

Abstract
The Saccharomyces cerevisiae proteins Tel1p and Mec1p are involved in telomere length regulation and cellular responses to DNA damage. The closest relative of these proteins is the human Ataxia Telangiectasia Mutated (ATM) protein, a wortmannin-sensitive protein kinase that primarily phosphorylates serines in an SQ motif. We constructed yeast strains containing functional epitope-tagged versions of Tel1p and Mec1p. We showed that immunoprecipitated Tel1p and Mec1p were capable of in vitro phosphorylation of the mammalian protein PHAS-I (Phosphorylated Heat and Acid Stable protein). These activities are sensitive to wortmannin. Tel1p phosphorylates serine in an SQ motif in PHAS-I. Mutations in the kinase domains of Tel1p and Mec1p result in loss of in vitro kinase activity and the in vivo phenotypes associated with the null tel1 and mec1 mutations.
The Saccharomyces cerevisiae Tel1 and Mec1 proteins regulate telomere length (1–3) and cellular responses to DNA damage (4–8). Strains with the tel1 mutation have very short, but stable, telomeres (1). Although strains with the mec1 mutation have telomeres that are only slightly shorter than wild type, strains with both tel1 and mec1 mutations have very short telomeres and a senescent phenotype (3). Genetic studies demonstrate that Tel1p is in the same pathway of telomere length-regulation as the Mre11, Rad50, and Xrs2 proteins (9), whereas Mec1p is in a different pathway (3).
Strains with the mec1 mutation are sensitive to DNA-damaging agents (such as x-rays, UV light, and methylmethane sulfonate) and agents that lead to S phase arrest (hydroxyurea; refs. 4–8). Although tel1 cells are not sensitive to DNA-damaging agents or hydroxyurea, one extra copy of TEL1 can compensate for much of the damage sensitivity of mec1 strains (7, 8). These results suggest that Tel1p and Mec1p have functionally redundant roles in the repair of certain types of DNA damage. In addition to its role in DNA repair and telomere regulation, Mec1p has an essential cellular function, because null mec1 mutants are inviable (4, 5). This lethality can be suppressed by genetic alterations (such as a mutation in the SML1 gene) that elevate nucleotide pools (10).
Mec1p is a checkpoint protein, because strains with a mutation in mec1 fail to arrest the cell cycle in response to DNA damage or the inhibition of DNA synthesis (4, 11). In response to DNA damage or S phase arrest, Mec1p-dependent phosphorylation is observed for a number of checkpoint/DNA replication proteins such as Rad53p (8, 11). In the absence of Mec1p, Te11p-dependent phosphorylation is observed for several of these proteins (8). These results suggest, but do not prove, that Tel1p and Mec1p are protein kinases with the ability to phosphorylate similar substrates.
Tel1p and Mec1p are related structurally to a variety of mammalian proteins involved in DNA repair, maintenance of genomic stability, and telomere length regulation. Tel1p and Mec1p have a C-terminal domain of approximately 400 amino acids that is shared with a number of protein kinases including the mammalian proteins ATM, ATR, DNA-PK, and mTOR, and the yeast protein kinases Rad3 (Schizosaccharomyces pombe) and Tor1p (S. cerevisiae) (2, 7, 12). Although the C-terminal domain has sequence similarities to domains observed in phosphatidylinositol-3 kinases (13), all of the kinases described above phosphorylate proteins rather than lipids. Several of the mammalian kinases have overlapping substrate specificity; p53 is phosphorylated by both ATR (14) and DNA-PK (15) in addition to ATM (16). These same three kinases also are capable of phosphorylating the mammalian translation repressor, PHAS-I (Phosphorylated Heat and Acid Stable protein) (16, 17).
The sites phosphorylated by ATM and related mammalian proteins have been mapped for a number of their substrates. Many of these sites represent phosphorylation of serine or threonine followed by a glutamine (18). In addition, ATM (16, 17), DNA-PK (17), ATR (17), and Tor1p (19) are inhibited by wortmannin, a fungal metabolite that covalently modifies a lysine within the kinase domain of phosphoinositide 3-kinases (20).
The structural similarity of Tel1p and Mec1p with the ATM family of kinases also is reflected in similar cellular roles. For example, as observed with yeast mec1 cells, human AT cells are sensitive to DNA-damaging agents and have a defect in the checkpoint response (11, 21). In addition, AT cells, as observed with yeast tel1 cells, have chromosomes with short telomeres (21).
Based on the observations described above, it is assumed that Tel1p and Mec1p are protein kinases. In the experiments described in Results, we directly demonstrate that both Tel1p and Mec1p are wortmannin-sensitive protein kinases capable of in vitro phosphorylation of PHAS-I. In an independent study, Paciotti et al. (22) demonstrated recently that Mec1p phosphorylated the yeast checkpoint protein Ddc2p in vitro and in vivo.
Materials and Methods
Plasmid and Strain Constructions.
