

Abstract
Developmental hearing impairments compromise sound discrimination, speech acquisition, and cognitive function; however, the adjustments of functional properties in the primary auditory cortex (A1) remain unknown. We induced sensorineural hearing loss (SNHL) in developing gerbils and then reared the animals for several days. The intrinsic membrane and synaptic properties of layer 2/3 pyramidal neurons were subsequently examined in a thalamocortical brain slice preparation with whole-cell recordings and electron microscopic immunocytochemistry. SNHL neurons displayed a depolarized resting membrane potential, an increased input resistance, and a higher incidence of sustained firing. They also exhibited significantly larger thalamocortically and intracortically evoked excitatory synaptic responses, including a greater susceptibility to the NMDA receptor antagonist AP-5 and the NR2B subunit antagonist ifenprodil. This correlated with an increase in NR2B labeling of asymmetric synapses, as visualized ultrastructurally. Furthermore, decreased frequency and increased amplitude of miniature EPSCs (mEPSCs) in SNHL neurons suggest that a decline in presynaptic release properties is compensated by an increased excitatory response. To verify that the increased thalamocortical excitation was elicited by putative monosynaptic connections, minimum amplitude ventral medial geniculate nucleus-evoked EPSCs were recorded. These minimum-evoked responses were of larger amplitude, and the NMDAergic currents were also larger and longer in SNHL neurons. These findings were supported by significantly longer AP-5-sensitive durations and larger amplitudes of mEPSCs. Last, the amplitudes of intracortically evoked monosynaptic and polysynaptic GABAergic inhibitory synaptic responses were significantly smaller in SNHL neurons. These alterations in cellular properties after deafness reflect an attempt by A1 to sustain an operative level of cortical excitability that may involve homeostatic mechanisms.
Keywords: homeostasis, synaptic plasticity, GABAA, NMDA receptor, mEPSC, disuse
Introduction
Hearing impairments produce significant changes in the human cortex and may permanently diminish auditory perceptual skills and language acquisition (Mody et al., 1999; Vernon-Feagans, 1999; Psarommatis et al., 2001; Kidd et al., 2002; Emmorey et al., 2003; Iverson, 2003). The effects of sensorineural hearing loss (SNHL) are particularly severe in children with the longest periods of auditory deprivation (Sharma et al., 2002). Recordings from hearing-impaired animals have also revealed many profound changes in auditory processing (Syka, 2002). Unilaterally deafened animals display increased acoustically evoked activity in the ipsilateral inferior colliculus (IC) and auditory cortex, likely attributable to a change in the balance between existing excitatory and inhibitory inputs (Kitzes and Semple, 1985; McAlpine et al., 1997; Mossop et al., 2000). Processing after SNHL has also been examined in cats using electrical stimulation with cochlear prosthetic devices. Although some findings suggest that auditory deprivation leads to decreased synaptic drive, particularly to layer 5 (Klinke et al., 1999; Kral et al., 2000), there are also signs of increased excitability. For example, electrode-evoked thresholds are lower, spatial tuning curves are broader, and cochleotopy appears imprecise after 2 weeks of deafness (Raggio and Schreiner, 1999, 2003).
These in vivo studies do not distinguish between changes that may have occurred in the brainstem (Snyder et al., 2000; Moore et al., 2002) and those that have occurred locally in the cortex, nor do they identify alterations to synaptic and intrinsic membrane properties that may attend deafness. For example, our previous work in the auditory brainstem shows that unilateral hearing loss not only decreases synaptic inhibition but also enhances NMDA receptor (NMDA-R)-mediated excitation (Kotak and Sanes, 1996). Similarly, in the inferior colliculus, bilateral SNHL decreases the inhibitory postsynaptic conductance and increases EPSC amplitude (Vale and Sanes, 2002). These findings are consistent with experiments performed on invertebrate circuits and dissociated cortical neurons showing that synapses and ion channels are regulated by electrical activity (Marder and Prinz, 2002). In general, such changes tend to resist the manipulation. Thus, when cultures are grown in activity blockers, it is found that excitatory synaptic and sodium currents increase and inhibitory synaptic and potassium currents decrease (Rao and Craig, 1997; Desai et al., 1999; Murthy et al., 2001; Burrone et al., 2002; Kilman et al., 2002).
The present studies were designed to assess both intrinsic and synaptic properties in the primary auditory cortex (A1) after bilateral hearing loss. Only one previous study has examined alterations in biophysical properties of cochlear nucleus (CN) neurons after deafness (Francis and Manis, 2000); none have examined A1 neurons. By manipulating hearing in vivo and examining neuronal properties in the auditory thalamocortical brain slice preparation (Cruikshank et al., 2002), we provide evidence that membrane and synaptic properties become scaled to favor excitability. These results point to a homeostatic response after cochlear damage and may significantly affect cortical processing.
Materials and Methods
Surgery for SNHL. All protocols were reviewed and approved by the New York University Institutional Animal Care and Use Committee. Cochlear ablations were performed using procedures similar to those described previously (Sanes et al., 1992; Vale and Sanes, 2002). Gerbil (Meriones unguiculatus) pups at postnatal day 10 (P10) were anesthetized with the halogenated ethyl methyl ether methoxyflurane (Metofane). Anesthetic induction occurred within 10 min and produced complete elimination of responses to nociceptive stimuli. A small hole was made in the cochlear wall, and the contents were rapidly removed with a forceps. A piece of Gelfoam was then placed in the cavity, and the wound was closed. Ablations were performed bilaterally. After surgery, animals were warmed on a heating pad and returned to the litter when respiration and motor activity had recovered. Animals were reared for 3-13 d with their parents under conditions identical to those for normal pups. Before each brain slice experiment, successful ablations were confirmed by opening the inner wall of the cochlea under a dissecting microscope and observing the absence of cochlear tissue and the presence of a Gelfoam insert. Thus, the recordings were not performed blind. The age of surgery was chosen based on the work of Tierney and Moore (1997) showing that anteroventral cochlear nucleus cell number is unaffected by cochlear ablation at P9.
Thalamocortical brain slice recordings and pharmacology. Brain slices were generated from P13-P23 animals in a manner similar to that reported for the mouse (Cruikshank et al., 2002). This horizontal thalamocortical slice preparation retains much of the connectivity between the ventral medial geniculate nucleus (MGv) and the primary auditory cortex. The artificial CSF (ACSF) contained the following (in mm): 125 NaCl, 4 KCl, 1.2 KH2PO4, 1.3 MgSO4 26 NaHCO3, 15 glucose, 2.4 CaCl2, and 0.4 l-ascorbic acid, pH 7.3 (when bubbled with 95% O2-05% CO2). ACSF was superfused in the recording chamber at 5 ml/min at 32°C. Whole-cell current-clamp recordings were obtained from layer 2/3 pyramidal neurons in A1 (PC-501A; Warner Instruments, Hamden, CT). Data were acquired from neurons with VREST of less than or equal to -50 mV and overshooting action potentials. The internal patch solution contained the following: 127.5 mm potassium gluconate, 0.6 mm EGTA, 10 mm HEPES, 2 mm MgCl2, 5 mm KCl, 2 mm ATP, 0.3 mm GTP, and 5 mm phosphocreatine, pH 7.2, and 0.5% biocytin (Sigma, St. Louis, MO). Bipolar platinum wire stimulating electrodes were placed over the MGv and either layer 5/6 or layer 2/3 to stimulate the thalamocortical and intracortical pathways, respectively. These stimulating electrodes were fabricated from 0.004-inch-diameter Teflon-coated platinum wires (A-M Systems, Carlsborg, WA) that were inserted into a ≈2-inch-long double-barrel glass electrode. The tips of the wires were bared of the Teflon coat (the exposed tip was 0.002 inches in diameter). Intracortically evoked monosynaptic IPSPs were recorded in the presence of ionotropic glutamate receptor blockers, whereas mixed monosynaptic and polysynaptic IPSPs were recorded in the presence of AP-5 alone (below).
Standard pharmacological agents were used to block the following receptors: NMDA-R (AP-5, 50 μm; Tocris Cookson, Ballwin, MO), AMPA-R (DNQX, 20 μm; Sigma), GABAA-R (bicuculline methoiodide, 10 μm; Sigma), GABAB-R [SCH-50911 ((2S)(+)5,5-dimethyl-2-morpholineacetic acid)] (20 μm; Tocris Cookson), and NR2B subunit-containing NMDA-R (ifenprodil, 20 μm; Sigma). A 6 min application time was given for each of the drugs, except for ifenprodil, in which case recordings were made after 18 min of application. For NR2B subunit-mediated currents and miniature EPSCs (mEPSCs) two changes were made: (1) Mg2+-free ACSF was used, and (2) for the internal solution, K-gluconate was replaced by equimolar Cs-gluconate (Sigma) to block potassium channels and QX-314 [2(triethylamino)-N-(2,6-dimethyl-phenyl) acetamine] (5 mm; Alamone Labs, Jerusalem, Israel) was added to block sodium channels. The NR2B subunit-mediated currents were recorded at VHOLD of -50 mV as inward currents, in voltage-clamp mode, in the presence of DNQX (20 μm) and glycine (10 μm). mEPSCs were recorded in Mg+2-free ACSF in the presence of glycine (10 μm), bicuculline (10 μm), and tetrodotoxin (500 nm). mEPSCs were recorded for 15 min after superfusion of the modified ACSF. At least 10 sweeps of 20 s duration were acquired for each neuron at VHOLD of -70 mV. Similarly, recordings were obtained in the presence of bath-applied AP-5 (50 μm), and the change in mEPSC duration was calculated.
