

Abstract
A novel substituted cysteine accessibility method (SCAM) reagent was developed for monoamine uptake transporters. The new reagent, MTS-MPP+, was a derivative of the neurotoxin and transporter substrate MPP+. MTS-MPP+ labeled cysteine residues introduced into the serotonin transporter protein. Although it did not prove to be a substrate, as is MPP+, it appears to label cysteine residues lining the permeation pore of the transporter more readily than currently-available nonspecific SCAM reagents.
1. Introduction
Methanethiosulfonate cysteine scanning reagents have proven very valuable in mapping out protein structure by thiol-disulfide interchange.1–11 Unfortunately, most methanethiosulfonate reagents are relatively nonspecific, and their reactivity depends on solution accessibility of cysteine residues within the protein. For ongoing studies of monoamine transporters in our laboratory, we envisioned that a methanethiosulfonate reagent derived from a substrate for the transporter might prove useful. In particular, N-methylphenylpyridinium, MPP+, is a substrate for all biogenic amine (norepinephrine, dopamine, and serotonin) transporters.12–15 We speculated that if the reagent retained its substrate characteristics, it might be useful to map the permeation pathway through the transporter using the substituted cysteine accessibility method.8,10 A good deal of prior work in our laboratories with phenethylamine type substrates at monoamine transporters had established that chemical substitution at the 4-position was particularly well tolerated by the serotonin transporter (SERT). We hypothesized that a substituent in that position of MPP+ might similarly provide a molecule that was a substrate at the SERT. Thus, we synthesized 4-(1-methylpyridinium)phenylmethanethiosulfonate iodide (MTS-MPP+) 6. This report describes the synthesis of this new reagent, and a comparison with a conventional MTS reagent [2-(trimethylammonium)ethyl]methanethiosulfonate (MTSET) that lacks specificity for any features of the transporter.
2. Chemistry
The synthesis for the MTS-MPP+ reagent is shown in scheme 1. The initial biaryl compound was readily produced by a palladium-mediated Suzuki coupling reaction. Synthesis of the methylmethanesulfonate thioester then proved to be problematic due to facile dimerization of the thiol intermediate 3. We found, however, that dimer (4) could easily be converted into thioester 5.16 Thus, 3 was completely dimerized to 4, which was then derivatized as the methanesulfonate 5, as shown in Scheme 1.
Scheme 1.
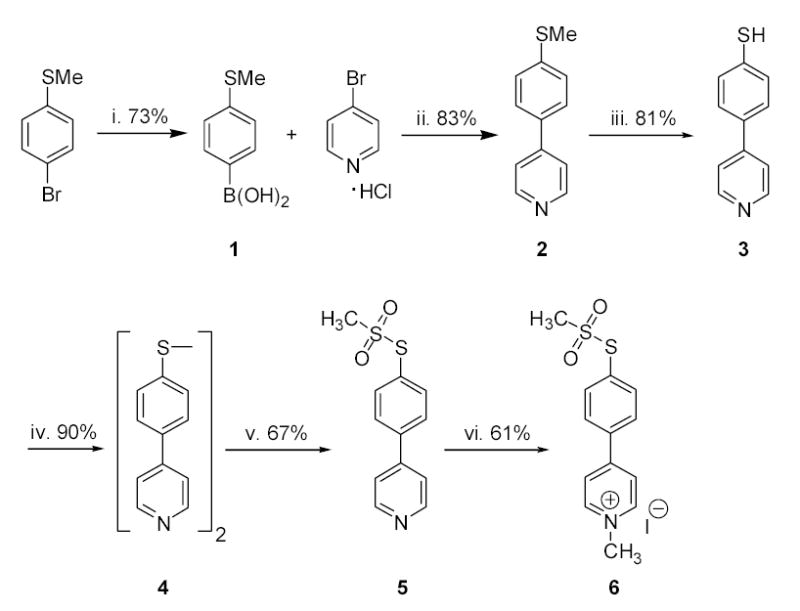
Reagents and conditions: (i) tert-BuLi, (iPrO)3B; (ii) KOH, (Bu)4NBr, (Ph3P)4Pd, DME, H2O; (iii) tert-BuSNa, DMF; (iv) tert-BuOOH, V(acac)2; (v) AgNO3, CH3SO2Na; (vi) CH3I, THF.