The genotypes of the yeast strains used in our study are shown in Table 1. These strains are isogenic with W303a (23, 24). We used the PCR epitope-tagging procedure developed by Schneider et al. (25) to prepare a yeast strain (KRY22) encoding a functional Tel1p with a hemagglutinin (HA) epitope. The plasmid pMPY-3xHA contains a URA3 gene flanked by 3xHA repeats. We PCR-amplified these sequences by using the primers TEL1 +2,339 HA forward (5′- CTCAAACAGGAACTTCTGCCATCAATTACTTCGAAGCCTCTTCAGAAGACACTACCAGGGAACAAAAGCTGG-3′) and TEL1 +2,448 HA reverse (5′-GGAGTGCAGAGGCTTCTGAAATCTACCTCC-AATTGTGTACGGACTATTATTCTGCTGTAGGGC-GAATTGGG-3′). The underlined segments are homologous to the pMPY-3xHA, and the remainder of the primers represent TEL1 sequences. These primers were designed to insert the 3xHA-URA3-3xHA sequences between amino acids 798 and 799 of Tel1p. The resulting DNA fragment was transformed into W303a, and Ura+ transformants were purified. Because the URA3 gene is flanked by duplicated 3xHA epitope sequences, recombination between these duplications results in Ura− derivatives that retain one copy of the 3xHA epitope. We selected for such recombinants by using 5-fluoro-orotate. By Southern analysis, we showed that strains with the HA-tagged allele of TEL1 (TEL1-HA) had telomeres that were elongated relative to those in tel1 strains but slightly shorter (≈50 bp) than those in wild-type strains. In other strains, Tel1p was epitope-tagged after amino acids 2 or 2,787, resulting in functional and nonfunctional Tel1 proteins, respectively (data not shown).
Table 1.
Strain names, strain constructions, and relevant genotypes
| Strain name | Strain construction or reference | Relevant genotype* |
|---|---|---|
| W303a | Ref. 23 | Wild-type |
| W1588-4c | Provided by R. Rothstein | RAD5 |
| JMY48 | Transformation of W303a with plasmid pKR5 | Wild-type, plasmid-borne TEL1-HA |
| JMY73 | Ref. 3 | sml1∷HIS3 |
| KRY22 | Transformation of W303a with PCR fragment to introduce HA tag into TEL1† | TEL1-HA |
| MD78 | Transformation of JMY73 with PCR fragment to introduce HA tag into MEC1† | sml1∷HIS3 MEC1-HA |
| MD85 | Two-step transplacement of MD78 with pMD81† | sml1∷HIS3 mec1-HA(KD) |
| JMY300-1a | Ref. 3 | α tel1∷ura3 mec1-21 |
| RCY278-5b | Provided by R. Craven | α RAD5‡ |
| JMY303 | Ref. 3 | a/α tel1∷ura3/TEL1 mec1-21/MEC1 |
| sml1∷HIS3/SML1 | ||
| JMY303-2a | Spore of JMY303 | tel1∷ura3 |
| KRY20a | Ref. 3 | ura3Δ∷HIS3 tel1∷ura3 ade3 |
| JMY103 | Transformation of KRY20a with plasmid pJM8 | tel1∷ura3, plasmid-borne tel1-HA(KD) |
| JMY303-1c | Spore of JMY303 | α mec1-21 |
| JMY303-3a | Spore of JMY303 | α sml1∷HIS3 tel1∷ura3 |
| JMY303-8a | Spore of JMY303 | α tel1∷ura3 |
| JMY303-8d | Spore of JMY303 | mec1-21 sml1∷HIS3 |
| JMY312 | Cross of MD78 and JMY303-8a | a/α tel1∷ura3/TEL1 sml1∷HIS3/SML1 |
| MEC1-HA/MEC1 | ||
| JMY312-6d | Spore derived from JMY312 | sml∷HIS3 tel1∷ura3 MEC1-HA |
| JMY312-8c | Spore derived from JMY312 | sml∷HIS3 tel1∷ura3 MEC1-HA |
| JMY313 | Cross of MD85 and JMY303-3a | a/α mec1-HA(KD)/MEC1 tel1∷ura3/TEL1 |
| sml1∷HIS3/sml1∷HIS3 | ||
| JMY313-9a | Spore derived from JMY313 | mec1-HA(KD) sml1∷HIS3 tel1∷ura3 |
| JMY314 | Cross of RCY278-5b and MD85 | a/α mec1-HA(KD)/MEC1 rad5-535/RAD5 |
| sml1∷HIS3/SML1§ | ||
| JMY314-3d | Spore derived from JMY314 | mec1-HA(KD) sml1∷HIS3 rad5-535 |
| JMY314-5a | Spore derived from JMY314 | α mec1-HA(KD) sml1∷HIS3 RAD5 |
Most of the strains in the study are isogenic (except for changes introduced by transformation) with W303a (a leu2-3,112 his3-11,15 ura3-1 ade2-1 trp1-1 rad5-535 can1-100). All genes that differ from those of the progenitor genotype are listed.
Details of these constructions are in Materials and Methods.
This strain also contains an insertion of URA3 near the telomere of chromosome XVL (TELXVL∷URA3; [24]).
This diploid is heterozygous for the TELXVL∷URA3 marker.