Minimal amplitude MGv-evoked EPSCs were recorded in normal ACSF under voltage-clamp conditions at VHOLD of -70 and -20 mV. First, at VHOLD of -70 mV, incremental stimulus intensities were delivered to the MGv at 0.5-0.2 Hz until an evoked EPSC was discernible (putative AMPAergic EPSC) (see Fig. 6 A). Stimulation at this intensity produced 50-80% failures in eliciting EPSCs. The intensity varied some-what between preparations. Increasing the minimal intensity by ∼5% decreased the failure rate without changing the EPSC amplitude. Additional increases in the stimulus intensity above this level evoked increments in EPSC size with potential polysynaptic involvement (see Fig. 6 A). Therefore, the minimal stimulation intensity was kept constant throughout each recording. The failures were not included in the analysis of EPSCs.
Figure 6.
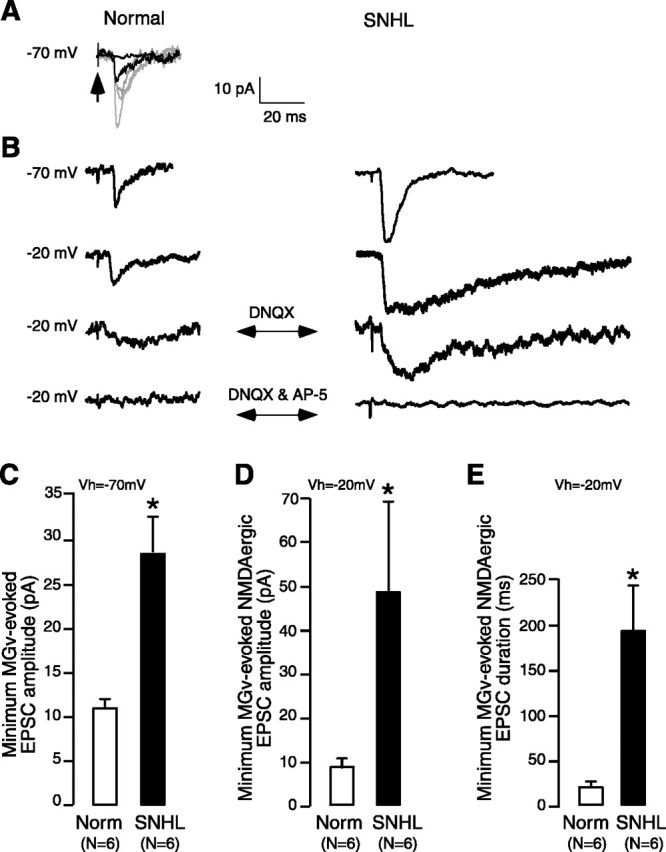
NMDA receptor-mediated minimum-evoked thalamocortical excitatory currents are stronger in SNHL neurons. A, Minimum MGv-evoked excitatory currents were recorded in voltage clamp at VHOLD of -70 mV in the presence of bicuculline and glycine. They were isolated by stimulating MGv (arrow, stimulus artifact) at 0.2-0.5 Hz in incremental intensities, resulting in larger EPSCs at each stimulus step (grayt races); the intensity at which minimum EPSC was recorded was then chosen for successive recordings (dark EPSC). This stimulation intensity produced a failure rate of ≥50% (dark trace at baseline is a failure). B, Left, In a normal neuron, a minimum MGv-evoked EPSC is shown (first trace). Holding the neuron at -20 mV revealed the initial fast AMPAergic as well as a delayed NMDAergic component (second trace). Addition of DNQX eliminated the AMPAergic component and revealed the NMDAergic EPSC (third trace). This was then blocked by AP-5. B, Right, Note the comparatively larger minimum MGv-evoked EPSC in an SNHL neuron (top). Holding the cell at -20 mV shows the initial AMPAergic component and a much longer (than normal) NMDAergic EPSC component (second trace). Addition of DNQX eliminated the AMPAergic component, revealing the long NMDAergic EPSC (third trace). Addition of AP-5 eliminates the NMDA receptor-mediated EPSC (bottom trace). C-E, Bar graphs of minimum MGv-evoked EPSCs recorded from six normal and six SNHL neurons show the following in the SNHL neurons: C, that the amplitude of mean minimum evoked EPSCs at VHOLD of -70 mV is significantly larger; D, that the amplitude of pure NMDAergic currents are significantly larger; and E, that the duration of pure NMDAergic currents are significantly longer (asterisks indicate a significant difference; for p values, see Results). Each neuron was obtained from a different animal.
Data collection and analysis. Data were collected using a Macintosh G4 personal computer running a custom-designed IGOR (version 3.14; WaveMetrics, Lake Oswego, OR) macro called SLICE. The data on firing properties and evoked synaptic potentials/currents were analyzed offline using a second IGOR macro called SLICE ANALYSIS.
A customized macro was added to the SLICE ANALYSIS software for off-line measurement of mEPSC amplitude and frequency. The macro used a semiautomated procedure to identify events, which used a threshold value based on the SD of the acquired trace. To ensure that all events were included, the macro also permitted the user to manually append events that were not identified by the automated process. In practice, our threshold value was no less than 8 pA, and smaller events were therefore excluded from our analyses. The amplitude of each event was calculated by the macro, and the frequency was calculated using a statistical package (JMP; SAS Institute, Cary, NC). The mEPSC durations were calculated manually on events that were ≥30 pA. This amplitude criterion was chosen such that events were well above the baseline noise and the full duration could be easily ascertained. Durations were calculated from all mEPSC recordings.
Both the data acquisition and analysis macros are available with complete documentation on-line at http://www.cns.nyu.edu/~sanes/slice_software/. Statistical tests (ANOVA, Student's t test, and Wilcoxon's) were performed using statistical software (JMP; SAS Institute). Biocytin-filled neurons were processed with a DAB-ABC protocol, and labeled neurons (SNHL, n = 30 slices; normal, n = 36 slices) were used to confirm the location of each recording, a pyramidal morphology, and an intact dendritic arborization.
Electron microscopic immunocytochemistry. Two normal and two SNHL animals (cochlear ablation performed at P10) were perfused transcardially with 4% paraformaldehyde/1% glutaraldehyde at P17. A1 region was cut into 80 μm free-floating sections and processed for electron microscopy using the osmium-free method described by Phend et al. (1995). They were infiltrated with the resin (Electron Microscopy Sciences, Fort Washington, PA), flat embedded, capsule embedded, and cut into 80 nm ultrathin sections. Sections from the four animals were simultaneously immunolabeled for NR2B using postembedding gold method (Fujisawa and Aoki, 2003). The final concentration of rabbit anti-NR2B (Upstate Biotechnology, Lake Placid, NY) was 67 μg/ml. The sections were counterstained with Reynold's lead citrate (Electron Microscopy Sciences) for 1 min. Under the electron microscope (1200XL; JEOL, Peabody, MA), asymmetric synapses within layer 2/3 in A1 were examined.
The gold labels were categorized into the following groups according to the ultrastructural position: within axon “terminal” bouton but away from synapse, on/near “presynaptic” membrane, “at postsynaptic density” (PSD), “near PSD,” on “extrasynaptic” membrane, and within “spine/dendrite.” An additional category called “synaptic” pooled the data from presynaptic, at PSD, and near PSD categories. The tallied gold counts from the two animals of the same condition were pooled together as “normal” and “SNHL” data. The average number of gold particles per synapse encountered and its SEM were calculated for each category. The electron microscopic examination and analyses were performed blind to the experimental condition. Data from normal and SNHL conditions were compared using Student's t test (two-tailed) (SPSS software; SPSS, Chicago, IL).
Results
SNHL alters passive and active membrane properties
The data were collected from 164 neurons recorded in 143 brain slices from experiments performed in an approximately equal number of normal and SNHL neurons. An intact thalamocortical projection to A1 was verified by recording a robust MGv-evoked extracellular response (Cruikshank et al., 2002). The cortex was first observed at 100× magnification, in which the laminar structure is clear, and the recording electrode position was established to be within layer 2/3. Neurons were visually identified under infrared-differential interference contrast optics at 400× magnification, and whole-cell recordings were then obtained. The search strategy excluded neurons in sparsely populated layer 1 and targeted pyramidal neurons that are densely packed in layer 2/3. A blind analyses of over 53 randomly chosen biocytin-filled pyramidal neurons showed that there was no difference in the soma depth from the pial surface between normal and SNHL neurons (normal, 402 ± 16 μm vs SNHL, 392 ± 18 μm; Wilcoxon's test; χ2 = 0.14; df = 52; p = 0.7). Therefore, there was no bias in recording site depth between the two groups. The depth range of these cell bodies varied between 125 and 575 μm from the pial surface.