The final step was quaternization of the pyridine nitrogen. When carried out in more polar solvents such as nitromethane, both the pyridine nitrogen and the aryl sulfur were methylated, leading to decomposition of the methanesulfonate ester and production of N-methyl-quaternized 2. In THF, however, the quaternization of the pyridine nitrogen proceeded more rapidly, affording the N-methypyridinium salt, which was insoluble in THF and precipitated upon formation. This simple strategy prevented formation of the undesired S-methyl side product. It might also be noted that this route is very attractive for the production of a radiolabeled version of the reagent when [3H]CH3I is used for this last quaternization step. Thus, alkylated transporters could be identified readily simply by quantifying tritium irreversibly bound to the membrane fraction of cells expressing transporter, potentially avoiding the additional effort required for functional characterization.
3. Results and Discussion
MTS-MPP+ inhibition of [3H]5-HT uptake at human serotonin transporter (hSERT) is less potent than MPP+
We anticipated that MTS-MPP+ 6, an MPP+ analog with a methanethiosulfonate group attached to the phenyl ring, might serve as a selective substrate for SCAM studies. Because we wished to take advantage of the methanethiosulfonate moiety on MTS-MPP+, we first assessed whether MTS-MPP+ would interact with hSERT. The ability of MTS-MPP+ to inhibit [3H]5-HT uptake was compared to MPP+ at the C109A/hSERT mutant. The C109A mutant was used in all studies because C109 is the only endogenous cysteine in SERT sensitive to modification by methanethiosulfonates, and the 5-HT uptake activity and pharmacology of C109A/hSERT is similar to wild-type hSERT.17,18 MTS-MPP+ was significantly less potent at inhibiting [3H]5-HT uptake than MPP+ (Figure 1, Ki = 48 ± 26 μM vs. 1300 ± 100 μM, n = 2).
Figure 1. MTS-MPP+ inhibition of [3H]5-HT uptake at SERT is less potent than MPP+.
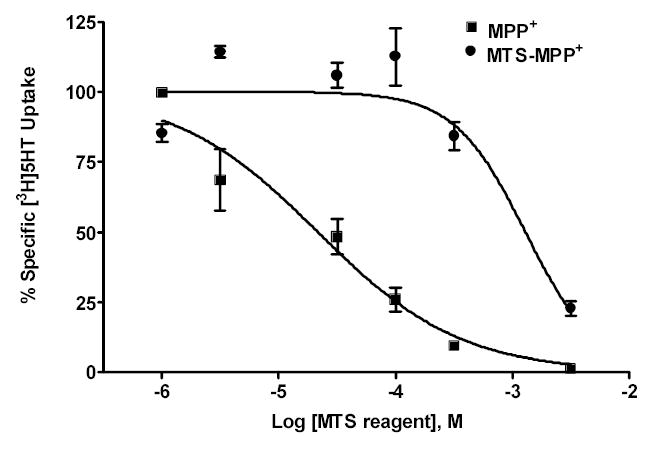
Dose-response of MTS-MPP+ vs. MPP+ for [3H]5-HT uptake inhibition at C109A/hSERT. C109A/hSERT cDNA was transiently transfected into HEK cells and uptake inhibition performed as described in the Experimental Section. Ki values (mean ± S.D.) were MPP+ 48 ± 26 μM vs. MTS-MPP+ 1300 ± 100 μM. Data represent two experiments performed in triplicate. R2 values for each curve fit were MPP+ 0.916 and MTS-MPP+ 0.877.
MTS-MPP+ is not a substrate at hSERT
MTS-MPP+ does inhibit [3H]5-HT uptake at C109A/hSERT, albeit with a higher Ki value than MPP+. Without a radiolabel, however, the ability of MTS-MPP+ to be transported by SERT cannot be directly assessed in standard uptake assays. Thus, the ability of MTS-MPP+ to function as a substrate was determined by evaluating the MTS-MPP+-induced current in hSERT-expressing oocytes. Surprisingly, unlike MPP+ (Figure 2A), MTS-MPP+ was incapable of inducing inward current in C109A/hSERT, but instead, caused an outward current consistent with blockade of the transporter-associated "leak" current that is indicative of ligand binding without translocation (Figure 2B). Transporter-associated currents for hSERT have been well characterized such that substrates are known to induce inward currents.19 In addition to substrate-activated currents, hSERT also exhibits a constitutive "leak" current that can be blocked by transporter antagonists. This block of the "leak" current is revealed as an apparent outward current upon application of a transporter blocker.19,20 Although MTS-MPP+ was not a potent blocker of 5-HT transport (Fig. 1), the ability of MTS-MPP+ to block hSERT-associated "leak" currents at lower micromolar concentrations (i.e., 30 μM) suggests specific interactions with the transporter are occurring at concentrations below the Ki value for 5-HT uptake inhibition.
