggest that acute hypoxia may specifically increase mitochondrial ROS generation, which subsequently activates PKC, particularly PKCɛ, contributing to hypoxia-induced increase in [Ca2+]i and contraction in PASMCs.
Keywords: Hypoxia, protein kinase C, reactive oxygen species, mitochondria, intracellular calcium, pulmonary artery smooth muscle cells
Hypoxic pulmonary vasoconstriction (HPV) is observed in isolated lungs, pulmonary arteries, and pulmonary artery smooth muscle cells (PASMCs). The pulmonary circulation differs from the systemic circulation in response to oxygen tension; pulmonary arteries constrict to physiological hypoxia (~ 20–60 mmHg PO2), whereas systemic arteries vasodilate. The mechanisms for these opposing responses to hypoxia appear to lie within the vascular SMCs. Hypoxia increases intracellular Ca2+ concentration ([Ca2+]i) and contracts PASMCs. In contrast, SMCs from systemic arteries display decreased [Ca2+]i and relax in response to hypoxia. The response of PASMCs to acute hypoxia involves calcium entry through voltage-dependent and store-operated Ca2+ channels, as well as Ca2+ release from the sarcoplasmic reticulum [1–8]. Hypoxia-dependent changes in reactive oxygen species (ROS) concentration have been proposed to mediate HPV by several laboratories, although the details of this hypothesis differ greatly [9; 10]. However, the signaling pathways underlying artery-specific, acute hypoxic vasoconstriction remain to be fully elucidated.
Protein kinase C (PKC) comprises at least 12 isoforms and appears to be involved in HPV in isolated lungs [11–16]. Although the role of individual PKC isoforms in acute hypoxic responses remains unclear, PKCɛ is likely to be important since this isoform regulates myocardial responses to hypoxic injury [17] and PKCɛ gene depletion protects against acute HPV by ~50% in perfused lungs [15]. So far, no biochemical evidence indicates the involvement of PKCɛ in hypoxic responses specific in pulmonary arteries; the mechanisms for how it may be involved are uncertain. In this study, thus, we sought to examine whether: 1) acute hypoxia could differentially affect total PKC and PKCɛ activity in mouse pulmonary and systemic (mesenteric) arteries; 2) mitochondria-derived ROS contributed to hypoxic effect on PKCɛ activity in PASMCs; and 3) PKCɛ was involved in hypoxia-induced increases in [Ca2+]i in PASMCs.
Materials and Methods
Reagents
Fura-2/AM, and 5,6-chloromethyl-2,7-dichlorodihydrofluorescein diacetate (CM-DCF-DA) were obtained from Molecular Probes (Eugene, OR); PKCɛ translocation peptide inhibitor from Calbiochem (La Jolla, CA); monoclonal antibodies to PKCɛ, PKC inhibitor, Protein G-coated beads from Santa Cruz (Santa Cruz, CA); and rotenone, myxothiazol, hydrogen peroxide, phorbol 12-myristate 13-acetate (PMA), GÖ6976, myeline basic protein(4–14), phosphatidyl serine, diolein, histone IIIs from Sigma (St. Louis, MO).
Animals
PKCɛ−/− mice were purchased from the Jackson Laboratory (Bar Harbor, ME); Swiss-Webster mice from Taconic (Germantown, NY). Glutathione peroxidase-1 (Gpx1) overexpression mice were generated and maintained as described previously [18]. All animal experiments were approved by the Institutional Animal Care and Use Committee of Albany Medical College. To examine the effects of pharmacological reagents, control experiments were carried out in cells or tissues from the same mice. For experiments using transgenic and knockout mice, control cells and tissues were obtained from wild-type mice with the same genetic background, age and sex.
Cell and Tissue preparation
Freshly isolated mouse resistance pulmonary and mesenteric artery SMCs (PASMCs and MASMCs) and tissues were prepared as described previously [7]. Briefly, mice were euthanized by sodium pentobarbital (150 mg/kg, i.p.). Arteries were carefully dissected free of endothelium and connective tissues in ice-cold, normoxic physiological saline solution (PSS, gassed with 20%O2, 5%CO2 and 75%N2). For PKC activity assay, the tissues were subjected to lysis. To obtain isolated cells, arteries were cut into small pieces, incubated in nominally Ca2+-free PSS containing papain and dithioerythritol for 15 min (37°C), then in low-Ca2+ (100 μM) PSS containing type H and II collagenase for 10–15 min (37°C), and finally in ice-cold low-Ca2+ PSS for 15 min. Single cells were harvested by gentle trituration and kept on ice for daily use. To obtain acute hypoxic responses, cells or arteries were exposed to hypoxic PSS solution (bubbled with 1%O2, 5%CO2 and 94%N2) for 5 min. In control experiments, cells or tissues were treated identically, but not exposed to hypoxic PSS. Unless indicated otherwise, reagents were applied to cells or tissues for 10 min before the start of hypoxia.
Total PKC activity assay
Total PKC activity was determined from combined cytosolic and particulate protein fractions, as reported previously [19]. In brief, after lysis in a modified RIPA buffer, the samples were centrifuged to clear insoluble debris and incubated for 10 min at 30°C in reaction buffer with 2 μCi γ-32P ATP (PerkinElmer Life Sciences) in the presence and absence of 1 μM PKC inhibitor. After incubation, 25 μl of reaction samples were spotted onto P81 filter paper, and washed 5 times in 75 mM phosphoric acid and once in ethanol. After drying, 32P incorporation was determined by scintillation counting with a Beckman LS6500 scintillation counter. The background activity obtained in the presence of the PKC inhibitor was subtracted from each value to obtain the specific PKC activity of each sample.