Strains with plasmid-borne TEL1 genes also were used in our study. We previously constructed plasmids with the wild-type TEL1 gene or a tel1 gene with four mutant substitutions in the kinase domain (2). The plasmid pSP21 has the wild-type TEL1 gene inserted as a KpnI fragment into the SmaI site of the ADE3- and URA3-containing high-copy-number vector pTSV31A (2). The plasmid pSP-KD is identical to pSP21 except that the TEL1 gene has four mutant substitutions in the kinase domain (G–D at position 2,611, D–A at position 2,612, N–K at position 2,616, and D–E at position 2,631). Strains with pSP-KD have short telomeres. To prepare a plasmid-borne HA-tagged TEL1 gene, we transformed NheI- and BsiWI-treated pSP21 into the strain with the HA-tagged chromosomal copy of TEL1 (KRY22). The resulting gap-repair event resulted in a plasmid (pKR5) with a plasmid-borne HA-tagged TEL1 gene; Southern analysis indicated that this plasmid complemented the short-telomere phenotype of tel1 strains, indicating that the plasmid-borne TEL1-HA gene was functional.
We also constructed a plasmid with an HA-tagged TEL1 gene with mutations in the kinase domain. The plasmid pKR5 was treated with NheI and religated to delete an NheI fragment containing the kinase domain and DNA sequences downstream of TEL1 (pJM7). A 1.4-kb DNA fragment that included the NheI sites and the kinase region was PCR-amplified from pSK-KD by using the primers TEL1 +7,400 forward (5′-GTAATGACGATTTGAGACAAGATGCAATTATGG-3′) and TEL1 +8,841 reverse (5′-CGATAACAGAGCTAGCTTTATTCCATTAGACAACTT-3′). This DNA fragment was treated with NheI and inserted into the NheI site of pJM7 to generate the plasmid pJM8, which contains the tel1-HA(KD) allele of TEL1.
Similar methods were used to construct a yeast strain (MD78) encoding an HA-tagged Mec1p. The primers used to PCR-amplify pMPY-3xHA sequences were MEC1 +2,161 HA forward (5′-CTCGATATTTTCCAGAGATATATCATCCCTTATGCAATTATTCAATATAGGGAACAAAAGCTGGAGCTC-3′) and MEC1 +2,259 HA reverse (5′-TGTATCGCCATCACACATAATCTTAGCAATTTCACTTAGCACATCGCTCTTCTGTAGGGCGAATTGGGTACC-3′). These primers are designed to insert the epitope between amino acids 736 and 737 of Mec1p. The PCR-generated DNA fragment was used to transform JMY73 (an sml1 derivative of W303a) and, as described above, in two steps we obtained a strain with an HA-tagged version of MEC1 (MEC1-HA). This strain (MD78) had wild-type sensitivity to hydroxyurea, as would be expected if the HA-tagged Mec1p was fully functional.
A yeast strain with an HA-tagged version of Mec1p with mutant substitutions in the kinase domain also was generated. The plasmid pMD81 was treated with KpnI and a two-step transplacement was performed with strain MD78 (MEC1-HA sml1∷HIS3). The plasmid pMD81 had a 1.2-kb fragment containing the kinase domain of MEC1 with two mutant substitutions, D-to-A at position 2,224 and N-to-K at position 2,229. This plasmid was generated by a series of three PCR reactions. Primers A [5′-GACACTATAGTGATCACACTAGAGTCTAACTCAATGAG-3′, MEC1 sequences upstream of the introduced mutations plus an introduced BclI site (underlined)] and B [5′-CTTTACCCGTCTGTATATCTAGTAATATCTTTTCACAGTGCCTAGCACCTAGACCTAATATATGGCCAAC-3′; MEC1 sequences with two introduced mutations in kinase domain (underlined)] were used in one PCR, and primers C [5′-GTTGGCCATATATTAGGTCTAGGTGCTAGGCACTGTGAAAAGATATTACTAGATATACAGACGGGTAAAG-3′, the complement of primer B with the introduced mutations (underlined)] and D [5′-TTCAGACAGGTGATCACGTCTGTTGCCGAAAATGGTG-3′, containing MEC1 sequences downstream of the introduced mutations plus an introduced BclI site (underlined)] were used in a second PCR. The products of these reactions then were used as templates for a third reaction by using primers A and D. The net result of these manipulations was a 1.2-kb fragment containing MEC1 sequences with the mutant alterations near the middle of the fragment. This fragment was treated with BclI and inserted into the BamHI site of YIp5 to create pMD81.
Kinase Assays/Western Blots.
To isolate protein for the kinase assays and the Western blot, we grew strains containing HA-tagged Te1lp or Mec1p to mid-log phase (OD600 = 0.5). The preparation of protein extracts, immunoprecipitations, and kinase assays were done as described for the Tor1p kinase (19). In brief, protein extracts were prepared by sonication of oxalyticase-generated spheroplasts (Enzogenetics, Corvallis, OR). To extracts containing 2 mg of total protein in lysis buffer (50 mM Hepes, pH 7.4/100 mM KCl/0.1 mM EDTA/0.2% Tween-20/1 mM DTT/20 μM β-glycerophosphate/20 μM NaF/100 μM Na3VO4/1 mM PMSF), we added 1 μl of HA.11 antibody (Babco, Richmond, CA) and incubated the mixture on a rotator at 4° for 2 h. Protein A sepharose CL-4B beads (45 μl; equilibrated in lysis buffer) were added and the incubation continued for 2 h. The beads then were washed and resuspended in 45 μl of kinase buffer (10 mM Hepes, pH 7.4/50 mM NaCl/10 mM MnCl2/1 mM DTT). Kinase reactions contained 15 μl of washed beads, 11.5 μl of kinase buffer, 1.5 μl of 200 μM unlabeled ATP, 10 μCi of γ32P-labeled ATP (4,500 Ci/mmol; 1 Ci = 37 GBq), and 1 μl of PHAS-I (1 μg/μl; Stratagene). The reactions were performed at 30° for 30 min. In studies involving wortmannin, the beads with the immunoprecipitated proteins were incubated for 30 min at room temperature with wortmannin that was diluted in kinase buffer lacking DTT. Kinase assays with the treated beads then were performed as described above.