Figure 2. Unlike MPP+, MTS-MPP+ does not activate inward current at hSERT.
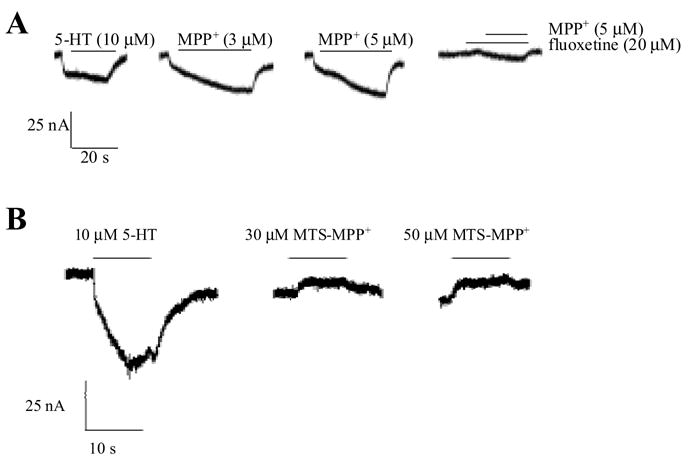
MPP+ (A) or MTS-MPP+ (B) were applied to C109A/hSERT expressing oocytes as described in the Experimental Section. Oocytes were clamped at −120 mV and perfused with drug solutions as indicated. MTS-MPP+ did not induce SERT-associated inward currents but blocked C109A/hSERT leak current. Data are representative of six oocytes from two separate batches.
MTS-MPP+ significantly inhibits G498C and W103C mutants compared to C109A/hSERT
A SERT homology model based on the crystal structure of the Aquifex aeolicus leucine transporter21 allowed us to identify locations for several residues that appear to reside at the entrance to the substrate permeation pathway (Figure 3). Our laboratory has observed that the hSERT G498C mutation was sensitive to inactivation by MTSET (M. Torres-Altoro and E. Barker, unpublished results). To explore whether MTS-MPP+ could interact with residues at the entrance to the permeation pore, G498C/hSERT was treated with increasing concentrations of either MTSET or MTS-MPP+ (Figure 4A). MTS-MPP+ inactivated [3H]5-HT uptake at the G498C mutation with a potency comparable to the non-selective reagent MTSET.
Figure 3. Approximate location of residues G498 and W103 in a homology model of the SERT.
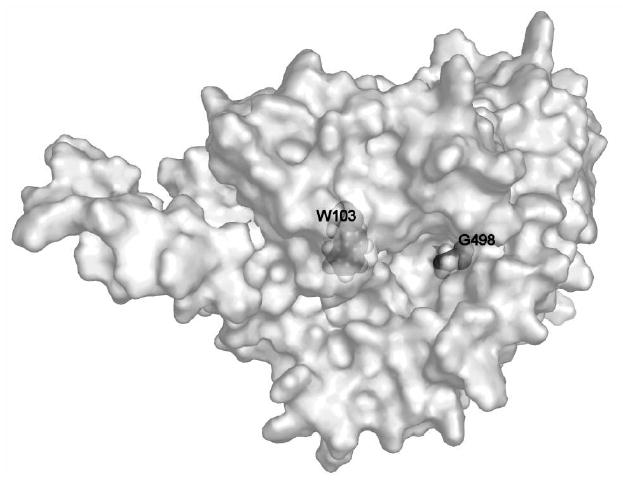
The view is looking down into the proposed entrance to the substrate permeation pathway, approximately in the center of the protein, just to the left of G498, which is evident on the exposed surface of the protein. W103, by contrast, is buried slightly within the protein and we hypothesize only becomes accessible when a ligand binds within the permeation pathway.
Figure 4. MTS-MPP+ inactivates [3H]5-HT uptake at the hSERT G498C and W103C mutants.