Immunoprecipitation and PKCɛ activity assay
PKCɛ activity was determined by an immune complex kinase assay [19]. After lysis and centrifugation, samples were incubated overnight at 4°C with monoclonal anti-PKCɛ antibody. Immune complexes were collected on protein G beads and then washed three times with modified RIPA buffer and once with 250 mM sucrose buffer. The protein G beads were incubated for 10 min at 30°C in reaction buffer with 2 μCi γ-32P ATP. The 32P incorporation was determined by scintillation counting.
Western blot analysis
Proteins in tissue lysates were solubilized in SDS-PAGE buffer, and heated for 5 min at 95°C. Then samples were resolved, transferred to PVDF membrane (Bio-Rad), and immunoblotted. After being blocked in 5% nonfat dry milk for overnight at 4°C, membranes were incubated with a primary antibody for 1 h, washed 3 times for 10 min with 20 mM Tris-150 mM NaCl-0.2% Tween 20, incubated with an HRP-conjugated secondary antibody for 1 h, and then washed again. Proteins were detected by chemiluminescent substrate (Santa Cruz). The PKCɛ and β-actin levels were quantified by using Multi Gauge software (version 3.0; Fujifilm). The protein level of PKCɛ in each sample was normalized to the level of β-actin.
Measurement of single cell contraction
Contraction in single freshly isolated cells was examined by measuring cell shortening with a laser scanning confocal microscope (LSM510, Carl Zeiss, Thornwood, NY). Transmitted light x-y images were continuously taken every 30 s. Cell length in each image was determined at its long axis.
Measurement of intercellular reactive oxygen species
Freshly isolated cells were loaded with 5 μM CM-DCF/DA at 35 °C for 30 min, and then were perfused with pre-warmed, dye-free bath solution for 15 min to washout extracellular dye and to allow the conversion of intracellular dye into its non-ester form. The dye was excited at 488 nm with an LSM510 laser scanning confocal microscope. DCF fluorescence at 510 nm was collected and expressed in arbitrary units (a.u.) after background subtraction.
Measurement of [Ca2+]i.
[Ca2+]i were measured by a dual excitation wavelength fluorescence method using the IonOptix fluorescence photometric system (Milton, MA), as described previously [7]. Freshly isolated cells were loaded with 5 μM fura-2/AM. The dye was excited at 340 and 380 nm wavelengths (Xenon 75W arc lamp). The emission fluorescence was detected at 510 nm.
Statistical analysis
Data were expressed as mean ± SE of at least three independent experiments. Student’s t-test was used for determining the significance of differences between two groups, whereas one-way ANOVA for multiple comparisons. P < 0.05 was accepted as a level of statistical significance.
Results
Hypoxia increases total PKC and PKCɛ in endothelium-denuded pulmonary, but not, mesenteric arteries