Western Blot Analysis.
The relative levels of Tel1p-HA and Mec1p-HA were examined by Western blots. The HA-tagged proteins were immunoprecipitated from 8 mg of total protein as described above. Washed beads (45 μl) containing the immunoprecipitated proteins were resuspended in SDS/PAGE loading buffer, heated at 95° for 10 min, and then loaded onto a 5% SDS/PAGE gel. After electrophoresis, the proteins were transferred onto a Hybond-C membrane (Amersham Pharmacia). The membrane was preincubated with TBST buffer (20 mM Tris, pH 7.8/150 mM NaCl/0.1% Tween-20) containing 5% (vol/vol) powdered milk for 2 h at 4°. The HA.11 antibody was then added (1:1,000 dilution) in fresh TBST/powdered milk and the incubation continued for 2 h at 4°. The membrane then was washed three times with TBST and incubated with a 1:1,000 dilution (in TBST/powdered milk buffer) of the anti-mouse secondary antibody-horseradish peroxidase conjugate (Amersham Pharmacia) for 1 h at 4°. The blot then was washed three times in TBST, developed with the enhanced chemiluminescence reagent ECL (Amersham Pharmacia), and exposed to film.
Southern Analysis of Telomere Length.
Details of this analysis have been published previously (3). Yeast DNA was treated with PstI and the resulting fragments were separated by gel electrophoresis. The fragments were transferred to Hybond N+ nylon membranes and hybridized to a probe (derived from pYT14) that contained telomeric DNA and a portion of the Y′ subtelomeric repeat.
Sequence Analysis of Mec1–21.
The MEC1 gene was sequenced (using a set of 11 primer pairs spaced evenly through the gene) in two strains (JMY303–2b and JMY303–6c; ref. 3) containing the mec1–21 mutation (6, 8) and the isogenic wild-type strain W303a. A single mutant substitution was detected, a G-to-A at position 2,644 (G882S).
Results
Tel1p- and Mec1p-Dependent Phosphorylation of PHAS-I.
To investigate whether Tel1p and Mec1p are protein kinases, we constructed strains in which the Tel1p or Mec1p protein was tagged with an HA epitope. In the strain JMY48 (Table 1), the HA-tagged TEL1 gene is on a high-copy plasmid. tel1 strains with this plasmid had telomeres of nearly wild-type length, indicating that the HA-tagged version of Tel1p was functional (K. Ritchie and T.D.P., unpublished data). We used antibodies directed against the HA tag to immunoprecipitate Tel1p (details in Materials and Methods). The immunoprecipitated Tel1p efficiently phosphorylated the protein substrate PHAS-I (Fig. 1a, lane 1), a target of other members of the ATM-kinase family. No kinase activity was detected when a mock immunoprecipitation (no HA-specific antibody) was performed in extracts derived from the same strain (Fig. 1a, lane 2). More importantly, no phosphorylation of PHAS-I was observed when immunoprecipitates were derived from a strain (JMY103) in which the plasmid-borne HA-tagged TEL1 gene had four mutant substitutions (G2611D, D2612A, N2616K, and D2631E) in the kinase domain (Fig. 1a, lane 3). We have shown that these mutant substitutions result in a short-telomere phenotype (2). Kinase activity also was detectable when TEL1-HA was present as a single chromosomal copy (KRY22; Fig. 1a, lane 4).
Figure 1.

Kinase activities of Tel1p and Mec1p. (a) Kinase activity of immunoprecipitated HA-tagged Tel1p on PHAS-I. Products of a kinase reaction using labeled ATP were analyzed by SDS/PAGE. Strains from which immunoprecipitates were derived: lane 1, JMY48 (high-copy-number plasmid with TEL1-HA); lane 2, mock immunoprecipitate (no anti-HA antibody) from JMY48; lane 3, JMY103 [high-copy number plasmid with tel1-HA(KD)]; lane 4, KRY22 (TEL1-HA in single chromosomal copy); and lane 5, JMY303–2a (tel1 strain, no epitope-tagged Tel1p). (b) Kinase activity of immunoprecipitated HA-tagged Mec1p on PHAS-I. Strains from which immunoprecipitates were derived: lane 1, JMY312–8c (tel1 sml1∷HIS3 MEC1-HA); lane 2, JMY313–9a [tel1 mec1-HA(KD) sml1∷HIS3]; and lane 3, mock immunoprecipitate (no HA-antibody) from JMY312–8c. (c) Western analysis of HA-tagged Tel1p and Mec1p proteins. Immunoprecipitated HA-tagged Tel1p or Mec1p were examined by Western analysis using HA.11 antibody and a secondary antibody-horseradish peroxidase conjugate. Lane 1, JMY48 (high-copy number plasmid with TEL1-HA); lane 2, JMY103 [high-copy number plasmid with tel1-HA(KD)]; lane 3, JMY312–8c (MEC1-HA); and lane 4, JMY313–9a [mec1-HA(KD)]. Wt, wild type; kd, kinase dead.