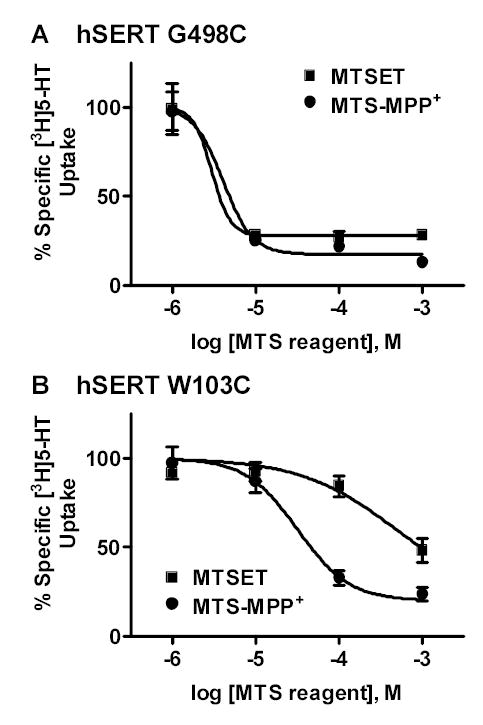
G498C/hSERT, or W103C/hSERT transiently expressed in HEK-293 cells were treated with increasing concentrations of either MTSET or MTS-MPP+ as described in the Experimental Section. (A) G498C/hSERT showed a decrease in [3H]5-HT uptake following treatment with MTS-MPP+ and MTSET. The potencies of MTS-MPP+ and MTSET were similar (Log EC50 values: MTS-MPP+ −5.4 ± 0.2 vs. MTSET −5.5 ± 0.1). (B) W103C/hSERT was inactivated by MTS-MPP+ at lower concentrations than MTSET (Log EC50 values: MTS-MPP+ −4.5 ± 0.1 vs. MTSET −3.3 ± 0.2). Data represent the mean ± SEM from three separate experiments performed in duplicate. R2 values for each curve fit were: G498C, MTSET 0.999, MTS-MPP+ 0.995; W103C, MTSET 0.996, MTS-MPP+ 0.999.
Based on our homology model of hSERT, W103 in transmembrane helix I is predicted to be in the region lining the permeation pathway. The W103C mutant also has been shown to be sensitive to inactivation by MTSET.17,22 Because W103 resides deeper in the protein region surrounding the pore region than G498, however, we predicted that MTS-MPP+, which presumably binds to SERT at sites within the substrate permeation pathway, would inactivate the W103C mutant with greater potency than the non-selective reagent MTSET. Indeed, MTS-MPP+ inactivated the W103C mutant with a ten-fold greater potency than MTSET (Fig. 4B). These data are consistent with the hypothesis that MTS-MPP+ reacts more rapidly with residues in or near the substrate permeation pathway, in contrast to MTSET, which simply interacts with any freely accessible cysteine residue.
We speculated that both MTSET and MTS-MPP+ would be capable of reacting with residues such as G498C and W103C that are located in the aqueous region of the permeation pathway. Even though MTS-MPP+ proved not to be a substrate, we envisioned that its structural similarity to MPP+ would allow it to be more concentrated in the permeation pathway, and hence would produce more rapid covalent labeling of those residues deeper in the pore.
By contrast, with its non-specific interactions and relatively small size, MTSET would not be concentrated in the permeation pathway and would have free access to cysteine residues in any region that was exposed to the aqueous environment. These interactions of MTSET could alter transporter function by disrupting conformations required for substrate translocation. We believe the experiments with the G498C and W103C mutants give results consistent with this reasoning. G498C is predicted to reside at the surface of the pore and is thus freely accessible to both MTSET and MTS-MPP+. W103C is predicted to lie deeper within the permeation pathway, and reacts more potently with MTS-MPP+ than MTSET, possibly even becoming more accessible as a result of the functional processes involved in substrate binding.
4. Conclusions
We have synthesized a novel analog of MPP+ that contains a reactive methanethiosulfonate group similar to commercially-available methanethiosulfonate compounds. Synthesis of an MTS-containing MPP+ molecule should allow for better understanding of specific residues that interact with MPP+. The ability of this compound, MTS-MPP+, to inhibit [3H]5-HT uptake at C109A/hSERT versus MPP+ was determined to characterize the interaction of this molecule with SERT. Unfortunately, MTS-MPP+ was not as potent as MPP+ at inhibiting [3H]5-HT uptake. Furthermore, an inability to induce current at C109A/hSERT suggests that, unlike MPP+, MTS-MPP+ does not function as a substrate. In fact, MTS-MPP+ appeared to act as an antagonist, as outward movement of current was evident in the presence of MTS-MPP+, suggesting the blockade of the SERT "leak" current.23
A more potent interaction of MTS-MPP+ with a residue that lines the actual permeation pathway was observed when compared to the non-selective reagent MTSET. Alternative explanations for our results could include that the more lipophilic nature of MTS-MPP+ or its more linear conformation compared to the spherical MTSET might allow more ready access to the W103C mutant. Additional studies of the MTS-MPP+ reagent at other biogenic amine transporters as well as in comparisons to other non-specific MTS reagents are warranted. Regardless, it therefore seems possible that the differential reactivity to MTS-MPP+ versus a nonspecific SCAM reagent such as MTSET may be useful in identifying residues that line the extracellular face of the substrate permeation pathway.
