

Abstract
We determined whether the implantation of human pancreatic cancer cells into the pancreas of nude mice can be used to select variants with increasing metastatic potential. COLO 357 line fast-growing cells were injected into the spleen or pancreas of nude mice. Hepatic metastases were harvested, and tumor cells were reinjected into the spleen or pancreas. This cycle was repeated several times to yield cell lines L3.6sl (spleen to liver) and L3.6pl (pancreas to liver). The variant cells produced significantly higher incidence and number of lymph node and liver metastases than the parental cells. Their increased metastatic potential was associated with increased expression (mRNA and protein) of the proangiogenic molecules basic fibroblast growth factor, vascular endothelial growth factor, and interleukin-8. The metastatic cells also exhibited increased motility and invasiveness, which were associated with increased expression of collagenase type IV (MMP-9) and decreased expression of E-cadherin. Collectively, the data show that the orthotopic implantation of human pancreatic cancer cells in nude mice is a relevant model with which to study the biology of pancreatic cancer metastasis and to select variant cell lines with enhanced metastatic potential.
Keywords: pancreatic cancer, orthotopic, metastasis, selection, IL-8
Introduction
Cancer of the exocrine pancreas is a major unsolved health problem with approximately 27,000 deaths per year in the United States and 50,000 deaths per year in Western Europe [1,2]. It is characterized by extensive local invasion as well as early lymphatic and hematogenous metastasis. Due to the inability to detect pancreatic cancer at an early stage, its aggressiveness, and the lack of effective systemic therapies, only 1% to 4% of all patients with adenocarcinoma of the pancreas will be alive 5 years after diagnosis [3–6]. Because most deaths from pancreatic cancer are caused by metastasis, improvements in treatment require a better understanding of the biology of this process.
The process of metastasis consists of sequential and selective steps that include proliferation, induction of angiogenesis, detachment, motility, invasion into the circulation, aggregation and survival in the circulation, cell arrest in distant capillary beds and extravasation into the organ parenchyma, response to local growth factors, induction of angiogenesis, and proliferation [7]. The outcome of metastasis is determined by multiple interactions between metastatic tumor cells and host factors [7,8]. To produce clinically relevant metastases, tumor cells must complete all steps in the metastatic cascade [7]. Thus, the failure to produce a metastasis can be due to different single or multiple deficiencies [9].
The process of metastasis is highly selective and its outcome depends on the interaction of metastatic cells that preexist within a heterogeneous primary neoplasm and a variety of host factors [7,8]. Critical to our studies has been the development of relevant in vivo models for cancer metastasis [10,11], which have produced conclusive evidence that the outcome of metastasis is regulated by the interaction of unique tumor cells with homeostatic mechanisms [7,8]. Indeed, studies from our laboratory and others have shown that malignant human tumors implanted into orthotopic organs are highly vascularized, grow progressively, and produce distant metastasis, whereas the implantation of the same tumors at an ectopic organ does not lead to extensive angiogenesis or production of metastasis [12–20]. Similarly, the isolation of multiple variants (from biologically heterogeneous neoplasms) that differ in metastatic properties has greatly advanced our understanding of the genetic and epigenetic determinants of cancer metastasis [7–9].
Two general methods have been used to select metastatic variants in vivo. In the first, tumor cells are implanted into orthotopic organs, and spontaneous metastases from distant organs are isolated and reinjected into orthotopic organs. In the second method, tumor cells are introduced into the circulation to produce lesions in distant organs, or experimental metastases. Spontaneous metastases then occur subsequent to completion of all steps of the metastatic process, whereas experimental metastases reflect the ability of tumor cells to survive in the circulation, arrest in a distant capillary bed, and grow in a distant organ parenchyma [7–11].
Unfortunately, such models do not exist for pancreatic cancer. The purpose of the present study was, therefore, to develop an orthotopic model for metastasis of human pancreatic cancer, to select (in vivo) variant lines with increasing metastatic capacity, and to begin detailed characterization of their properties. To do so, we used the COLO 375 human pancreatic cancer cell line originally established by Morgan et al. [21] from a celiac axis lymph node that had been partially replaced by neoplastic foci of well-differentiated, mucin-containing pancreatic ducts. Vezeridis et al. subsequently injected the fast-growing (FG) variant line of the COLO 375 cells into the spleen of nude mice. The in vivo isolated cells designated as L3.3 produced liver lesions at a higher incidence than the original COLO 375 cells [22].