To determine whether acute hypoxia differentially activated PKC in pulmonary and mesenteric (systemic) arteries, we assayed PKC activity in lysates from hypoxia-treated and untreated tissues. As shown in Fig. 1A, hypoxic exposure for 5 min caused an approximately 3 fold increase in total PKC activity in pulmonary arteries, but had no effect in mesenteric arteries. Hypoxia also resulted in an increase in PKCɛ activity by 3.2-fold in pulmonary, but not mesenteric arteries. As a positive control, the PKC activator PMA was found to increase PKCɛ activity in both pulmonary and mesenteric arteries (Fig. 1B). Furthermore, Western blot analysis revealed that PKCɛ protein expression was comparable in resistance pulmonary and mesenteric arteries (Fig. 1C). Thus, differential Ca2+ and contractile responses of pulmonary and mesenteric (systemic) arteries to hypoxia may lie proximally to PKCɛ activation.
Fig. 1.
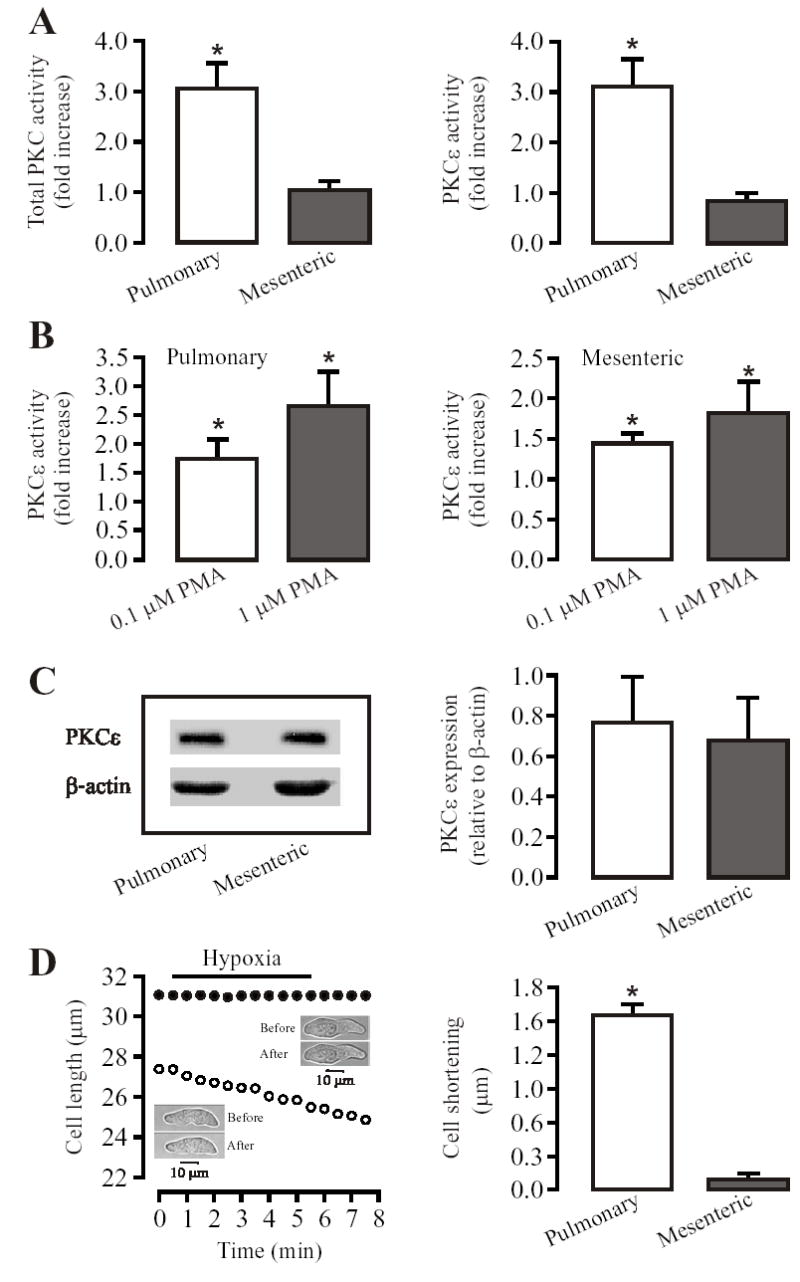
Acute hypoxia increases total PKC and PKCɛ activity in mouse resistance pulmonary, but not in mesenteric arteries. (A): Hypoxic exposure for 5 min caused an increase in the activity of total PKC and PKCɛ in pulmonary, but not mesenteric arteries. (B): The PKC activator PMA concentration-dependently augmented the activity of PKCɛ in both pulmonary and mesenteric arteries. (C): PKCɛ protein expression, determined by using SDS-PAGE and immunobloted with anti-PKCɛ antibody, was comparable in pulmonary and mesenteric arteries. Bar graph shows quantification of PKCɛ expression levels normalized to β-actin levels. All data are presented as means ± SE from 3 or 4 independent experiments. *P < 0.05 compared with control (without hypoxia or PMA). (D): Hypoxia for 5 min resulted in a significant contraction (shortening) in PASMCs, but not in MASMCs. Recording traces show that the effect of hypoxia on cell length in a PASMC (open circles) and MASMC (closed circles). Insets show that transmitted light images were taken before and after hypoxia for 5 min. Bar graph summarizes hypoxia-induced cell shortening in PASMCs and MASMCs. Data are presented as means ± SE from 21 PASMCs and 25 MASMCs in 4 independent experiments. *P < 0.05 compared with before hypoxia (normoxia).
Consistent with differential activation of PKC, hypoxia for 5 min could result in a significant contraction in freshly isolated PASMCs, but not in MASMCs under the same conditions used for measuring PKC activity (Fig. 1D). The mean length of PASMCs was shortened by 1.6 ± 0.1 μm following hypoxic stimulation (P < 0.05), whereas the mean length of MASMCs was unchanged (29.0 ± 0.5 μm before hypoxia vs 28.9 ± 0.5 μm after hypoxia, P > 0.05).
Inhibition of mitochondrial ROS generation prevents hypoxia-induced PKCɛ activation in pulmonary arteries
Possible involvement of mitochondrial ROS in hypoxic activation of PKC in pulmonary arteries was examined by using the electron transport chain (ETC) inhibitors rotenone and myxothiazol. Hypoxic increases in total PKC and PKCɛ activity were significantly attenuated following pretreatment with rotenone (12.7 μM) or myxothiazol (10 μM) for 10 min (Fig. 2A). To complement the effect of mitochondrial inhibitors, we studied whether Gpx1 gene overexpression to enhance mitochondrial H2O2 degradation could block hypoxic activation of PKCɛ activity. Our data indicate that the hypoxic increase in PKCɛ activity was greatly reduced in pulmonary arteries from Gpx1 overexpression mice (Fig. 2B).
Fig. 2.