5. Experimental
5.1 General
All reagents were commercially available and were used without further purification unless otherwise indicated. Anhydrous THF was obtained by distillation from benzophenone-sodium under nitrogen immediately before use. Melting points were determined using a Thomas-Hoover apparatus and are uncorrected. 1H NMR spectra were obtained with a Bruker AXR300 (300 MHz) instrument. Chemical shifts are reported in δ values (ppm) relative to an internal reference of TMS. Chemical ionization mass spectra (CI-MS), using isobutene as the carrier gas, were obtained with a Finnigan 4000 spectrometer. Electrospray ionization analyses were carried out on a Finnigan MAT LCQ Classic (ThermoElectron Corp, San Jose, CA) mass spectrometer. Elemental analyses were performed by the Purdue University Microanalysis Laboratory and are within ±0.4% of the calculated values. Thin-layer chromatography was performed using J.T. Baker flex silica gel IB2-F, plastic-backed sheets with fluorescent indicator, visualizing with UV light at 254 nm and plates developed with ethyl acetate, unless otherwise noted. When indicated, compounds were purified by column chromatography over using silica gel 60, 230–400 mesh (J.T.Baker). All reactions were carried out under an inert atmosphere of argon unless otherwise indicated.
5.1.1 4-Methylthiophenylboronic acid (1)
A solution of 4-bromothioanisole (5.0 g, 24.6 mmol) in 100 mL of dry THF was cooled to −78 °C. A solution of 2.5 M n-BuLi in hexane (34.0 mL, 85 mmol) was slowly added. After 15 min stirring, triisopropoxy borate (5.64 g, 6.90 mL, 30 mmol) was added in one portion. The mixture was stirred at −78 °C for 4 h, then warmed to RT and stirred overnight. The reaction was quenched by addition of 50 mL of water, the THF was removed by rotary evaporation, and the product was extracted into Et2O. The organic layer was washed with brine, dried, and evaporated to yield 2.56 g of crude product (72%). 1H NMR (300 MHz, CD3COCD3): δ 7.85 (d, 2H, J = 8.3 Hz), 7.25 (d, 2H, J = 7.50 Hz), 7.00 (s, 2H, OH), 2.60 (s, 3H, SCH3). Mp: 211–212 °C; lit24 mp 206–207 °C; CI-MS (m/z) 169 (M+ H).
5.1.2 4-(4-Methylthiophenyl)pyridine (2)
Boronic acid 1 (2.0 g, 11.9 mmol) and 4-bromopyridine hydrochloride (2.3 g, 11.9 mmol) were dissolved in 50 mL DME. To this stirring mixture was added KOH (3.4 g, 60 mmol) and (Bu)4NBr (0.5 g, 1.5 mmol) in 30 mL of water. The mixture was stirred and degassed by sparging with Ar for 20 min at RT. Tetrakis-(triphenylphosphine)palladium catalyst (0.5 g, 0.43 mmol) was added and the mixture was heated at 80 °C for 48 h. The reaction was then cooled to RT, the solvents were removed under vacuo, the residue was taken up into CH2Cl2, washed with water and brine, dried (anhydrous Na2SO4), filtered, and the solvent evaporated to yield the product. The crude base was then purified by column chromatography over silica, using ethyl acetate:hexane (1:9) to yield the product as white crystals 1.50 g (63%). 1H NMR (300 MHz, CDCl3): δ 8.63 (d, 2H, J = 6.07 Hz), 7.55 (d, 2H, J = 8.36 Hz), 7.47 (d, 2H, J = 6.09 Hz), 7.33 (d, 2H, J = 8.37 Hz), 2.51 (s, 3H, SCH3). Mp: 112–113 °C; CI-MS (m/z) 202 (M+ H). Anal. (C12H11NS) C, H, N, S.