The mean resting membrane potential of neurons from the SNHL animals displayed a small but significant depolarization when compared with the value from normal animals (Fig. 1A) (mean ± SEM; normal, -63.4 ± 0.8 mV vs SNHL, -59.9 ± 0.8 mV; t test; t = 3; df = 100; p = 0.002). SNHL also led to a significant increase in the mean input resistance (Fig. 1B) (mean ± SEM; normal, 277 ± 13.5 MΩ vs SNHL, 409 ± 29 MΩ; t test; t = 4; df = 92; p < 0.001). A linear fit between resting potential and input resistance did not reveal a significant correlation in either normal (R2 = 0.01; p > 0.05) or SNHL (R2 = 0.02; p > 0.05) neurons. Furthermore, we compared certain properties of adapting only neurons after hearing loss. This analysis showed a trend similar to that seen in the entire neuron population (resting potential: normal adapting, 64 ± 0.8 vs SNHL adapting, 59.4 ± 0.9 mV, t test, t = 3.4, df = 73, p < 0.0008; input resistance: normal adapting, 271 ± 14 vs SNHL adapting, 452 ± 29 MΩ, t test, t = 4.5, df = 67, p < 0.0001). Whereas there was no effect of age on VREST, there was a small effect for RINPUT. Therefore, a linear fit was performed for data from normal and SNHL neurons that revealed a very small effect of age on RINPUT of SNHL neurons (R2 = 0.11; p < 0.05) but not normal neurons (R2 = 0.06; p > 0.05).
Figure 1.

SNHL increases membrane excitability. The resting membrane potential of SNHL neurons is significantly depolarized (A), and the input resistance is significantly higher (B) compared with neurons recorded in normal animals. In this and subsequent figures, each open symbol represents the measurement from one neuron (n values shown on the x-axis). Filled symbols are mean ± SEM. C, Examples of neurons that respond to suprathreshold current injection with an onset (top), sustained (middle trace), and adapting (bottom trace) response. Five suprathreshold pulses (10 pA, 1500 ms) were injected, and firing pattern was established if the fifth pulse did not alter the firing pattern. D, Bar graphs of firing patterns in normal and SNHL neurons show percentile distribution. After SNHL, onset-type neurons were not observed, and there was a significant rise in the sustained and a decrease in adapting neuron patterns, suggesting greater excitability. E, Linear fit of number of spikes by depolarizing current injection. Five 10 pA, 1500 ms depolarizing current steps were injected, and the resultant spikes were counted. The fit shows no significant difference between the stimulus-response characteristics of normal and SNHL adapting neurons; the SNHL sustained neuron fits had a greater correlation coefficient, similar to the correlation in sustained neuron fits of normal neurons (see Results). Therefore, the characteristics of firing patterns did not change; rather, the incidence of sustained firing increased and adapting neurons decreased in SNHL neurons, as shown in D.
The firing patterns of A1 neurons were characterized in response to suprathreshold depolarizing pulses (1500 ms) and fell into three broad categories: (1) onset (phasic) neurons that fired a single action potential; (2) sustained neurons that fired at a high rate with little or no adaptation; and (3) adapting (regular spiking) neurons that fired at a relatively lower rate exhibited a significant degree of spike frequency adaptation (Fig. 1C). Whereas 15% of normal neurons were onset type, this response was not found in SNHL cases. Furthermore, the adapting pattern decreased by 13% (Fig. 1D), whereas the incidence of sustained type of firing increased by 30% after SNHL.
Table 1 presents a comparison of additional measures, including spike threshold and half-spike width. These measures strongly suggest that all included recordings were obtained from pyramidal neurons and that differences in firing patterns were induced by hearing loss. For example, there were no significant differences between the half-spike width (p = 0.7; df = 15), input resistance (p = 0.4; df = 15), or resting membrane potential (p = 0.11; df = 15) between the adapting and sustained SNHL neurons. Likewise, there was no difference between the half-spike widths of normal and SNHL neurons (p = 0.14; df = 20). In contrast, the spike threshold (i.e., magnitude of voltage deflection from the resting potential before the neurons fired) and the threshold current (i.e., the amount of current required to elicit a threshold response) were significantly lower in SNHL neurons compared with normals. Additional comparisons between the resting potentials and input resistance between all adapting neurons (normal vs SNHL) showed a highly significant difference as seen in the entire population of normal versus SNHL neurons (Table 1). To compare the stimulus-response curves, a correlation coefficient was obtained for injected current (five current steps of 1500 ms in 10 pA increments) and number of elicited spikes for each firing pattern in Table 1. SNHL sustained neurons exhibited a stronger correlation (R2 = 0.6) compared with adapting neurons (normal, R2 = 0.42; SNHL, R2 = 0.43). The stimulus-response curve for normal sustained firing neurons is not plotted in Figure 1E (R2 = 0.59). The maximum instantaneous firing frequency in response to depolarizing current injections (during the first 100 ms) for adapting neurons was 25 Hz (n = 6), whereas that for sustained neurons was 50 Hz (n = 6). The stimulus-response plots (Fig. 1E) for normal and SNHL adapting neurons showed a similar increment in firing rate. Therefore, hearing loss led to a net increase in the incidence of the sustained firing pattern and a net decrease in the adapting and onset patterns.
Table 1.
Membrane properties of adapting and sustained firing neurons
|
Group |
Resting potential (mV) |
Input resistance (MΩ) |
Half-spike width (ms) |
Threshold current to elicit spike (pA) |
Spike threshold (ΔVm in mV) |
|---|---|---|---|---|---|
| Normal adapting (n = 11) | −64±2.2 | 265±26 | 1.8±0.18 | 58±5 | 22±1.5 |
| SNHL adapting (n = 10) | −61±1.4 | 410±29 | 2.2±0.19 | 36±2 | 16±1.4 |
| SNHL sustained (n = 6) |
−59.8±1.4 |
375±17 |
2.4±0.37 |
20±3.6 |
12.8±1.7 |
Statistical comparisons of membrane properties of ∼100 recorded neurons appear in Figure 1 and Results. Properties of a subset adapting and sustained neurons were additionally compared (columns 1, 2). Furthermore, ∼25% of neurons from each group (left column) were blindly selected to compare the half-spike width, spike thresholds (in picoamperes, subthreshold current injected before the cell spiked and the resultant Vm). Statistical analyses: half-spike width (not different) among the three groups, one-way ANOVA, p = 0.22, df = 26; spike thresholds (in picoamperes), normal adapting versus SNHL adapting, t test, t = 3.4, df = 20, p < 0.002; SNHL adapting versus SNHL sustained, t test, t = 3.5, df = 15, p < 0.003; spike threshold (Vm: the difference between the VREST and spike threshold); normal adapting versus SNHL adapting, t test, t = 2.6, df = 20, p< 0.01; SNHL adapting versus SNHL sustained (not different), p = 0.19. For frequency-intensity (F-I) correlation coefficients, see Results and Figure 1E.
To confirm that nonpyramidal cells were not included in our sample, biocytin filled neurons were examined after each experiment under 40-600× magnification to identify the dendritic structure and to confirm the presence of spines. This qualitative examination of normal and SNHL neurons indicated the presence of intact basal and apical dendrites with spines in each recovered neuron included in this study (Fig. 2).
Figure 2.

Layer 2/3 spiny pyramidal neurons. A selection of nine biocytin-filled layer 2/3 neurons with different firing patterns reveal cell and dendritic architecture. A, A normal adapting neuron. B, A normal sustained neuron. C, A normal onset neuron. D, Dendritic apical spines from the neuron shown in B. E, An SNHL sustained neuron. F, SNHL adapting (left) and sustained (right) neurons recorded in the same brain slice. G, Normal adapting (left) and onset (right) neurons recorded in the same slice. H, An SNHL adapting neuron. Scale bar: A-C, E-H, 100 μm; D, 10 μm.
SNHL enhances synaptic excitation
MGv stimulation was used to evoke EPSPs in layer 2/3 pyramidal neurons (Fig. 3A). A comparison of maximum EPSP amplitudes between normal and SNHL neurons showed no difference, both before and after treatment with the NMDA receptor blocker AP-5 (mean ± SEM before AP-5 application: normal, 10.9 ± 1.5 mV vs SNHL, 12.1 ± 1.7 mV, t test, t = 0.54, df = 26, p = 0.5; mean ± SEM after AP-5 application: normal, 5.7 ± 1 mV vs SNHL, 4.2 ± 0.7 mV, t test, t = -1.1, df = 26, p = 0.2).
Figure 3.