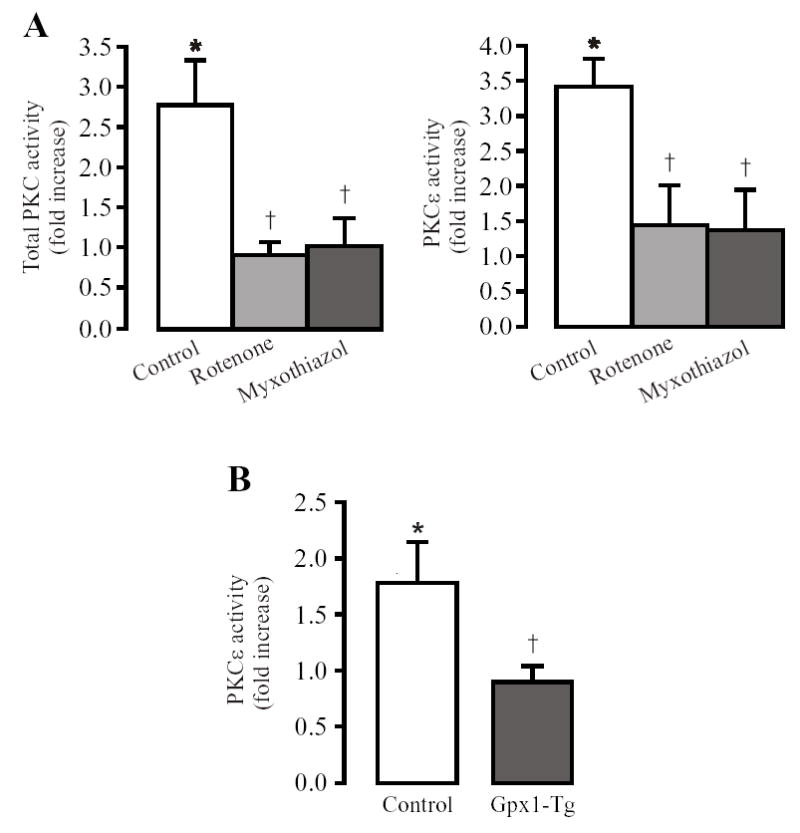
Inhibition of mitochondrial ROS generation blocks hypoxic increases in total PKC and PKCɛ activity in mouse resistance pulmonary arteries. (A): Inhibition of mitochondrial ROS generation with rotenone (12.7 μM) or myxothiazol (10 μM) for 10 min blocked hypoxia-induced increases in total PKC and PKCɛ activity. (B): Gpx1 overexpression (Gpx1-Tg) to enhance mitochondrial H2O2 metabolism prevented hypoxia-induced increase in PKCɛ activity. Data are presented as means ± SE from 3 independent experiments. *P < 0.05 compared with normoxia; †P < 0.05 compared with control (hypoxia alone or non-transgenic).
Exogenous H2O2, mimicking hypoxia, increases PKCɛ activity in pulmonary arteries
A possible role of ROS in hypoxic increase in PKCɛ activity was further investigated by application of exogenous H2O2. As shown in Fig. 3A, H2O2 (5.1 μM and 51 μM) increased PKC-ɛ activity comparably in both pulmonary and mesenteric arteries. The maximal increases in PKCɛ activity in response to H2O2 and hypoxia were similar in pulmonary artery. Furthermore, H2O2 rescued the inhibitory effect of rotenone on hypoxic activation of PKCɛ (Fig. 3B). Together, these results point towards the increased ROS generation as a potential mechanism underlying hypoxic activation PKCɛ in pulmonary arteries.
Fig. 3.
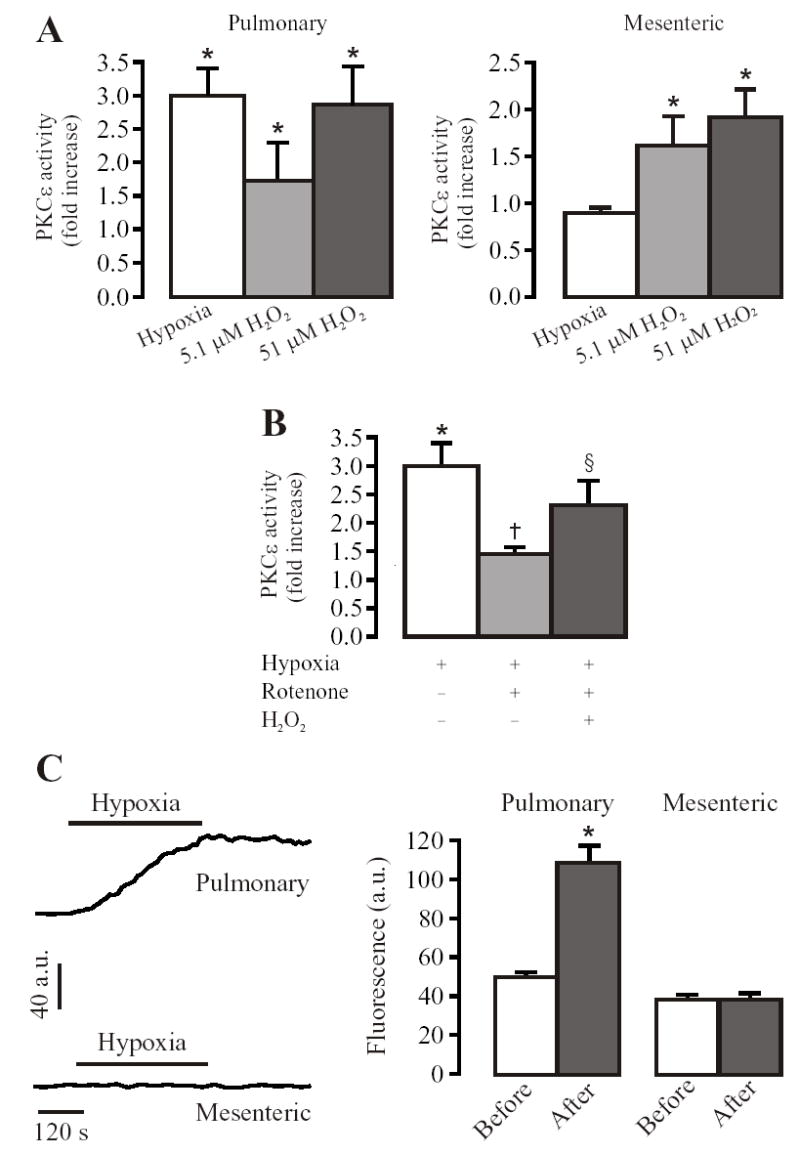
Exogenous H2O2 increases PKCɛ activity in mouse resistance pulmonary and mesenteric arteries. (A): Application of H2O2 for 5 min concentration dependently increased the activity of PKCɛ in pulmonary and mesenteric arteries. Data are presented as means ± SE from 3 independent experiments, *P < 0.05 compared with normoxia or untreated with H2O2. (B): H2O2 (5.1 μM) reversed the inhibition of hypoxia-induced increase in PKCɛ activity by pretreatment with rotenone (12.7 μM) for 10 min in pulmonary arteries. Data are presented as means ± SE from 3 independent experiments. *P < 0.05 compared with normoxia, †P < 0.05 compared with control (hypoxia alone), §P < 0.05 compared with hypoxia plus rotenone. (C): Acute hypoxia caused an increase [ROS]i in PASMCs, but not in MASMCs. Representative recordings show that hypoxic exposure for 5 min resulted in a large increase in DCF fluorescence ([ROS]i) in a PASMC, but not MASMC. Bar graph summarizes the effect of hypoxia on DCF fluorescence in PASMCs and MASMCs. Data are presented as means ± SE from 21 cells in 5 independent experiments. *P < 0.05 compared with before hypoxia (normoxia).