5.1.3 4-(4-Thiophenyl)pyridine (3)
To an oven-dried 250 mL flask, connected to a condenser and flushed with Ar, was added 1.0 g (5.0 mmol) of 2, followed by 1.12 g (10.0 mmol) of commercial tert-BuSNa powder. Dry DMF (50 mL) was injected via syringe while stirring vigorously. The mixture was placed in an oil bath preheated to 160 °C and stirred for 5 h. The reaction was allowed to cool to room temperature, maintaining the Ar atmosphere. The reaction was quickly poured over 100 g of crushed ice and stirred until the ice had melted. The pH was adjusted to 7 and the mixture was extracted with 3 x 70 mL CH2Cl2. This organic solution was washed repeatedly with water to remove DMF, then dried, filtered, and the solvent evaporated to afford the product as dark white crystals 0.75 g (81.1%). 1H NMR (300 MHz, CDCl3): δ 8.65 (d, 2H, J = 6.18 Hz), 7.55–7.30 (m, 6H), 3.55 (s, 1H, SH). Mp: 171–172 °C, lit25 mp 171 °C; CI-MS (m/z) 188 (M+ H).
5.1.4 4-(4-Pyridyl)phenyldisulfide (4)
Following the method of Raghavan et al.,26 a solution of 3 (1.0 g, 5.3 mmol) in dry CH2Cl2 (10 mL) was cooled to −15 °C. Vanadyl acetylacetonate (0.5 mol%) and tert-butylhydroperoxide (1.0 mL of ca. 5.5M in decane; 5.30 mmol) were then added. The reaction was stirred overnight, with gradual warming to room temperature. The mixture was then diluted with CH2Cl2 and washed with water, 0.2 N NaOH, water, and brine. The organic layer was dried, filtered and the solvent evaporated to provide the product as yellow crystals; 1.0 g (50%). 1H NMR (300 MHz, CDCl3): δ 8.66 (dd, 4H, J = 1.64 Hz, J′ = 6.18 Hz), 7.62 (m, 8H), 7.46 (dd, 4H, J = 1.64 Hz, J′ = 6.19 Hz). CI-MS (m/z) 373 (M+ H). Mp: 171–172 °C. Anal. (C22H16N2S2) C, H, N, S.
5.1.5 4-Pyridyl-(4-phenylmethanethiosulfonate) (5)
This step was based on the procedure of Bentley et al.16 To a solution of 4 (0.2 g, 0.53 mmol) in 50% acetone-H2O (5 mL) was added a solution of silver nitrate (100 mg, 0.60 mmol) and sodium methanesulfinate (60 mg, 0.60 mmol) in 50% acetone-H2O (5 mL). The mixture was heated at reflux for 1 h, during which the mixture became bright yellow. The product mixture was filtered and the filtrate was extracted with 3 x 50 mL of CH2Cl2. The combined organic extracts were dried, filtered, and evaporated to yield yellow crystals; 100 mg (71.4%). 1H NMR (300 MHz, CDCl3): δ 8.72 (dd, 2H, J = 1.52 Hz, J′ = 6.10 Hz), 8.84 (m, 2H), 7.74 (m, 2H), 7.51 (dd, 2H, J = 1.67 Hz, J′ = 6.15 Hz), 3.43 (s, 3H, SO2Me). Mp: 133–135 °C; CI-MS (m/z) 373 (M+ H).
5.1.6 1-Methylpyridinium-4-(4-phenylmethanethiosulfonate) iodide (6)
To a solution of 5 (50 mg, 0.19 mmol) in dry THF (0.5 mL) was added dropwise a solution of methyl iodide (26.7 mg, 0.011 mL, 0.19 mmol) in 0.5 mL of dry THF. The mixture was stirred for 24 h at room temperature as the pyridinium salt precipitated. The product was collected by filtration, washed with THF (5 mL), and air dried. IR 1315, 1132 cm−1 (S-SO2). 1H NMR (300 MHz, D2O): δ 8.75 (d, 2H, J = 5.69 Hz), 8.25 (d, 2H, J = 5.51 Hz), 7.97 (m, 4H), 4.32 (s, 3H, NMe), 3.37 (s, 3H, SO2Me). Mp: 164–167 °C; ESI-MS (m/z) 280 (M+). Anal. (C13H14INO2S2) C, H, N, I.