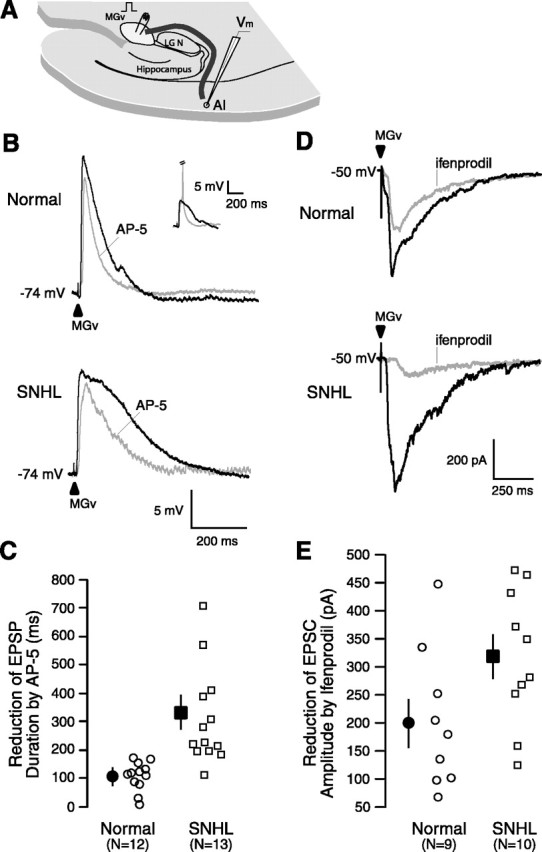
SNHL augments NMDA receptor function. A, A Schematic of the thalamocortical brain slice showing the position of a stimulating electrode in the MGv (square pulse), the pathway (dark line) from MGv to A1, and a recording electrode within A1 (Vm). The approximate distance the afferents travel from the MGv around the lateral geniculate (LGN) and hippocampus, radiating to the recording site in layer 2/3, is ∼5.5 mm. B, Maximum EPSP evoked by stimulating MGv (arrowhead). Note the significant duration (in milliseconds) reduction by the NMDA receptor antagonist AP-5 and a greater reduction in an SNHL neuron. Resting membrane potentials (in millivolts) indicated at the left of the traces. Inset shows MGv-evoked EPSP and EPSP-elicited spike (clipped); in all cases, the subthreshold maximum EPSPs were analyzed. C, Scatter plot of the magnitude of reduction in EPSP duration by AP-5 between normal and SNHL neurons shows a significantly greater AP-5-sensitive NMDA receptor-mediated component among SNHL cases. D, EPSCs evoked by stimulating at MGv (arrowhead). Note the reduction in amplitude by the NR2B subunit-specific antagonist ifenprodil and a greater reduction in an SNHL neuron. We do not rule out an effect of ifenprodil on presynaptic NMDA receptors. E, Scatter plot of the magnitude of reduction in EPSC amplitude by ifenprodil between normal and SNHL neurons shows a significantly greater ifenprodil-sensitive NR2B subunit component in SNHL neurons.
In contrast to maximum EPSP amplitude, the EPSP duration was significantly longer in SNHL neurons. Figure 3B illustrates that the reduction in EPSP duration by the NMDA receptor blocker AP-5 was significantly greater after SNHL. The mean EPSP duration (milliseconds ± SEM) of normal and SNHL neurons was significantly different before the application of AP-5 (normal, 238 ± 22 ms vs SNHL, 412 ± 69 ms; Wilcoxon's test; χ2 = 5.89; p = 0.02). Furthermore, the absolute reduction in EPSP duration by application of AP-5 was significantly greater in SNHL neurons (normal, 110 ± 13 ms vs SNHL, 315 ± 44 ms; Wilcoxon's test; χ2 = 16.62; p < 0.0001). However, the duration of the remainder EPSP, presumably carried by AMPA receptors, was not different between normal and SNHL neurons (normal, 129 ± 20 ms vs SNHL, 123 ± 31 ms).
A similar trend was found for intracortically evoked EPSPs. SNHL led to longer EPSP durations and greater reduction by AP-5 than in normals (mean ± SEM reduction in duration of intracortically evoked EPSPs after AP-5 treatment: normal, 70 ± 14 ms vs SNHL, 190 ± 39 ms; t test; t = 2.8; df = 17; p = 0.01). There was no significant difference in the maximum amplitude of intracortically evoked EPSPs before or after AP-5 treatment.
To test whether the extended EPSP duration in SNHL neurons was attributable to a change in the subunit composition of NMDA receptors, MGv-evoked maximum EPSCs were recorded after blocking sodium and potassium channels in an Mg+2-free ACSF (VHOLD of -50 mV). The NMDA receptor-mediated component was recorded in the presence of DNQX (20 μm), bicuculline (10 μm), and glycine (10 μm). In these experiments, we chose a stimulus intensity that produced an initial (before ifenprodil) EPSC amplitude of 400-500 pA for both normal and SNHL neurons (mean ± SEM EPSC amplitude: normal, 437 ± 89 pA vs SNHL, 474 ± 80 mV; Wilcoxon's test; χ2 = 3.84; p = 0.05). We maintained this stimulus intensity throughout the experiments. In the absence of the preceding AMPAergic EPSC component (DNQX in bath), we measured the reduction in EPSC amplitude by the antagonist to the NR2B subunit-containing NMDA receptors, ifenprodil, as a function of change in NR2B subunits.
The reduction of EPSC amplitude by the antagonist specific for NR2B subunit-containing NMDA receptors, ifenprodil, was significantly greater in SNHL neurons (Fig. 3D,E) (mean ± SEM reduction after application of ifenprodil: normal, 201 ± 41 pA vs SNHL, 318 ± 38 pA; t test; t = 2.1; df = 18; p = 0.05). The remaining EPSC amplitudes, presumably carried by the NR2A subunit, were not significantly different between normal and SNHL neurons. In two normal and two SNHL neurons recorded from four different slices, these remainder EPSCs were completely and reversibly abolished by AP-5 (data not shown), demonstrating that the analyzed EPSCs in this experiment were exclusively NMDAergic.
To assess whether increased sensitivity of EPSCs to ifenprodil in SNHL neurons was attributable to increased expression of NR2B subunits at synapses in layer 2/3, postembedding immunogold staining was performed to examine the localization of these subunits at synapses using an electron microscope (Fig. 4). Gold particles reflecting immunolabeling for NR2B that resided in the vicinity of asymmetric synapses were counted and grouped into the following mutually exclusive categories: (1) on presynaptic membrane, (2) at PSD, (3) near PSD, (4) on extrasynaptic membrane, and (5) within spine/dendrite or within axon terminal. For each category, the average number of gold particles per synapse was more numerous for the SNHL neurons (Table 2). The difference was most pronounced at the PSD and over the total synaptic region (i.e., the average of the sum of gold particles in categories 1-3).
Figure 4.
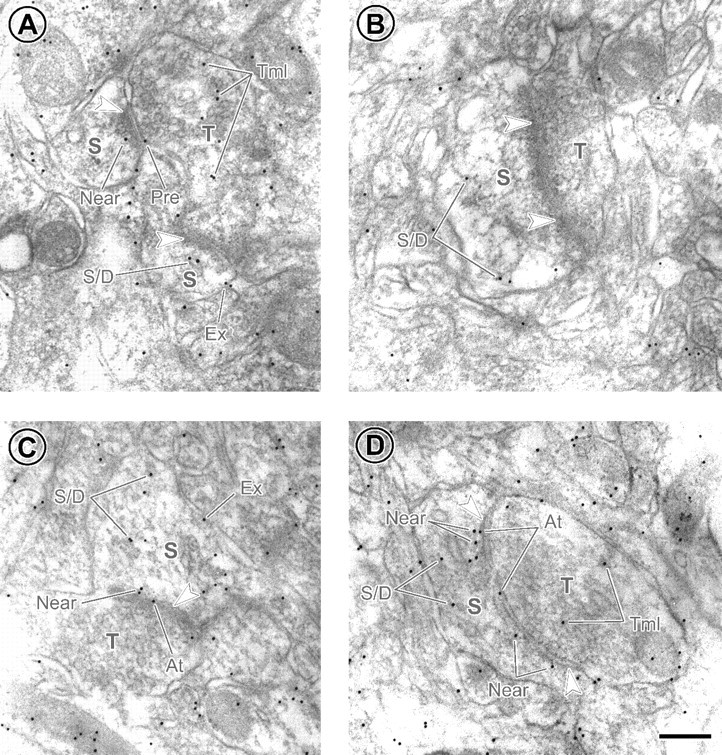
Greater occurrence of immunolabeling of NR2B subunits at synapses from SNHL animals. A, B, Electron micrographs from normal brain. C, D, Micrographs from SNHL brain. Arrowheads indicate PSDs of asymmetric synapses. S and T represent postsynaptic spine and presynaptic axon terminal, respectively. Black dots are 10 nm immunogold particles labeling for NR2Bs. The categories of labeling are as follows: Pre, on presynaptic membrane; At, at PSD; Near, near PSD; Ex, on extrasynaptic membrane; S/D, within postsynaptic spine or dendrite away from the synapse; Tml, within axon terminal away from the synapse. Scale bar, 200 nm.
Table 2.