Hypoxia induces an increase in [ROS]i in pulmonary, but not mesenteric arteries
Since differential generation of mitochondrial ROS may underlie the observed heterogeneity in PKC responses to hypoxia in pulmonary and mesenteric arteries, we measured intracellular ROS generation in freshly isolated PASMCs and mesenteric artery SMCs (MASMCs) using CM-DCF/DA as a probe. Hypoxia for 5 min increased DCF fluorescence by ~2 times in PASMCs, indicating a significant increase in [ROS]i. The mean DCF fluorescence was increased to 108.12 ± 9.04 a.u. from a resting level of 49.9 ± 2.8 a.u.. Restoration of normoxia only caused a slight decrease in the DCF signal, which might reflect the irreversible oxidization of the dye. In MASMCs, no significant change in DCF fluorescence was observed during hypoxia exposure (Fig. 3C). These data further demonstrate the differential sensitivity of ROS-related hypoxia-sensing mechanisms in pulmonary and mesenteric (systemic) arteries.
PKCɛ is involved in hypoxia-induced increases in [Ca2+]i in PASMCs
In order to test the involvement of PKCɛ in hypoxic increase in [Ca2+]i, we used pharmacological and genetic approaches to block PKCɛ. In freshly isolated PASMCs, hypoxia challenge for 5 min resulted in a significant increase in [Ca2+]i. A mean increase in [Ca2+]i by hypoxia was 344 ± 27 nM. Pretreatment of the cells with a PKCɛ translocation inhibitory peptide (10 μM) for 10 min decreased hypoxia-induced increases in [Ca2+]i by approximately 40%, whereas GÖ6976 (an inhibitor of classic PKC isozymes and PKCμ, 100 nM) had no effect (Fig. 4A). Moreover, hypoxic increase in [Ca2+]i was significantly attenuated in PKCɛ−/− mouse PASMCs. The mean hypoxic increases in [Ca2+]i were 80±11 nM in PKCɛ−/− cells and 213±15 nM in PKCɛ+/+ cells (P < 0.05, Fig. 4B). Together, these results implicate the importance of PKCɛ (a “novel” PKC isoform) as a signaling element in hypoxic increase in [Ca2+]i in PASMCs.
Fig. 4.
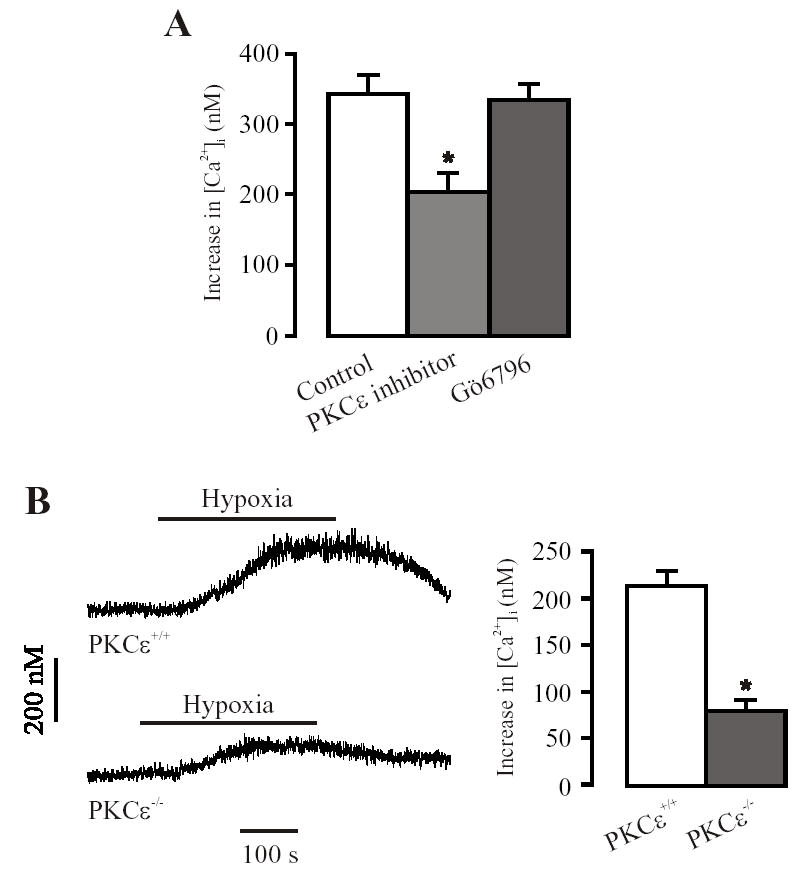
PKCɛ is involved in hypoxia-induced increase in [Ca2+]i in freshly isolated mouse resistance PASMCs. (A): PKCɛ translocation inhibitory peptide (10 μM) reduced hypoxia-induced increase in [Ca2+]i, whereas GÖ6976 (1 00 nM) had no effect. Data are presented as means ± SE from 18 cells in 5 independent experiments. *P < 0.05 compared with control (hypoxia alone). (B): Original recordings show hypoxia-induced increase in [Ca2+]i in a control (PKCɛ+/+) and PKCɛ gene deletion (PKCɛ−/−) PASMC. (C): Summary of the effect of PKCɛ gene deletion on hypoxic increase in [Ca2+]i. Data are presented as means ± SE from 23 cells in 4 independent experiments. *P < 0.05 compared with control (PKCɛ+/+).