A comparable series of experiments also were performed with HA-tagged Mec1p. When immunoprecipitates were prepared from JMY312–8c (MEC1-HA tel1∷ura3 sml1∷HIS3) and used in a kinase reaction, we observed phosphorylation of PHAS-I (Fig. 1b, lane 1). Because the immunoprecipitates are derived from a strain with the tel1 mutation, this activity cannot represent coimmunoprecipitated Tel1p kinase activity. Most of the kinase activity (>90%) was lost when extracts were prepared from a strain [JMY313–9a; mec1-HA(KD) tel1∷ura3 sml1∷HIS3] in which the HA-tagged MEC1 gene contained two mutations in conserved amino acids in the kinase domain (D2224A and N2229K; Fig. 1b, lane 2). As described below, strains with these mutations have the same phenotype as null mec1 mutants.
The kinase activity observed in immunoprecipitates containing HA-tagged Tel1p and Mec1p, and the loss of this activity in strains with point mutations in the kinase domains of these proteins, strongly argues that Tel1p and Mec1p are protein kinases. To ensure that the loss of kinase activity observed in the immunoprecipitates derived from the tel1-HA(KD) and mec1-HA(KD) strains did not reflect unstable or poorly immunoprecipitable proteins, we examined the relative levels of the HA-tagged wild-type and kinase-dead versions of Tel1p and Mec1p. No significant differences were detectable (Fig. 1c).
Because the ATM protein kinase family members are sensitive to the phosphatidylinositol-3 kinase-inhibitor wortmannin (16, 17, 19), we examined the sensitivity of the Tel1p and Mec1p kinase activities to this drug. Both Tel1p and Mec1p were sensitive, although Tel1p was less sensitive than Mec1p (Fig. 2). The sensitivity of Tel1p was intermediate to that observed previously for ATM and ATR, whereas the sensitivity of Mec1p was roughly similar to that seen for ATM (17).
Figure 2.

Wortmannin sensitivity of the Tel1p and Mec1p kinases. Immunoprecipitates from strains with either TEL1-HA (JMY48) or MEC1-HA (JMY312–8c) were treated with wortmannin (details in Materials and Methods) before kinase reactions were performed. Activities are normalized to a value of 1 for immunoprecipitates incubated in the absence of wortmannin. Tel1p and Mec1p activities are represented by ● and □, respectively. Error bars indicate standard deviations (total of three experiments).
Target(s) of Tel1p Kinase in PHAS-I.
The commercially available PHAS-I substrate used in our experiments contained an N-terminal 20-aa tag (MGSSHHHHHHSSGLVPRGSH) in addition to the 117 aa of the rat PHAS-I protein (26). Because this tag contains a thrombin-cleavage site (after Arg-17), we determined whether the tag was necessary for phosphorylation and whether there was a phosphorylation site within the tag. As shown in Fig. 3a, if the intact PHAS-I is phosphorylated and then cleaved by thrombin, most of the radioactivity is retained in the 120-aa proteolytic fragment, indicating that at least some of the phosphorylation sites are not within the tag. As shown in Fig. 3b, if the tagged PHAS-I protein is cleaved before phosphorylation, it is a very poor substrate for the Tel1p kinase. These results indicate that the 20-aa tag is required for PHAS-I to act as a good substrate for Tel1p, but the tag is not a unique target site for the kinase. One possibility is that the highly charged tag helps PHAS-I associate with Tel1p. Alternatively, the tag might allow PHAS-I to form a different tertiary or quaternary structure, creating a better Tel1p substrate.
Figure 3.
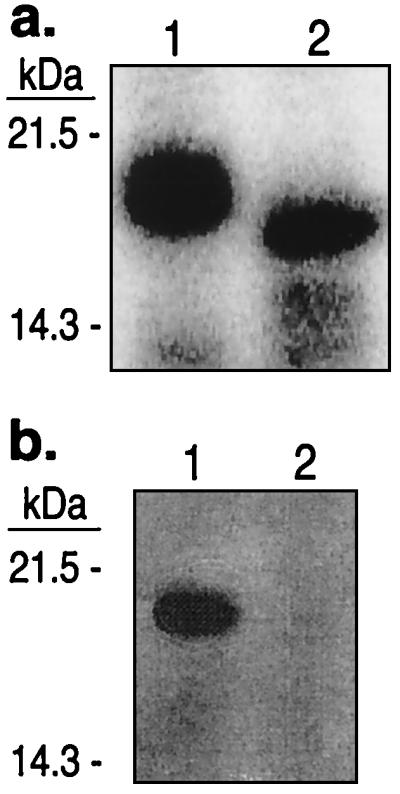
Requirement of 20-aa “tag” (MGSSHHHHHHSSGLVPRGSH) for PHAS-I to function as a substrate for the Tel1p kinase. (a) PHAS-I was phosphorylated by an immunoprecipitate derived from a strain with TEL1-HA (JMY48); the final two washes of the immunoprecipitate were done in the absence of PMSF. A portion of the sample was treated with thrombin (Roche Molecular Biochemicals) to remove the N-terminal 17 aa; both the untreated (lane 1) and treated (lane 2) samples were examined by gel electrophoresis. (b) Kinase reactions with immunoprecipitated Tel1p were done with two samples. One sample (lane 1) contained untreated PHAS-I. The sample in lane 2 was treated with thrombin before performing the kinase reaction. Thrombin was inactivated by addition of PMSF.