Average ± SEM number of NR2B-labeling immunogold particles per synapse in layer 2/3 of A1
|
|
Terminal |
Presynaptic |
At PSD |
Near PSD |
Synaptic |
Extrasynaptic |
Spine/Dendrite |
|---|---|---|---|---|---|---|---|
| Normal (n = 153) | 1.95 ± 0.19 | 0.20 ± 0.05 | 0.12 ± 0.04 | 0.16 ± 0.05 | 0.48 ± 0.08 | 0.18 ± 0.04 | 0.97 ± 0.15 |
| SNHL (n = 175) | 2.85 ± 0.22 | 0.51 ± 0.06 | 0.56 ± 0.07 | 0.38 ± 0.05 | 1.45 ± 0.11 | 0.60 ± 0.10 | 1.48 ± 0.18 |
|
p values |
0.0022 |
0.0001 |
<0.0001 |
0.0018 |
<0.0001 |
0.0001 |
0.0336 |
n denotes the number of synapses analyzed for each condition. Synaptic category was calculated by summing the values in presynaptic, at PSD, and near PSD categories. p values were obtained by performing nonpaired Student's t test, two-tailed.
If the maximum EPSP amplitude is determined primarily by the AMPAergic component (Fig. 3), there could have been a masking effect attributable to the depolarized resting membrane potential (Fig. 1) and decreased firing threshold (Table 1). Therefore, a series of voltage-clamp recordings were performed. First, to examine whether such postsynaptic alterations are accompanied by a presynaptic change, mEPSCs were recorded in nominal Mg2+-ACSF and in the presence of blockers of Na+ and K+ channels and GABAA receptors under voltage-clamp conditions (see Materials and Methods). Analyses showed a significant decrease in the frequency and an increase in the peak amplitude of mEPSCs in SNHL neurons (Fig. 5) (mEPSC frequency, mean ± SEM: normal, 3.3 ± 0.2 Hz vs SNHL, 1.9 ± 0.4 Hz, t test, t = 2.9, df = 7, p = 0.02; mEPSC amplitude, mean ± SEM: normal, 12.7 ± 1.4 pA vs SNHL, 20 ± 2.6 pA, t test, t = 2.6, df = 7, p = 0.03). Furthermore, the duration of total mEPSCs, and reduction in duration by AP-5, was greater in SNHL neurons than in normals (Fig. 5) (total mEPSC duration, mean ± SEM: normal, 51 ± 4.7 vs SNHL, 100.9 ± 4.4, t test, t = 7.6, p < 0.0001; mEPSC duration after AP-5, mean ± SEM: normal, 34.5 ± 3 ms vs SNHL, 46 ± 7 ms; t test, t = 2.8, p < 0.01). Statistical comparisons confirmed that AP-5 significantly reduced mEPSC duration within both the normal and the SNHL groups (data not shown).
Figure 5.
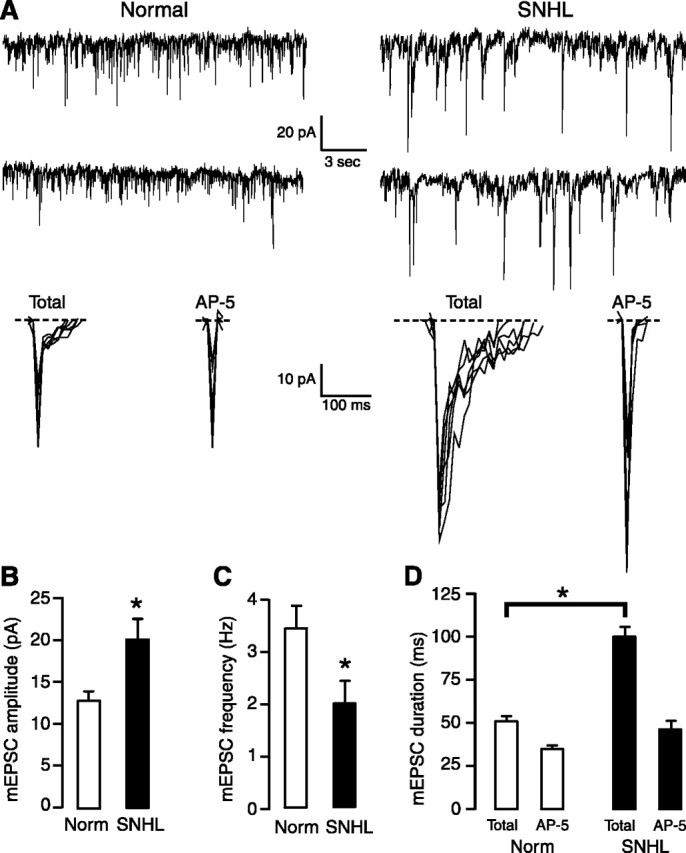
Frequency and amplitude of mEPSCs, respectively, decreases and increases in SNHL neurons. A, The left panel shows two sweeps of mEPSCs recorded for 20 s each in a normal (left) and an SNHL neuron (right) at a holding potential of -70 mV. Each recording was acquired ∼2 min apart. Note that amplitudes of mEPSCs in an SNHL neuron (right) appear to be larger, whereas the frequency of mEPSCs have decreased. At the bottom panel of A, expanded mEPSCs from a normal (left) and an SNHL neuron are shown, both before and after AP-5 treatment. The traces indicate that the amplitude, duration, and AP-5 sensitivity in the SNHL neuron are greater. B-D, Bar graphs summarizing the mean amplitude and duration of mEPSCs recorded from five normal and three SNHL neurons. B, The amplitude of mean mEPSCs was significantly larger in SNHL neurons. C, The mean frequency of mEPSCs declined significantly in SNHL neurons. D, The total mEPSC duration was significantly greater in SNHL neurons. Asterisks (and bar in D) indicate that the differences are significant (for p values, see Results). Each neuron was obtained from a different animal.
To assess whether putative monosynaptic connections from the MGv to layer 2/3 pyramidal neurons exhibit NMDAergic alterations and whether there was a parallel change in AMPAergic function, minimum MGv-evoked EPSCs (≥50% failure rates) were recorded in voltage clamp with normal ACSF, bicuculline (10 μm), and glycine (10 μm) in the bath (Fig. 6A). The results showed that the amplitude of minimum EPSCs at VHOLD of -70 was significantly larger in SNHL neurons (minimum MGv-evoked amplitudes at VHOLD of -70 mV, mean ± SEM: normal, 11 ± 2 pA vs SNHL, 28 ± 5 pA; Wilcoxon's test; χ2 = 5; p = 0.025). To examine the NMDAergic component, cells were held at -20 mV. Comparison of such EPSC durations at this holding potential showed that SNHL neuron EPSCs were significantly longer (minimum MGv-evoked EPSC duration at VHOLD of -20 mV, mean ± SEM: normal, 22 ± 3 ms vs SNHL, 182 ± 40 ms; Wilcoxon's test; χ2 = 8.3; p = 0.003). Finally, after blockade of AMPA receptors by DNQX, the pure NMDA receptor-mediated minimum-evoked EPSC currents revealed at VHOLD of -20 mV were significantly larger and longer in SNHL neurons (minimum MGv-evoked amplitude in the presence of DNQX at VHOLD of -20 mV, mean ± SEM: normal, 8 ± 1.8 pA vs SNHL, 49 ± 19 pA, Wilcoxon's test, χ2 = 7.4, p = 0.006; minimum MGv-evoked duration in the presence of DNQX at VHOLD of -20 mV, mean ± SEM: normal, 17 ± 2 ms vs SNHL, 191 ± 38 ms, Wilcoxon's test, χ2 = 8.3, p = 0.003) (Fig. 6). In all neurons (n = 6 each normal and SNHL neurons), the EPSCs were abolished by AP-5, demonstrating that they were pure minimum-evoked MGv-evoked NMDAergic currents (Fig. 6B). For each recorded neuron, we calculated the maximum difference in response latency as a measure of variability in minimum-evoked EPSCs. We compared this measure of variability between normal and SNHL neurons and found no significant difference (normal, 1.5 ± 0.3 ms vs SNHL, 1.1 ± 0.2 ms; n = 12; p = 0.3).
SNHL reduces synaptic inhibition
Because the resting potential of layer 2/3 pyramidal neurons in normal animals is close to ECl (data not shown), it was often difficult to observe the evoked IPSPs. Furthermore, cortical stimulation produces a highly variable mixed excitatory-inhibitory synaptic response (Cruikshank et al., 2002). To circumvent these issues, recorded neurons were held at -55 mV to enhance the driving force for IPSPs, and excitatory ionotropic transmission was blocked partially with AP-5. This procedure revealed polysynaptic IPSPs. In three each normal and SNHL neurons, these IPSPs could then be blocked by the sequential addition of GABAB and GABAA receptor antagonists, respectively, demonstrating that they were GABAergic (data not shown). In another set of experiments, the AMPA receptor blocker DNQX was also added with AP-5; this strategy blocked recruitment of intracortical excitatory neurons that would drive inhibitory interneurons. However, because such monosynaptic IPSPs were not recorded consistently, IPSP data were first examined with AP-5 application alone.
Figure 7A is a schematic of the stimulation and recording site for monosynaptic IPSPs. The results showed that the amplitudes of both monosynaptic IPSPs (Fig. 7B,C; DNQX and AP-5 in the bath) and polysynaptic IPSPs (data not shown; AP-5 alone in the bath) were significantly reduced in SNHL neurons when compared with those in normal neurons (maximum monosynaptic IPSP amplitude, mean ± SEM: normal, -9.2 ± 1.5 mV vs SNHL, -4.2 ± 1.0 mV, t test, t = 2.6, df = 16, p = 0.02; maximum polysynaptic IPSP amplitude, mean ± SEM: normal, -7.8 ± 0.7 mV vs SNHL, -4.4 ± 0.5 ms, t test, t = 3.5, df = 25, p = 0.001).