Discussion
In the present study we have demonstrated: 1) acute hypoxic activation of total PKC and PKCɛ in PASMCs, 2) involvement of mitochondrial ROS in hypoxic PKCɛ activation in PASMCs, 3) PKCɛ involvement in acute hypoxia-induced increase in [Ca2+]i in PASMCs, and 4) differential responsiveness of the hypoxia-sensing pathway in pulmonary and mesenteric arteries underlying heterogeneous hypoxic responses. These data provide extensive experimental support for the concept that mitochondrial ROS-dependent activation of PKCɛ is involved in the signal transduction pathways leading to HPV.
Studies using pharmacological inhibitors have shown that PKC is involved in HPV in isolated lungs and pulmonary arteries [11–16]. However, no biochemical evidence is available to support the importance of PKC in hypoxic responses in vascular SMCs; the contribution of individual isoforms of PKC is unclear. Our biochemical findings indicate that acute hypoxia causes an increase in total PKC and PKCɛ activity in pulmonary, but not in systemic (mesenteric) arteries, whereas the PKC activator PMA leads to an increase in PKCɛ activity, and PKCɛ protein expression is comparable in both arteries. These results not only extend previous pharmacological studies, but also provide evidence that hypoxic activation of PKC, particularly PKCɛ, is unique to pulmonary arteries and may explain the differential hypoxic responses (e.g., contraction in pulmonary, but not in mesenteric arteries). In agreement with this view, we have found that hypoxia causes a significant contraction in freshly isolated mouse PASMCs, but has no effect in MASMCs.
Michelakis et al [20] have reported that opposing responses of the pulmonary and systemic vascular beds to hypoxia are, at least in part, due to the differences in mitochondrial functions in perfused lungs. Other investigators have further shown that increases in mitochondrial ROS formation at the sites prior to the ubisemiquinone site of complex III is one of the initial steps in the signal cascade underlying HPV [21–24]. In this study, we examined the effects of mitochondrial complex I inhibitor rotenone and the complex III inhibitor myxothiazol on the hypoxic increases in total PKC and PKCɛ activity in pulmonary arteries. Our results indicate that rotenone and myxothiazol both significantly prevent hypoxia-induced increases in total PKC and PKCɛ activity. Thus, mitochondrial ETC complex molecules prior to the complex III semiubiquinone site may work as a functional unit to mediate hypoxic activation of PKC, particularly PKCɛ, by increasing mitochondrial ROS generation in pulmonary arteries.
Although a role for antioxidant enzymes in preventing HPV in pulmonary arteries has been implicated in some studies [25–27], their effects on hypoxia-induced PKC activation were not reported. We have found that Gpx1 overexpression to increase the conversion of H2O2 into H2O in mitochondria blocks hypoxic increase in PKCɛ activity in pulmonary arteries, further suggesting that the role of the increased generation of mitochondrial ROS (particularly H2O2) in hypoxic responses. To provide further evidence for the role of H2O2 in PKCɛ activation during hypoxic stimulation, we examined the effect of H2O2 on PKCɛ activity in normoxic condition, and have found that exogenous application of H2O2 mimics hypoxic responses, causing an increase in PKCɛ activity in pulmonary arteries. In mesenteric arteries, H2O2, although not hypoxia, also significantly activates PKCɛ. Inhibition of the ETC complexes with rotenone and myxothiazol significantly prevent hypoxic increase in PKCɛ activity in pulmonary arteries; and the inhibitory effect of rotenone was rescued by exogenous H2O2. Taken together, PKCɛ may be activated by mitochondrial ROS, particularly H2O2, during acute hypoxia.
The potential importance of mitochondrial ROS in the heterogeneity of PKC responses to hypoxia in PASMCs and MASMCs were further examined by measuring intracellular ROS using CM-DCF/DA as a probe, which is a sensitive probe for detecting intracellular ROS, particularly H2O2 in living cells, since this dye becomes fluorescent after it is oxidized through an intracellular enzymatic process that requires H2O2, and has much less leakage from the cells than DCF/DA [28]. Hypoxia exposure results in a significant increase in DCF fluorescence in freshly isolated mouse PASMCs, but not in MASMCs, indicating that hypoxia increases [ROS]i in PASMCs only. Michelakis et al have reported the opposing responses of the pulmonary and renal vascular beds to hypoxia: inhibition of voltage-dependent K+ currents and contractions in PASMCs, whereas voltage-dependent K+ currents and relaxations in renal artery SMCs, which are, at least in part, due to potential diversity of mitochondrial function in the attributed vasculature [20]. These results are consistent with our finding, and further suggest the differential sensitivity of the hypoxia-sensing mechanisms between pulmonary and systemic arteries.