To map the phosphorylation sites within PHAS-I, we sent a gel-purified Tel1p-phosphorylated sample of PHAS-I to the Harvard Microchemistry Facility (Boston) for analysis. Tryptic digest of these proteins were analyzed by microcapillary reverse-phase HPLC nano electrospray tandem mass spectrometry on a Finnigan LCQ quadrupole ion trap mass spectrometer. This analysis detected phosphorylation of Ser-131 in the tagged version of PHAS-I; the sequence of the peptide with the phosphorylated serine is RAGGEES*QFEMDI (asterisk indicates the phosphorylated serine). Because this serine is followed by a glutamine, Tel1p recognizes serine in an SQ motif, as observed for most of the other ATM kinase family members.
Several other points concerning the phosphorylation sites recognized by Tel1p and Mec1p should be mentioned. First, our analysis does not exclude the possibility of other targets in PHAS-I in addition to Ser-131. For example, there are two other SQ sites in PHAS-I in addition to Ser-131. Second, our analysis does not exclude the possibility of Tel1p and Mec1p recognizing other motifs, such as TQ, in other proteins.
Requirement of the Kinase Domains of Tel1p and Mec1p for in Vivo Functions.
In another study (2), we showed that strains with four mutations in the kinase domain of TEL1 (G2611D, D2612A, N2616K, and D2631E) had short telomeres. This conclusion was confirmed for the HA-tagged version of TEL1 [tel1-HA(KD)] (Fig. 4a, lanes 3 and 4). We also showed that the mec1–21 allele resulted in slightly shortened telomeres, but strains with both the mec1–21 and sml1 mutation had telomeres of wild-type length (3); mec1–21 is a hypomorphic allele that is viable in SML1 strains (6, 8). Because (as described below) mec1-HA(KD) is inviable unless SML1 is mutated, we monitored telomere length in the double-mutant mec1-HA(KD) sml1. Telomeres were of wild-type length in the double mutant (Fig. 4a, lane 7). Strains with the genotype tel1 mec1-HA(KD) sml1 had telomeres that were slightly shorter than those observed in the tel1 single-mutant strains (Fig. 4a, lane 8), indicating that the mec1-HA(KD) allele affects telomere length.
Figure 4.

Phenotypes of strains with mutations in the kinase domains of Tel1p and Mec1p. (a) Assay for telomere length. DNA was isolated from strains of various genotypes and treated with PstI. The resulting fragments were separated by gel electrophoresis and hybridized to a telomeric-specific probe as described in Materials and Methods. The dispersed band near the bottom of the gel represents the terminal-DNA fragments of chromosomes with Y′-containing telomeres. The strain name and genotype (indicated in parentheses) in each lane are: lanes 1 and 5, W303a (wild-type); lane 2, JMY303–2a (tel1); lanes 3 and 4, JMY103 [tel1 + plasmid-borne TEL1-HA(KD)]; lane 6, JMY73 (sml1); lane 7, JMY314–3d [mec1-HA(KD) sml1 rad5]; and lane 8, JMY313–9a [mec1-HA(KD) sml1 tel1∷ura3]. (b) Assay for growth. Spore colonies derived from the diploids JMY313 and JMY314 were streaked onto plates containing rich growth medium and incubated at 30o for 2 days. The two tel1 mec1-HA(KD) sml1 strains are JMY313–3a and JMY313–9a, and the two mec1-HA(KD) sml1 strains are JMY314–3d and JMY314–5a. (c) Assay of response to DNA-damaging agents. Serial dilutions of six strains were prepared on four plates containing rich growth medium (yeast extract/peptone/dextrose). One plate was untreated without any DNA-damaging agent; one was treated for 10 sec with UV light derived from a germicidal lamp; one contained 10 mM hydroxyurea (HU); and one contained 2 μg/ml streptonigrin. The strains used in the study (row 1 at the tops of the plates) were: row 1, W303a; row 2, W1588–4c (identical to W303a except RAD5 instead of rad5–535); row 3, JMY303–1c; row 4, JMY303–8d; row 5, JMY314–3d; and row 6, JMY314–5a (identical to JMY314–3d except RAD5). Cells were grown for 2 days at 30°C.
Null mec1 mutations result in lethality, although this lethality can be suppressed by the sml1 (suppressor of mec1 lethality) mutation (10). To determine whether the mec1-HA(KD) allele resulted in lethality, we sporulated a diploid (JMY314) that was heterozygous for mec1-HA(KD) and sml1∷HIS3. Of 15 tetrads examined, 5 had four viable spores, 3 had three viable spores, and 7 had two viable spores, which was the pattern of spore viability expected for a synthetic lethal interaction between unlinked mutations. Of the 43 viable spores in these tetrads, 13 were MEC1 SML1, 17 were MEC1 sml1∷HIS3, 13 were mec1-HA(KD) sml1∷HIS3, and none were mec1-HA(KD) SML1. We conclude that the mec1-HA(KD) allele, like mec1 null alleles, results in lethality, but this lethality can be suppressed by sml1.