5.2 SERT expression and electrophysiology in oocytes
hSERT contains one native cysteine, C109, that is reactive with MTS reagents, thus all studies were performed with the hSERT C109A mutant.27 hSERT C109A/pcDNA3.1+ was linearized and transcribed in vitro using the AmpliCap™ T7 High Yield Message Maker Kit (Eppicentre, Madison, WI). Following injection of SERT cRNA, Xenopus laevis oocytes were maintained at 18 °C in Ca2+-Ringer’s solution. Three or four days post-injection oocytes were subjected to two-electrode voltage-clamp. Recording solutions consisted of room temperature Ca2+-Ringer’s solution with concentrations of 5-HT, fluoxetine, MTS-MPP+ or MPP+ (Sigma), as indicated. Transporter-associated currents were recorded by clamping the oocyte membrane potential at −120 mV and perfusing the oocytes with 5-HT or drug for fifteen to thirty seconds and then washing with Ca2+-Ringer’s solution for at least forty seconds. Perfusion was controlled by gravity. For MTS-MPP+ experiments, MTS-MPP+ was made fresh in deionized water and then diluted in Ca2+-Ringer’s for perfusion. The oocyte was clamped at −120 mV for the duration of the experiment. Data were acquired digitally using Clampex 8.1 (Axon Instruments) and analyzed using Clampfit 8.1 (Axon Instruments) and SigmaPlot 5.0 (SPSS Science, Chicago, IL). Current averages and statistics were performed using Prism 3.0 (GraphPad Software, San Diego, CA). Water-injected oocytes were assayed in parallel with SERT-injected oocytes to determine nonspecific effects on currents.
5.3 [3H]5-HT uptake assays in HEK cells
HEK cells were maintained, plated, and transfected with C109A/hSERT cDNA using Lipofectamine 2000. Forty-eight hours post-transfection, cells were washed once with KRH/glucose buffer (120 mM NaCl, 4.7 mM KCl, 2.2 mM CaCl2, 10 pH 7.4). KRH/glucose (200 mM HEPES, 1.2 mM KH2PO4, 1.2 mM MgSO4, 1.8 g/L D-glucose, μL; 37 °C) was added to each well alongside 25 μL fluoxetine (nonspecific, 10 μM final), increasing concentrations of MPP+ or MTS-MPP+, or KRH/glucose (total) and incubated for ten minutes at 37 °C. Next, 25 μL of [3H]5-HT (Amersham, Piscataway, NJ, ~125 Ci/mmol; final assay concentration 20 nM) diluted in KRH/glucose buffer supplemented with 100 μM pargyline and 100 μM ascorbic acid was added to each well, followed by incubation for an additional ten minutes at 37 °C. Cells were then washed three times with cold KRH/glucose buffer, solubilized with scintillation cocktail (Microscint-20, Packard Instruments), and shaken overnight before determining accumulated tritium using a Packard TopCount NXT. Data were analyzed using the nonlinear regression curve fit for a sigmoidal dose-response curve (four parameter logistic equation) from GraphPad Prism v3.0 (GraphPad Software, San Diego, CA). Total specific accumulation of tritium equaled ~25,000 cpm which is <10% of the total available tritium in each assay (>400,000 cpm), thus, validating the use of this equation. The concentration of substrate (20 nM) used in our uptake inhibition assays is far below the Km value for 5-HT uptake (500–700 nM) making the conversion from IC50 to Ki values negligible (<4%).
For the MTS-MPP+ inactivation assay, C109A/hSERT, W103C/hSERT, or G498C/hSERT cDNAs were transfected into HEK cells as described above. Forty-eight hours post-transfection, the cells were washed once with PBS/CM buffer (137 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 8.1 mM Na2HPO4, 0.1 mM CaCl2, and 1.0 mM MgCl2). For the assay, 225 μL PBS/CM was added to each well plus 25 μL of increasing concentrations of MTS reagent or KRH/glucose (total and nonspecific wells) followed by incubation at room temperature for ten minutes. Following incubation with MTS-MPP+, cells were washed twice with PBS/CM. Next, 225 μL of 37 °C KRH/glucose (200 μL KRH/glucose + 25 μL 10 μM fluoxetine for nonspecific wells) were added to each well, as well as 25 μL of [3H]5-HT (20 nM final). The cells were incubated for ten minutes at 37 °C. Transport was terminated by washing twice with cold KRH/glucose. Cells were solubilized and data analyzed as described above.
Acknowledgments
This work was supported by NIDA grant DA02189 (D.E.N.) and NIMH grant MH60221 (E.L.B.).