An elevation of [Ca2+]i in SMCs is a critical element to develop HPV. Although there are many reports to suggest a pre-eminent role for inhibition of K+ channels by hypoxia, with subsequent membrane depolarization and Ca2+ entry via voltage-dependent Ca2+ channels [1], others have provided evidence for capacitative Ca2+ entry through store-operated channels [8; 29]. Intracellular Ca2+ release is also a critical event for the elevation of [Ca2+]i and constriction in PASMCs [2–7]. PKC and other protein kinases are implicated in hypoxic responses [30]. In this investigation, a specific PKCɛ translocation peptide inhibitor markedly blocks hypoxic increases in [Ca2+]i in freshly isolated PASMCs, whereas the selective classic PKC inhibitor GÖ6976 has no effect. Using gene knockout mice, Littler et al have shown that acute HPV in isolated lungs from PKCɛ null mice is blunted [15]. We have now confirmed this general result, specifically implicating the role of PKCɛ in HPV at the cellular level by providing evidence that hypoxic increases in [Ca2+]i are significantly attenuating in PASMCs from the PKCɛ null mice.
Acknowledgments
We thank Ms. Jodi Heim for technical assistance. This work was supported by AHA and NIH (Y.-X.W.), and EHS Center Grant (Y.-S.H.).
References
- 1.Mauban JR, Remillard CV, Yuan JX. Hypoxic pulmonary vasoconstriction: role of ion channels. J Appl Physiol. 2005;98:415–420. doi: 10.1152/japplphysiol.00732.2004. [DOI] [PubMed] [Google Scholar]
- 2.Jabr RI, Toland H, Gelband CH, Wang XX, Hume JR. Prominent role of intracellular Ca2+ release in hypoxic vasoconstriction of canine pulmonary artery. Br J Pharmacol. 1997;122:21–30. doi: 10.1038/sj.bjp.0701326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson HL, Dipp M, Thomas JM, Lad C, Galione A, Evans AM. ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase act as a redox sensor. a primary role for cyclic ADP-ribose in hypoxic pulmonary vasoconstriction. J Biol Chem. 2001;276:111 80–111 88. doi: 10.1074/jbc.M004849200. [DOI] [PubMed] [Google Scholar]
- 4.Dipp M, Nye PC, Evans AM. Hypoxic release of calcium from the sarcoplasmic reticulum of pulmonary artery smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2001;281:L318–L325. doi: 10.1152/ajplung.2001.281.2.L318. [DOI] [PubMed] [Google Scholar]
- 5.Ng LC, Wilson SM, Hume JR. Mobilization of sarcoplasmic reticulum stores by hypoxia leads to consequent activation of capacitative Ca2+ entry in isolated canine pulmonary arterial smooth muscle cells. J Physiol. 2005;563:409–419. doi: 10.1113/jphysiol.2004.078311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng YM, Mei QB, Wang QS, Abdullaev I, Lai FA, Xin HB, Kotlikoff MI, Wang YX. Role of FKBP12.6 in hypoxia- and norepinephrine-induced Ca2+ release and contraction in pulmonary artery myocytes. Cell Calcium. 2004;35:345–355. doi: 10.1016/j.ceca.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Zheng YM, Wang QS, Rathore R, Zhang WH, Mazurkiewicz JE, Sorrentino V, Singer HA, Kotlikoff MI, Wang YX. Type-3 ryanodine receptors mediate hypoxia-, but not neurotransmitter-induced calcium release and contraction in pulmonary artery smooth muscle cells. J Gen Physiol. 2005;125:427–440. doi: 10.1085/jgp.200409232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J, Shimoda LA, Weigand L, Wang W, Sun D, Sylvester JT. Acute Hypoxia Increases Intracellular [Ca2+] in Pulmonary Arterial Smooth Muscle by Enhancing Capacitative Ca2+ Entry. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1059–L1069. doi: 10.1152/ajplung.00448.2004. [DOI] [PubMed] [Google Scholar]
- 9.Waypa GB, Schumacker PT. Hypoxic pulmonary vasoconstriction: redox events in oxygen sensing. J Appl Physiol. 2005;98:404–414. doi: 10.1152/japplphysiol.00722.2004. [DOI] [PubMed] [Google Scholar]
- 10.Moudgil R, Michelakis ED, Archer SL. Hypoxic pulmonary vasoconstriction. J Appl Physiol. 2005;98:390–403. doi: 10.1152/japplphysiol.00733.2004. [DOI] [PubMed] [Google Scholar]
- 11.Orton EC, Raffestin B, McMurtry IF. Protein kinase C influences rat pulmonary vascular reactivity. Am Rev Respir Dis. 1990;141:654–658. doi: 10.1164/ajrccm/141.3.654. [DOI] [PubMed] [Google Scholar]
- 12.Barman SA. Potassium channels modulate canine pulmonary vasoreactivity to protein kinase C activation. Am J Physiol. 1999;277:L558–L565. doi: 10.1152/ajplung.1999.277.3.L558. [DOI] [PubMed] [Google Scholar]
- 13.Jin N, Packer CS, Rhoades RA. Pulmonary arterial hypoxic contraction: signal transduction. Am J Physiol. 1992;263:L73–L78. doi: 10.1152/ajplung.1992.263.1.L73. [DOI] [PubMed] [Google Scholar]