The mec1–21 allele is synthetically senescent with tel1; this senescent phenotype is likely to reflect a defect in telomere length regulation (3). We examined the growth of spore colonies (derived from JMY313 and JMY314) of the mec1-HA(KD) sml1 and tel1 mec1-HA(KD) sml1 genotypes. As shown in Fig. 4b, the triple-mutant spores grew very poorly even in the first subcloning. Subsequent subclonings exhibited a senescent phenotype similar to that observed previously for tel1 mec1–21 sml1 strains (3).
Strains with mec1 mutations are sensitive to a number of DNA-damaging agents and drugs such as hydroxyurea that cause S phase arrest (4–8). As shown in Fig. 4c, the mec1-HA(KD) strain was sensitive to UV light, hydroxyurea, and the radiomimetic drug streptonigrin. The mec1-HA(KD) sml1 strain was considerably more sensitive to UV and streptonigrin than were the mec1–21 or the mec1–21 sml1 strains. Because the mutant substitution in mec1–21 is located outside of the kinase domain (G882S), the strain with this substitution is not necessarily kinase-deficient. It is interesting to note that the presence of the sml1 mutation partially rescues the sensitivity of the mec1–21 mutants to UV irradiation and streptonigrin. Zhao et al. (10) found that the sml1 mutation did not rescue the damage sensitivity in a mec1–1 strain. In summary, for several different phenotypes, mec1-HA(KD) strains resemble strains with mec1-null alleles, indicating that the kinase activity of Mec1p is important for its biological functions.
Discussion
Our analyses show that Tel1p and Mec1p are protein kinases. The sensitivity of their kinase activity to wortmannin and the demonstration that Tel1p is capable of phosphorylating serine in an SQ motif indicate that the kinase activities of Tel1p and Mec1p are related to those of other ATM family members. In addition, we found that mutant substitutions in the kinase domains of Tel1p and Mec1p result in phenotypes similar to those found for null tel1 and mec1 alleles, indicating that the kinase activity is essential for the biological function of these proteins.
An important extension of these studies is to determine the in vivo protein targets of Tel1p and Mec1p and the mechanism by which phosphorylation of these targets is necessary for telomere-length regulation and checkpoint functions. Because Tel1p functions in the same pathway of telomere-length regulation as proteins of the MRX complex (Mre11p/Rad50p/Xrs2p; ref. 9), these proteins are obvious target candidates. We have found that Xrs2p is a target for in vitro phosphorylation of Tel1p (J.C.M., K. Trujillo, P. Sung, and T.D.P., unpublished data), but have not observed Tel1p-dependent phosphorylation of Xrs2p in vivo (K. Ritchie and T.D.P., unpublished data). Based on the Mec1p- and/or Tel1p-dependent phosphorylation observed in vivo for other checkpoint proteins (11), these proteins are obvious targets for direct phosphorylation by Tel1p and/or Mec1p. By using immunoprecipitates of epitope-tagged Mec1p, Paciotti et al. (22) showed that Ddc2p undergoes Mec1p-dependent phosphorylation in vivo and in vitro. They also found, as we did, that mutations in the kinase domain of MEC1 resulted in lethality unless they were suppressed by the sml1 mutation.
As discussed in the Introduction, Tel1p and Mec1p have partially overlapping functions in two different pathways: telomere-length regulation and DNA repair. The most straightforward interpretation of this functional overlap is that it reflects an overlap in the ability of these proteins to phosphorylate their targets. For example, DNA damage-induced phosphorylation of Rad53p is Mec1p-dependent, but an additional plasmid-borne copy of TEL1 can restore phosphorylation of Rad53p in a mec1 strain (8). The Tel1p and Mec1p share very limited homology outside of the C-terminal kinase domain. It is likely that the target preferences of the two proteins are not based on differences in the kinase domains, but rather on differences in the N-terminal regions where, presumably, the different targets are tethered to Tel1p and Mec1p to stabilize interactions with the kinase domain. Because Tel1p requires a highly charged 20-aa tag to efficiently phosphorylate PHAS-I, it will be interesting to determine whether other targets of Tel1p might have a highly positively charged domain required for its interaction with Tel1p.
The Mec1p-dependent phosphorylation of Rad53p (8) and other protein targets is induced by DNA damage. It would be of interest to determine whether the kinase activity of Mec1p is elevated in strains treated with various DNA-damaging agents. One common feature in the metabolism of telomeres and the repair of DNA damage is that both mechanisms involve DNA ends, directly or indirectly. Because of this consideration, and because Tel1p and Mec1p share homology with DNA-PK, the effect of DNA ends on the kinase activity of these proteins should also be examined.