Figure 7.
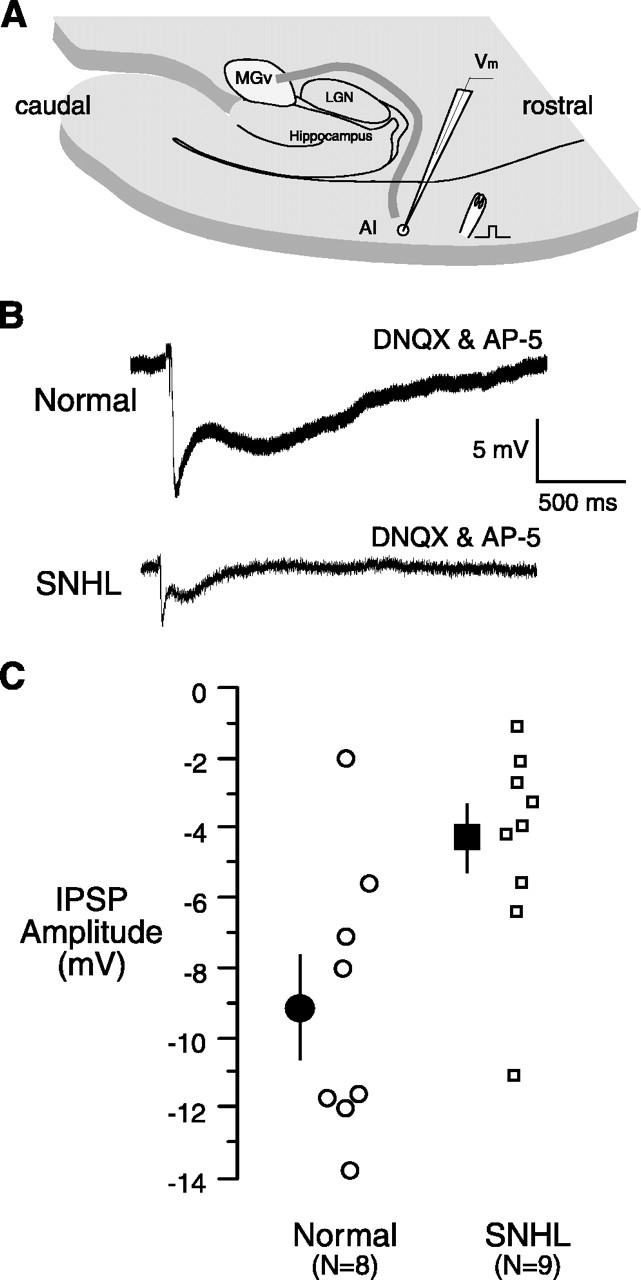
SNHL reduces monosynaptic IPSP amplitude. A, Schematic of the thalamocortical brain slice showing a stimulating electrode on layer 2/3 (square pulse) ∼1 mm rostral to the recording electrode. B, The maximum monosynaptic IPSP evoked by stimulating layer 2/3 is shown for a normal cell (top) and an SNHL cell (bottom). Note that the SNHL IPSP amplitude appears to be smaller. These recordings were obtained in the presence of blockers of the ionotropic glutamate receptors DNQX and AP-5. C, The plot of monosynaptic IPSP amplitudes from all recorded neurons shows a significant reduction for SNHL neurons.
Discussion
In vivo findings show altered A1 function after partial or complete deafness. However, these studies cannot discern whether the changes occurred in the brainstem nor which properties are modified. We identify several cellular properties in A1 influenced by hearing loss. We predicted that SNHL would disrupt the balance between excitation and inhibition, leading to altered excitability, based on previous observations from auditory brainstem showing several modifications in inhibitory and excitatory transmission after hearing loss (Kotak and Sanes, 1996, 1997; Vale and Sanes, 2000, 2002). Additionally, disruption of cortical activity by various manipulations, including loss of vision, tends to upregulate excitation and downregulate inhibition (Rao and Craig, 1997; Desai et al., 1999; Quinlan et al., 1999; Marder and Prinz, 2002; Maffei et al., 2004).
Hearing loss increases intrinsic excitability
Our findings support the concept that excitability increases after deprivation of afferent activity (Desai et al., 1999; Francis and Manis, 2000; Murthy et al., 2001; Maffei et al., 2004). Depolarization of VREST in A1 (Fig. 1) is similar to the depolarization observed in phasic neurons in the cochlear nucleus after SNHL (Francis and Manis, 2000). Increased input resistance (Fig. 1B) is also consistent with observations in the auditory brainstem (Kotak and Sanes, 1996; Francis and Manis, 2000). These alterations and decreased spike threshold (Table 1) may underlie increased firing in our preparation (Fig. 1C-E) and in vivo after cochlear damage (Salvi et al., 2000; Wang et al., 2002a). The lack of difference in the half-spike width between sustained and adapting neurons (Table 1) argues against the possibility that our sustained neurons are fast-spiking inhibitory cells in other cortices (McCormick et al., 1985; Agmon and Connors, 1992; Porter et al., 2001). It is possible that fast-spiking or intrinsic bursting patterns (McCormick et al., 1985; Agmon and Connors, 1992; Porter et al., 2001) are present in layer 2/3 of A1 in other developmental ages (>P23) or cortical layers (1, 4, 5, or 6). Moreover, characteristics of A1 neurons (Table 1, half-spike width of adapting or sustained neurons over 2 ms, maximum instantaneous firing up to 50 Hz) are not comparable with extremely fast-spiking cells (up to 800 Hz, spike duration of 0.16 ms) or chattering cells (up to 800 Hz, spike duration of 0.14 ms) (Gray and McCormick, 1996).
The slow firing rates (20-50 Hz) of the adapting or sustained neurons are likely to be associated with spectrotemporal processing of static or dynamic auditory cues that elicit low discharge rates (Semple and Scott, 2003). This may contrast with integration of image-specific synchronous oscillations in the visual cortex (for review, see Singer and Gray, 1995; Gray and McCormick, 1996).
The changes in voltage-gated channels or molecular mechanisms after hearing impairments that cause firing properties to alter remain unidentified. Because potassium and calcium currents regulate firing patterns, hearing loss may modify these channels. Thus, downregulation of a potassium leakage conductance after SNHL could depolarize VREST and increase input resistance, making the neurons electrically compact. Additionally, downregulation of a low- or high-threshold K+ conductance could underlie a lack of phase locking and heightened firing rates (Fig. 1) (Svirskis et al., 2002).
SNHL increases synaptic excitation and decreases inhibition
Longer EPSPs and larger reduction by AP-5 after SNHL demonstrates an enhanced NMDA receptor-mediated excitatory response (Fig. 3B,C). Furthermore, greater ifenprodil sensitivity of SNHL EPSCs (Fig. 3D,E) and higher NR2B-positive immunogold counts at synapses (Fig. 4, Table 2) demonstrate that increased postsynaptic glutamate sensitivity may compensate for the loss of cochlear activity. These findings agree with an augmented NMDA receptor function in evoked excitation in the lateral superior olive (LSO) and IC after hearing loss (Kotak and Sanes, 1996; Vale and Sanes, 2002) and increased NMDAergic EPSCs in the CN of deaf mutant mice (Oleskevich and Walmsley, 2002).
An augmentation of NR2B subunits (Figs. 3, 4) parallels the finding in visual cortex that high expression of synaptic NR2B subunits occurs after visual deprivation. In the visual cortex, the termination of the critical period, demonstrated by reduction in NR2B expression and increase in NR2A expression, can be delayed by sensory deprivation (Erisir and Harris, 2003). Our results support the notion that similar mechanisms operate during A1 development. NR2A and NR2B subunit expression at cortical and hippocampal synapses follow disparate trafficking and delivery rules, depending on activity (Rao and Craig, 1997; Quinlan et al., 1999; Barria and Malinow, 2002; Fong et al., 2002; Aoki et al., 2003; Fujisawa and Aoki, 2003).
The increase of mEPSC amplitude and duration in SNHL neurons suggests an upregulation of both AMPA and NMDA receptors (Fig. 5) (Myme et al., 2003). Furthermore, the parallel increase in DNQX-sensitive (AMPAergic) and AP-5-sensitive (NMDAergic) components in MGv-evoked minimum-evoked EPSCs (Fig. 6) demonstrates that the increased excitation is mediated, in part, by thalamic afferents. The lack of change in MGv-evoked maximum EPSP amplitude could be attributable to several confounding variables, including depolarized VREST (Fig. 1) and decreased spike threshold (Table 1).