References
- 1.Shaked Z, Szajewski RP, Whitesides GM. Biochemistry. 1980;19:4156. doi: 10.1021/bi00559a004. [DOI] [PubMed] [Google Scholar]
- 2.Bruice TW, Kenyon GL. J Protein Chem. 1982;1:47. [Google Scholar]
- 3.Stauffer DA, Karlin A. Biochemistry. 1994;33:6840. doi: 10.1021/bi00188a013. [DOI] [PubMed] [Google Scholar]
- 4.Javitch JA, Li X, Kaback J, Karlin A. ProcNatlAcadSciUSA. 1994;91:10355. doi: 10.1073/pnas.91.22.10355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Javitch JA, Fu D, Chen J, Karlin A. Neuron. 1995;14:825. doi: 10.1016/0896-6273(95)90226-0. [DOI] [PubMed] [Google Scholar]
- 6.Javitch JA, Fu D, Chen J. Molecular Pharmacology. 1996;49:692. [PubMed] [Google Scholar]
- 7.Javitch JA. AdvPharmacol. 1998;42:412. [Google Scholar]
- 8.Javitch JA. Methods Enzymol. 1998;296:331. doi: 10.1016/s0076-6879(98)96025-6. [DOI] [PubMed] [Google Scholar]
- 9.Javitch JA, Shi L, Simpson MM, Chen J, Chiappa V, Visiers I, Weinstein H, Ballesteros JA. Biochemistry. 2000;39:12190. doi: 10.1021/bi001069m. [DOI] [PubMed] [Google Scholar]
- 10.Javitch JA, Shi L, Liapakis G. Methods Enzymol. 2002;343:137. doi: 10.1016/s0076-6879(02)43131-x. [DOI] [PubMed] [Google Scholar]
- 11.Shi L, Simpson MM, Ballesteros JA, Javitch JA. Biochemistry. 2001;40:12339. doi: 10.1021/bi011204a. [DOI] [PubMed] [Google Scholar]
- 12.Kitayama S, Shimada S, Uhl GR. AnnNeurol. 1992;32:109. doi: 10.1002/ana.410320120. [DOI] [PubMed] [Google Scholar]
- 13.Inazu M, Kubota N, Takeda H, Oguchi K, Koizumi M, Kimura S, Matsumiya T. NeurochemInt. 2001;39:253. doi: 10.1016/s0197-0186(01)00015-8. [DOI] [PubMed] [Google Scholar]
- 14.Martel F, Keating E. Placenta. 2003;24:361. doi: 10.1053/plac.2002.0917. [DOI] [PubMed] [Google Scholar]
- 15.Dohi T, Kitayama S, Morioka N, Kumagai K, Mitsuhata C, Morita K, Kozai K, Lin Z, Uhl GR. Nihon Shinkei Seishin Yakurigaku Zasshi. 2004;24:43. [PubMed] [Google Scholar]
- 16.Bentley MD, Douglass IB, Lacadie JA. Journal of Organic Chemistry. 1972;37:333. [Google Scholar]
- 17.Henry LK, Adkins EM, Han Q, Blakely RD. J BiolChem. 2003;278:37052. doi: 10.1074/jbc.M305514200. [DOI] [PubMed] [Google Scholar]
- 18.Chen JG, Liu-Chen S, Rudnick G. Biochemistry. 1997;36:1479. doi: 10.1021/bi962256g. [DOI] [PubMed] [Google Scholar]
- 19.Mager S, Min C, Henry DJ, Chavkin C, Hoffman BJ, Davidson N, Lester HA. Neuron. 1994;12:845. doi: 10.1016/0896-6273(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 20.Lin F, Lester HA, Mager S. Biophys J. 1996;71:3126. doi: 10.1016/S0006-3495(96)79506-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Nature. 2005;437:215. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 22.White KJ, Kiser PD, Nichols DE, Barker EL. Protein Science. 2006 doi: 10.1110/ps.062386106. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mager S, Min C, Henry DJ, Chavkin C, Hoffman BJ, Davidson N, Lester HA. Neuron. 1994;12:845. doi: 10.1016/0896-6273(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 24.Brikh A, Morin C. Journal of Organometallic Chemistry. 1999;581:82. [Google Scholar]
- 25.Combellas C, Dellerue S, Mathey G, Thiebault A. Tetrahedron Letters. 1997;38:539. [Google Scholar]
- 26.Raghavan S, Rajender A, Joseph S, Rasheed MA. SynthCommun. 2001;31:1477. [Google Scholar]
- 27.Chen JG, Liu-Chen S, Rudnick G. Biochemistry. 1997;36:1479. doi: 10.1021/bi962256g. [DOI] [PubMed] [Google Scholar]