Acknowledgments
We thank M. Cardenas, J. Heitman, R. Abraham, Y. Sanchez, and J. Lawrence for reagents and/or for useful suggestions, W. Lane for information concerning phosphorylation sites, K. Ritchie for epitope tagging of Tel1p, and M. Dominska for help with strain constructions. We thank R. Craven and K. Ritchie for advice and/or comments on the manuscript. The research was supported by National Institutes of Health Grants GM52319 and GM24110 (to T.D.P.). J.C.M. was supported by a National Institutes of Health Genetics Training grant (5 T32 GM07092–26).
Abbreviations
- PHAS-1
phosphorylated heat and acid stable protein
- HA
hemagglutinin
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.250475697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.250475697
References
- 1.Lustig A J, Petes T D. Proc Natl Acad Sci USA. 1986;83:1398–1402. doi: 10.1073/pnas.83.5.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greenwell P G, Kronmal S L, Porter S E, Gassenhuber J, Obermaier B, Petes T D. Cell. 1995;82:823–829. doi: 10.1016/0092-8674(95)90479-4. [DOI] [PubMed] [Google Scholar]
- 3.Ritchie K B, Mallory J M, Petes T D. Mol Cell Biol. 1999;19:6065–6075. doi: 10.1128/mcb.19.9.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weinert T A, Kiser G L, Hartwell L H. Genes Dev. 1994;8:652–665. doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]
- 5.Kato R, Ogawa H. Nucleic Acids Res. 1994;22:3104–3112. doi: 10.1093/nar/22.15.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen J B, Zhou Z, Siede W, Friedberg E C, Elledge S J. Genes Dev. 1994;8:2416–2428. doi: 10.1101/gad.8.20.2401. [DOI] [PubMed] [Google Scholar]
- 7.Morrow D M, Tagle D A, Shiloh Y, Collins F S, Hieter P. Cell. 1995;8:831–840. doi: 10.1016/0092-8674(95)90480-8. [DOI] [PubMed] [Google Scholar]
- 8.Sanchez Y, Desany B, Jones W J, Liu Q, Wang B, Elledge S J. Science. 1996;271:357–360. doi: 10.1126/science.271.5247.357. [DOI] [PubMed] [Google Scholar]
- 9.Ritchie K B, Petes T D. Genetics. 2000;155:475–479. doi: 10.1093/genetics/155.1.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao X, Muller E G D, Rothstein R. Mol Cell. 1998;2:329–340. doi: 10.1016/s1097-2765(00)80277-4. [DOI] [PubMed] [Google Scholar]
- 11.Lowndes N F, Murguia J R. Curr Opin Genet Dev. 2000;10:17–25. doi: 10.1016/s0959-437x(99)00050-7. [DOI] [PubMed] [Google Scholar]
- 12.Savitsky K, Sfez S, Tagle D A, Ziv Y, Sartiel A, Collins F S, Shiloh Y, Rotman G. Hum Mol Genet. 1995;4:2025–2032. doi: 10.1093/hmg/4.11.2025. [DOI] [PubMed] [Google Scholar]
- 13.Toker A, Cantley L C. Nature (London) 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- 14.Tibbetts R S, Brumbaugh K M, Williams J M, Sarkaria J N, Cliby W A, Shieh S-Y, Taya Y, Prives C, Abraham R T. Genes Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lees-Miller S P, Sakaguchi K, Ullrich S J, Appella E, Anderson C W. Mol Cell Biol. 1992;12:5041–5049. doi: 10.1128/mcb.12.11.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banin S, Moyal L, Shieh S-Y, Taya Y, Anderson C W, Chessa L, Smorodinsky N I, Prives C, Reiss Y, Shiloh Y, et al. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 17.Sarkaria J N, Tibbetts R S, Busby E C, Kennedy A P, Hill D E, Abraham R T. Cancer Res. 1998;58:4375–4382. [PubMed] [Google Scholar]
- 18.Kim S-T, Lim D-S, Canman C E, Kastan M B. J Biol Chem. 1999;274:37538–37643. doi: 10.1074/jbc.274.53.37538. [DOI] [PubMed] [Google Scholar]
- 19.Alarcon C M, Heitman J, Cardenas M E. Mol Biol Cell. 1999;10:2531–2546. doi: 10.1091/mbc.10.8.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wymann M P, Bulgarelli-Leva G, Zvelebil M J, Pirola L, Vanhaesebroeck B, Waterfield M D, Panayotou G. Mol Cell Biol. 1996;16:1722–1733. doi: 10.1128/mcb.16.4.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shiloh Y. Annu Rev Genet. 1997;31:635–662. doi: 10.1146/annurev.genet.31.1.635. [DOI] [PubMed] [Google Scholar]
- 22.Paciotti V, Clerici M, Lucchini G, Longhese M P. Genes Dev. 2000;14:2046–2059. [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas B J, Rothstein R. Cell. 1989;56:619–630. doi: 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
- 24.Craven R J, Petes T D. Mol Cell Biol. 2000;20:2378–2384. doi: 10.1128/mcb.20.7.2378-2384.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider B L, Seufert W, Steiner B, Yang Q H, Futcher A B. Yeast. 1995;11:1265–1274. doi: 10.1002/yea.320111306. [DOI] [PubMed] [Google Scholar]
- 26.Hu C, Pang S, Kong X, Velleca M, Lawrence J C. Proc Natl Acad Sci USA. 1994;91:3730–3734. doi: 10.1073/pnas.91.9.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]