Activity deprivation alters the scaling of excitatory synaptic inputs at a network level in an age-dependent manner (Murthy et al., 2001; Desai et al., 2002; Maffei et al., 2004). Decreased frequency of mEPSCs in SNHL neurons are in agreement with reports showing that deprivation of activity also leads to a decrease in presynaptic release properties (Fig. 5) (Desai et al., 1999; Murthy et al., 2001). Moreover, in the cochlear nucleus of congenitally deaf mice and IC neurons of deafened gerbils, presynaptic properties have been shown to change (Oleskevich and Walmsley, 2002; Vale and Sanes, 2002). In our preparation, an increase of presynaptically localized NR2B subunits was also observed (Fig. 4). Presynaptic NR2B-containing NMDA receptors have been shown to enhance glutamate release (Berretta and Jones, 1996). It remains to be determined whether a subset of presynaptic glutamate receptors can augment the release of transmitter after hearing loss. Together, our observations are consistent with analyses of synapse physiology in the CN, LSO, and IC after SNHL or in congenitally deaf mice and support a homeostatic mechanism: excitatory synapses get stronger, whereas inhibitory synapses become weaker (Kotak and Sanes, 1996; Vale and Sanes, 2000, 2002; Oleskevich and Walmsley, 2002).
Enhanced synaptic excitation is accompanied by diminished inhibition in diverse experimental models. For example, disruption of activity in cultured cortical neurons decreases spontaneous IPSCs possibly by downregulation of GABAA receptors (Kilman et al., 2002). In vivo studies in the auditory system show that decreased inhibition after auditory deprivation complements greater excitation (Salvi et al., 2000). Specifically, weakened side-band inhibition after noise- and drug-induced hearing loss contributes to enhanced IC neuron discharge, as well as expansion and increased sensitivity of low-frequency tuning curve (Wang et al., 2002a). Based on pharmacological observations in vivo, a weakening of intracortical inhibition would affect A1 firing properties as well as their ability to encode sound frequency (Wang et al., 2002b). Increased firing may explain heightened sound-evoked activity in the ipsilateral IC and auditory cortex after partial deafening; the cause of the latter may be an imbalance between excitation and inhibition (Kitzes and Semple, 1985; Reale et al., 1987; McAlpine et al., 1997; Mossop et al., 2000). In our preparation, reduction in maximum IPSP amplitude suggests modulation of the fast initial IPSP component carried by bicuculline-sensitive GABAA receptors (Fig. 6). Our results on decreased synaptic inhibition are interpreted to mean that the strength of GABAergic synapses decreased after hearing loss. An alternative explanation is that the inhibitory interneurons have a lower threshold for activation after hearing loss. However, we used a stimulus level that produced maximal IPSP amplitude (i.e., no additional increase in amplitude could be obtained with increasing stimulus amplitude). Therefore, a decreased firing threshold of inhibitory interneurons is unlikely to explain the findings.
Implications for cortical processing and plasticity
In vivo recordings from animals with hearing loss reveal profound changes in auditory processing (Syka, 2002). Even studies from human cortex suggest that central physiology has changed after deafness (Ponton and Eggermont, 2001; Sharma et al., 2002). The regional blood flow measured in A2 is relatively greater in prelingually deaf cochlear implant users. Furthermore, speech tends to activate a larger fraction of temporal cortex in implanted patients (Hirano et al., 2000; Naito et al., 2000). An increase in sound-evoked discharges is consistently observed after cochlear damage (Kitzes and Semple, 1985; Szczepaniak and Moller, 1995; Rajan, 1998; Klinke et al., 1999; Rajan, 2001). Our findings indicate that three major cellular changes can account for increased excitability and support a homeostatic mechanism: passive membrane properties favor excitability, excitatory synapses become stronger, and inhibitory synapses become weaker after SNHL. Greater NMDAergic participation could overwhelm decreased inhibition during prostheses-evoked responses, thus recruiting an excitable A1. Furthermore, prolonged EPSP time courses could support temporal summation by virtue of the boosted NMDA/NR2B expression (Fig. 3), especially in association with enhanced intrinsic excitability (Fig. 1).
The processing of spectrotemporal and level-dependent properties of sound are dependent on synaptic and intrinsic properties of A1 neurons. Developing auditory brainstem neurons depend on cochlear-evoked activity for synaptic refinements and emergence of adult-like tonotopic maps (Sanes and Constantine-Paton, 1985; Kapfer et al., 2002). Our data support empirical and modeling work associated with various invertebrate and vertebrate preparations that have shown that ionic and synaptic mechanisms are capable of homeostasis (Marder and Prinz, 2002). The persistent excitability we observe after SNHL may ultimately limit the finer integrative computations performed by A1 neurons, unless prosthetic devices are used early in development to restore activity (Sharma et al., 2002).
Footnotes
This work was supported by National Institutes of Health Grants DC00540 (D.H.S.) and DC006864 (D.H.S. and V.C.K.), National Eye Institute Grant EY 13145 (C.A.), National Institute of Neurological Disorders and Stroke Grant NS041091 (C.A.), and Japan Foundation for Aging and Health Grant R6189 (C.A.). We thank Dr. Patricio Huerta for his useful advice.
Correspondence should be addressed to Dr. Vibhakar C. Kotak, Center for Neural Science, 4 Washington Place, New York University, New York, NY 10003. E-mail: kotak@cns.nyu.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/253908-11$15.00/0
References
- Agmon A, Connors BW (1992) Correlation between intrinsic firing patterns and thalamocortical synaptic responses of neurons in mouse barrel cortex. J Neurosci 12: 319-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki C, Fujisawa S, Mahadomrongkul V, Shah P, Nader K, Erisir E (2003) NMDA receptor blockade in intact adult cortex increases trafficking of NR2A subunits into spines, postsynaptic densities and axon terminals. Brain Res 963: 139-149. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R (2002) Subunit-specific NMDA receptor trafficking to synapses. Neuron 35: 345-353. [DOI] [PubMed] [Google Scholar]
- Berretta N, Jones RS (1996) Tonic facilitation of glutamate release by presynaptic N-methyl-d-aspartate autoreceptors in the entorhinal cortex. Neuroscience 75: 339-344. [DOI] [PubMed] [Google Scholar]
- Burrone J, O'Byrne M, Murthy VN (2002) Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature 420: 414-418. [DOI] [PubMed] [Google Scholar]
- Cruikshank SI, Rose HJ, Metherate R (2002) Auditory thalamocortical synaptic transmission in vitro. J Neurophysiol 87: 361-384. [DOI] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG (1999) Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat Neurosci 2: 515-520. [DOI] [PubMed] [Google Scholar]
- Desai NS, Cudmore RH, Nelson SB, Turrigiano GG (2002) Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci 5: 783-789. [DOI] [PubMed] [Google Scholar]
- Emmorey K, Allen JS, Schenker N, Damasio H (2003) A morphometric analysis of auditory brain regions in congenitally deaf adults. Proc Natl Acad Sci USA 100: 10049-10054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erisir A, Harris JL (2003) Decline of the critical period of visual plasticity is concurrent with the reduction of NR2B subunit of the synaptic NMDA receptor in layer 4. J Neurosci 23: 5208-5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong DK, Rao A, Crump FT, Craig AM (2002) Rapid synaptic remodeling by protein kinase C: reciprocal translocation of NMDA receptor and calcium/calmodulin-dependent kinase II. J Neurosci 15: 2153-2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis HW, Manis PB (2000) Effects of deafferentation on the electrophysiology of ventral cochlear nucleus neurons. Hear Res 149: 91-105. [DOI] [PubMed] [Google Scholar]
- Fujisawa S, Aoki C (2003) In vivo blockade of N-methyl-d-aspartate receptors induces rapid trafficking of NR2B subunits away from synapses and out of spines and terminals in adult cortex. Neuroscience 121: 51-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray CM, McCormick DA (1996) Chattering cells: superficial pyramidal neurons contributing to the generation of synchronous oscillation in the visual cortex. Science 274: 109-113. [DOI] [PubMed] [Google Scholar]
- Hirano S, Naito Y, Kojima H, Honjo I, Inoue M, Shoji K, Tateya I, Fujiki N, Nishizawa S, Konishi J (2000) Functional differentiation of the auditory association area in prelingually deaf subjects. Auris Nasus Larynx 27: 303-310. [DOI] [PubMed] [Google Scholar]
- Iverson P (2003) Evaluating the function of phonetic perceptual phenomena within speech recognition: an examination of the perception of /d/-/t/by adult cochlear implant users. J Acoust Soc Am 113: 1056-1064. [DOI] [PubMed] [Google Scholar]