In the present study, we show that orthotopic implantation of heterogeneous human pancreatic adenocarcinoma into nude mice results in rapid growth and production of lymph node and liver metastases. Cells isolated from the metastases were more metastatic because they expressed several genes that correlate with production of metastasis.
Materials and Methods
Animals
Male athymic nude mice (BALB/c background) were purchased from the Animal Production Area of the National Cancer Institute-Frederick Cancer Research and Development Center (Frederick, MD). The mice were housed and maintained in laminar flow cabinets under specific pathogen-free conditions in facilities approved by the American Association for Accreditation of Laboratory Animal Care and in accordance with current regulations and standards of the US Department of Agriculture, US Department of Health and Human Services, and the National Institutes of Health. The mice were used in accordance with institutional guidelines when they were 8 to 12 weeks old.
Pancreatic Cancer Cell Lines and Culture Conditions
The FG and L3.3 [21,22] human pancreatic cancer cell lines were maintained as monolayer cultures in Dulbecco's minimal essential medium (DMEM), supplemented with 10% fetal bovine serum (FBS), sodium pyruvate, nonessential amino acids, l-glutamine, and a 2-fold vitamin solution (Life Technologies, Rockville, MD). The cultures were incubated at 37°C in a mixture of 5% carbon dioxide and 95% oxygen. The cultures were tested and found to be free of Mycoplasma and the following pathogenic murine viruses: reovirus type 3, pneumonia virus, K virus, Theiler's encephalitis virus, Sendai virus, minute virus, mouse adenovirus, mouse hepatitis virus, lymphocytic choriomeningitis virus, ectromelia virus, and lactate dehydrogenase virus (assayed by M. A. Bioproducts, Walkersville, MD). The cultures were maintained for no longer than 12 weeks after recovery from frozen stocks.
Tumor Cell Injection Techniques
For in vivo injection, cells were harvested from culture flasks by a 2 to 3 minutes treatment with trypsin and were transferred to serum-free Hanks' balanced salt solution (HBSS). Only single-cell suspensions of greater than 90% viability (trypan blue exclusion) were used for injection.
Orthotopic injection Male nude mice were anesthetized with methoxyflurane. A small left abdominal flank incision was made and the spleen exteriorized. Tumor cells (1 x 106/40 µL HBSS) were injected subcapsularly in a region of the pancreas just beneath the spleen. We used a 30-gauge needle, a 1-mL disposable syringe, and a calibrated, push-button-controlled dispensing device to inject the tumor cell suspension (Hamilton Syringe Co, Reno, NV). A successful subcapsular intrapancreatic injection of tumor cells was identified by the appearance of a fluid bleb without intraperitoneal leakage. To prevent such leakage, a cotton swab was held for 1 minute over the site of injection. One layer of the abdominal wound was closed with wound clips (Auto-clip; Clay Adams, Parsippany, NJ). The animals tolerated the surgical procedure well, and no anesthesia-related deaths occurred.
Intrasplenic inoculation Tumor cells were injected into the spleen by methods previously described in detail [11,12]. Briefly, mice were anesthetized with methoxyflurane, a small left abdominal flank incision was created, and the spleen was exteriorized. Tumor cells (1 x 106/40 µL HBSS) were injected into the spleen with a 30-gauge needle. The spleen was returned to the abdomen, and the wound was closed in one layer with wound clips.
Intravenous injection Tumor cells (1 x 106 cells/200 µl HBSS) were injected into the lateral tail vein of unanesthetized nude mice with a 27-gauge needle.
Necropsy Procedure and Histopathological Studies1
Mice were euthanized by methoxyflurane. The presence of tumor lesions in the pancreas, spleen, lymph nodes, liver, lung, and other peritoneal organs was confirmed with a dissecting microscope. The number of visible liver and lung metastases, as well as enlarged regional lymph nodes, was determined with the aid of a dissecting microscope. Tumor lesions were harvested. Some were snap-frozen in liquid nitrogen for mRNA extraction; some were fixed in 10% buffered formalin and embedded in paraffin, and some were embedded in OCT compound (Miles, Inc, Elkhart, IN), snap-frozen in liquid nitrogen, and stored at -70°C.