- 14.Weissmann N, Voswinckel R, Hardebusch T, Rosseau S, Ghofrani HA, Schermuly R, Seeger W, Grimminger F. Evidence for a role of protein kinase C in hypoxic pulmonary vasoconstriction. Am J Physiol. 1999;276:L90–L95. doi: 10.1152/ajplung.1999.276.1.L90. [DOI] [PubMed] [Google Scholar]
- 15.Littler CM, Morris KG, Jr, Fagan KA, McMurtry IF, Messing RO, Dempsey EC. Protein kinase C-epsilon-null mice have decreased hypoxic pulmonary vasoconstriction. Am J Physiol Heart Circ Physiol. 2003;284:H1321–H1331. doi: 10.1152/ajpheart.00795.2002. [DOI] [PubMed] [Google Scholar]
- 16.Tsai BM, Wang M, Pitcher JM, Meldrum KK, Meldrum DR. Hypoxic pulmonary vasoconstriction and pulmonary artery tissue cytokine expression are mediated by protein kinase C. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1215–L1219. doi: 10.1152/ajplung.00179.2004. [DOI] [PubMed] [Google Scholar]
- 17.Gray MO, Zhou HZ, Schafhalter-Zoppoth I, Zhu P, Mochly-Rosen D, Messing RO. Preservation of base-line hemodynamic function and loss of inducible cardioprotection in adult mice lacking protein kinase C epsilon. J Biol Chem. 2004;279:3596–3604. doi: 10.1074/jbc.M311459200. [DOI] [PubMed] [Google Scholar]
- 18.Xiong Y, Shie FS, Zhang J, Lee CP, Ho YS. The protective role of cellular glutathione peroxidase against trauma-induced mitochondrial dysfunction in the mouse brain. J Stroke Cerebrovasc Dis. 2004;13:129–137. doi: 10.1016/j.jstrokecerebrovasdis.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 19.Ginnan R, Pfleiderer PJ, Pumiglia KM, Singer HA. PKCδ and CaMKIIδ2 Mediate ATP-Dependent Activation of ERK1/2 in Vascular Smooth Muscle. Am J Physiol Cell Physiol. 2004 doi: 10.1152/ajpcell.00202.2003. [DOI] [PubMed] [Google Scholar]
- 20.Michelakis ED, Hampl V, Nsair A, Wu X, Harry G, Haromy A, Gurtu R, Archer SL. Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ Res. 2002;90:1307–1315. doi: 10.1161/01.res.0000024689.07590.c2. [DOI] [PubMed] [Google Scholar]
- 21.Leach RM, Hill HM, Snetkov VA, Robertson TP, Ward JP. Divergent roles of glycolysis and the mitochondrial electron transport chain in hypoxic pulmonary vasoconstriction of the rat: identity of the hypoxic sensor. J Physiol. 2001;536:211–224. doi: 10.1111/j.1469-7793.2001.00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waypa GB, Chandel NS, Schumacker PT. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ Res. 2001;88:1259–1266. doi: 10.1161/hh1201.091960. [DOI] [PubMed] [Google Scholar]
- 23.Weissmann N, Ebert N, Ahrens M, Ghofrani HA, Schermuly RT, Hanze J, Fink L, Rose F, Conzen J, Seeger W, Grimminger F. Effects of mitochondrial inhibitors and uncouplers on hypoxic vasoconstriction in rabbit lungs. Am J Respir Cell Mol Biol. 2003;29:721–732. doi: 10.1165/rcmb.2002-0217OC. [DOI] [PubMed] [Google Scholar]
- 24.Paddenberg R, Ishaq B, Goldenberg A, Faulhammer P, Rose F, Weissmann N, Braun-Dullaeus RC, Kummer W. Essential role of complex II of the respiratory chain in hypoxia-induced ROS generation in the pulmonary vasculature. Am J Physiol Lung Cell Mol Physiol. 2003;284:L710–L719. doi: 10.1152/ajplung.00149.2002. [DOI] [PubMed] [Google Scholar]
- 25.Weissmann N, Grimminger F, Voswinckel R, Conzen J, Seeger W. Nitro blue tetrazolium inhibits but does not mimic hypoxic vasoconstriction in isolated rabbit lungs. Am J Physiol. 1998;274:L721–L727. doi: 10.1152/ajplung.1998.274.5.L721. [DOI] [PubMed] [Google Scholar]
- 26.Waypa GB, Marks JD, Mack MM, Boriboun C, Mungai PT, Schumacker PT. Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary arterial myocytes. Circ Res. 2002;91:719–726. doi: 10.1161/01.res.0000036751.04896.f1. [DOI] [PubMed] [Google Scholar]
- 27.BelAiba RS, Djordjevic T, Bonello S, Flugel D, Hess J, Kietzmann T, Gorlach A. Redox-sensitive regulation of the HIF pathway under non-hypoxic conditions in pulmonary artery smooth muscle cells. Biol Chem. 2004;385:249–257. doi: 10.1515/BC.2004.019. [DOI] [PubMed] [Google Scholar]
- 28.Halliwell B, Whiteman M. Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br J Pharmacol. 2004;142:231–255. doi: 10.1038/sj.bjp.0705776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robertson TP, Hague D, Aaronson PI, Ward JP. Voltage-independent calcium entry in hypoxic pulmonary vasoconstriction of intrapulmonary arteries of the rat. J Physiol. 2000;525:669–680. doi: 10.1111/j.1469-7793.2000.t01-1-00669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ward JP, Knock GA, Snetkov VA, Aaronson PI. Protein kinases in vascular smooth muscle tone--role in the pulmonary vasculature and hypoxic pulmonary vasoconstriction. Pharmacol Ther. 2004;104:207–231. doi: 10.1016/j.pharmthera.2004.08.009. [DOI] [PubMed] [Google Scholar]