- Kapfer C, Seidl AH, Schweizer H, Grothe B (2002) Experience-dependent refinement of inhibitory inputs to auditory coincidence-detector neurons. Nat Neurosci 5: 247-253. [DOI] [PubMed] [Google Scholar]
- Kidd Jr G, Arbogast TL, Mason CR, Walsh M (2002) Informational masking in listeners with sensorineural hearing loss. J Assoc Res Otolaryngol 3: 107-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilman V, van Rossum MC, Turrigiano GG (2002) Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABAA receptors clustered at neocortical synapses. J Neurosci 22: 1328-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzes LM, Semple MN (1985) Single-unit responses in the inferior colliculus: effects of neonatal unilateral cochlear ablation. J Neurophysiol 53: 1483-1500. [DOI] [PubMed] [Google Scholar]
- Klinke R, Kral A, Heid S, Tillein J, Hartmann R (1999) Recruitment of the auditory cortex in congenitally deaf cats by long-term cochlear electro-stimulation. Science 285: 1729-1733. [DOI] [PubMed] [Google Scholar]
- Kotak VC, Sanes DH (1996) Developmental influence of glycinergic transmission: regulation of NMDA receptor-mediated EPSPs. J Neurosci 16: 1836-1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotak VC, Sanes DH (1997) Deafferentation weakens excitatory synapses in the developing central auditory system. Eur J Neurosci 9: 2340-2347. [DOI] [PubMed] [Google Scholar]
- Kral A, Hartmann R, Tillein J, Heid S, Klinke R (2000) Congenital auditory deprivation reduces synaptic activity within the auditory cortex in a layer specific manner. Cereb Cortex 10: 714-726. [DOI] [PubMed] [Google Scholar]
- Maffei A, Nelson SB, Turrigiano GG (2004) Selective reconfiguration of layer 4 visual cortical circuitry by visual deprivation. Nat Neurosci 7: 1353-1359. [DOI] [PubMed] [Google Scholar]
- Marder E, Prinz AA (2002) Modeling stability in neuron and network function: the role of activity in homeostasis. BioEssays 24: 1145-1154. [DOI] [PubMed] [Google Scholar]
- McAlpine D, Martin RL, Mossop JE, Moore DR (1997) Response properties of neurons in the inferior colliculus of the monaurally-deafened ferret to acoustic stimulation of the intact ear. J Neurophysiol 78: 767-779. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Connors BW, Lighthall JW, Prince DA (1985) Comparative electrophysiology of pyramidal and sparsely spiny stellate neuron of the neocortex. J Neurophysiol 54: 782-806. [DOI] [PubMed] [Google Scholar]
- Mody M, Schwartz RG, Gravel JS, Ruben RJ (1999) Speech perception and verbal memory in children with and without histories of otitis media. J Speech Lang Hear Res 42: 1069-1079. [DOI] [PubMed] [Google Scholar]
- Moore CM, Vollmer, M Leake PA, Snyder RL, Rebscher SJ (2002) The effects of chronic intracochlear electrical stimulation on inferior colliculus spatial representation in adult deafened cats. Hear Res 164: 82-96. [DOI] [PubMed] [Google Scholar]
- Mossop JE, Wilson MJ, Caspary DM, Moore DR (2000) Down-regulation of inhibition following unilateral deafening. Hear Res 147: 183-187. [DOI] [PubMed] [Google Scholar]
- Murthy VN, Schikorski T, Stevens CF, Zhu Y (2001) Inactivity produces increases in neurotransmitter release and synapse size. Neuron 32: 673-682. [DOI] [PubMed] [Google Scholar]
- Myme CI, Sugino K, Turrigiano GG, Nelson SB (2003) The NMDA-to-AMPA ratio at synapses onto layer 2/3 pyramidal neurons is conserved across prefrontal and visual cortices. J Neurophysiol 90: 771-779. [DOI] [PubMed] [Google Scholar]
- Naito Y, Tateya I, Fujiki N, Hirano S, Ishizu K, Nagahama Y, Fukuyama H, Kojima H (2000) Increased cortical activation during hearing of speech in cochlear implant users. Hear Res 143: 139-146. [DOI] [PubMed] [Google Scholar]
- Oleskevich S, Walmsley B (2002) Synaptic transmission in the auditory brainstem of normal and congenitally deaf mice. J Physiol (Lond) 540: 447-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phend KD, Rustioni A, Weinberg RJ (1995) An osmium-free method of epon embedment that preserves both ultrastructure and antigenicity for post-embedding immunocytochemistry. J Histochem Cytochem 43: 283-292. [DOI] [PubMed] [Google Scholar]
- Ponton CW, Eggermont JJ (2001) Of kittens and kids: altered cortical maturation following profound deafness and cochlear implant use. Audiol Neurootol 6: 363-380. [DOI] [PubMed] [Google Scholar]
- Porter JT, Johnson CK, Agmon A (2001) Diverse types of interneurons generate thalamus-evoked feedforward inhibition in the mouse barrel cortex. J Neurosci 21: 2699-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psarommatis IM, Goritsa E, Douniadakis D, Tsakanikos M, Kontrogianni AD, Apostolopoulos N (2001) Hearing loss in speech-language delayed children. Int J Pediatr Otorhinolaryngol 58: 205-210. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Olstein DH, Bear MF (1999) Bidirectional, experience-dependent regulation of N-methyl-d-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc Natl Acad Sci USA 96: 1276-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raggio MW, Schreiner CE (1999) Neuronal responses in cat primary auditory cortex to electrical cochlear stimulation. III. Activation patterns in short- and long-term deafness. J Neurophysiol 82: 3506-3526. [DOI] [PubMed] [Google Scholar]
- Raggio MW, Schreiner CE (2003) Neuronal responses in cat primary auditory cortex to electrical cochlear stimulation. IV. Activation pattern for sinusoidal stimulation. J Neurophysiol 89: 3190-3204. [DOI] [PubMed] [Google Scholar]
- Rajan R (1998) Receptor organ damage causes loss of cortical surround inhibition without topographic map plasticity. Nat Neurosci 1: 138-143. [DOI] [PubMed] [Google Scholar]
- Rajan R (2001) Plasticity of excitation and inhibition in the receptive field of primary auditory cortical neurons after limited receptor organ damage. Cereb Cortex 11: 171-182. [DOI] [PubMed] [Google Scholar]
- Rao A, Craig AM (1997) Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron 19: 801-812. [DOI] [PubMed] [Google Scholar]
- Reale RA, Brugge JF, Chan JC (1987) Maps of auditory cortex in cats reared after unilateral cochlear ablation in the neonatal period. Brain Res 431: 281-290. [DOI] [PubMed] [Google Scholar]
- Salvi RJ, Wang J, Ding D (2000) Auditory plasticity and hyperactivity following cochlear damage. Hear Res 147: 261-274. [DOI] [PubMed] [Google Scholar]
- Sanes DH, Constantine-Paton M (1985) The development of stimulus following in the cochlear nerve and inferior colliculus of the mouse. Brain Res 354: 255-267. [DOI] [PubMed] [Google Scholar]
- Sanes DH, Markowitz S, Bernstein J, Wardlow J (1992) The influence of inhibitory afferents on the development of postsynaptic dendritic arbors. J Comp Neurol 321: 637-644. [DOI] [PubMed] [Google Scholar]
- Semple MN, Scott BH (2003) Cortical mechanisms in hearing. Curr Opin Neurobiol 13: 167-173. [DOI] [PubMed] [Google Scholar]
- Sharma A, Dorman MF, Spahr AJ (2002) A sensitive period for the development of the central auditory system in children with cochlear implants: implications for age of implantation. Ear Hear 23: 532-539. [DOI] [PubMed] [Google Scholar]
- Singer W, Gray CM (1995) Visual feature integration and the temporal correlation hypothesis. Annu Rev Neurosci 18: 555-586. [DOI] [PubMed] [Google Scholar]
- Snyder RL, Sinex DG, McGee JD, Walsh EW (2000) Acute spiral ganglion lesions change the tuning and tonotopicorganization of cat inferior colliculus neurons. Hear Res 147: 200-220. [DOI] [PubMed] [Google Scholar]
- Svirskis GS, Kotak VC, Sanes DH, Rinzel JH (2002) Enhancement of signal-to-noise ratio and phase locking for small inputs by low threshold out-ward current in auditory neurons. J Neurosci 22: 11019-11025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syka J (2002) Plastic changes in the central auditory system after hearing loss, restoration of function, and during learning. Physiol Rev 82: 601-636. [DOI] [PubMed] [Google Scholar]
- Szczepaniak WS, Moller AR (1995) Effects of l-baclofen and d-baclofen on the auditory system: a study of click-evoked potentials from the inferior colliculus in the rat. Ann Otol Rhinol Laryngol 104: 399-404. [DOI] [PubMed] [Google Scholar]
- Tierney TS, Moore DR (1997) Naturally occurring neuron death during postnatal development of the gerbil ventral cochlear nucleus begins at the onset of hearing. J Comp Neurol 387: 421-429. [PubMed] [Google Scholar]
- Vale C, Sanes DH (2000) Afferent regulation of inhibitory synaptic transmission in the developing auditory midbrain. J Neurosci 20: 1912-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale C, Sanes DH (2002) The effect of bilateral deafness on excitatory synaptic strength in the auditory midbrain. Eur J Neurosci 16: 2394-2404. [DOI] [PubMed] [Google Scholar]
- Vernon-Feagans L (1999) Impact of otitis media on speech, language, cognition, and behavior. In: Evidence-based otitis media (Rosenfeld RM, Bluestone CD, eds), pp 353-373. St. Louis, MO: Decker.
- Wang J, Ding D, Salvi RJ (2002a) Functional reorganization in chinchilla inferior colliculus associated with chronic and acute cochlear damage. Hear Res 168: 238-249. [DOI] [PubMed] [Google Scholar]
- Wang J, McFadden SL, Caspary D, Salvi R (2002b) Gamma-aminobutyric acid circuits shape response properties of auditory cortex neurons. Brain Res 944: 219-231. [DOI] [PubMed] [Google Scholar]