In Vivo Selection of Metastatic Variants from the L3.3 Human Pancreatic Cancer Cell Line
L3.3 cells were injected into the pancreas or spleen of nude mice. Experimental (spleen injection) or spontaneous (pancreas injection) liver metastases were harvested and treated with DNase and collagenase [12,13]. Cells were established in culture. Primary cultures were passaged in vitro 2 or 3 times, and then cells were harvested by trypsinization and injected into the spleen or pancreas of another set of nude mice. The selection cycle was repeated 3 times to yield lines designated as L3.6sl (spleen to liver selection) or L3.6pl (pancreas to liver selection).
Immunohistochemistry
Paraffin-embedded tissues were sectioned at 4 to 6 µm thickness, mounted on positively charged Superfrost slides (Fisher Scientific Co, Houston, TX), and allowed to dry overnight at room temperature. Sections were deparaffinized in xylene followed by a graded series of ethanol (100%, 95%, 80%), and rehydrated in phosphate-buffered solution, pH 7.5). Paraffin-embedded tissues were used for localization of vascular endothelial growth factor/vascular permeability factor (VEGF/VPF), basic fibroblast growth factor (bFGF), matrix metalloproteinase-9 (MMP-9), and proliferating cell nuclear antigen (PCNA) proteins. Sections analyzed for PCNA and MMP-9 were microwaved 5 minutes for antigen retrieval [23]. All other paraffin-embedded tissues were treated for 5 minutes with pepsin (Biomeda, Foster City, CA) at 37°C and then washed 3 times with PBS. To identify CD-31/PECAM-1 [24], frozen tissues were sectioned (8–10 µm), mounted on positively charged Plus slides (Fisher Scientific), and air-dried 30 minutes. Frozen tissues were fixed in cold acetone (5 min), acetone/chloroform 1:1 (5 min), and acetone (5 min) and washed 3 times with PBS.
After the specific pretreatment procedures, all samples were incubated with a 3% H2O2 in methanol solution for 12 minutes at room temperature to block endogenous peroxidases. Sections were then washed 3 times with PBS and then incubated for 20 minutes at room temperature in a protein-blocking solution consisting of PBS supplemented with 1% normal goat serum and 5% normal horse serum. The primary antibodies were diluted in protein blocking solution to the desired concentration and applied to the sections overnight at 4°C. The sections were then rinsed in PBS and incubated for 10 minutes in protein-blocking solution before the addition of peroxidase-conjugated secondary antibody. After incubation in secondary antibody for 1 hour at room temperature, the samples were washed and incubated with stable diaminobenzidine (DAB, Research Genetics, Huntsville, AL). Staining was monitored under a bright-field microscope, and the reaction was stopped by washing with distilled water. Sections were counterstained with Gill's 3 hematoxylin (Sigma Chemical Co, St. Louis, MO) and mounted with Universal Mount (Research Genetics). The concentration of primary and secondary antibodies was determined by using separate specimens. Control specimens exposed to secondary antibody alone showed no specific staining.
Quantification of Microvessel Density and Immunohistochemistry
For the quantification of microvessel density (MVD), frozen sections of the different pancreatic tumors were fixed and stained with antibodies against CD31/PECAM-1 [24]. For each tumor, four fields of 1 mm2 each were captured at 40 x magnification with highest intensity [25–27] by using a Sony 3-chip camera (Sony Corporation of America, Montvale, NJ) mounted on a Zeiss universal microscope (Carl Zeiss, Thornwood, NY) and Optimas Image Analysis software (Bioscan, Edmond, WA), installed in a Compaq computer with Pentium chip, a frame grabber, an optical disk storage system, and a Sony color printer.
For quantification of the staining intensity, the optical density (OD) at 200 x magnification of 100 VEGF/VPF, bFGF, and MMP-9 positive cells from each tumor tissue was measured by using the Optimas Image Analysis soft ware (Bioscan). The samples were not counterstained, so the OD was due solely to the product of the immunohistochemical (IHC) reaction [27]. To minimize changes in background and color intensities that may be encountered with fluctuation in illumination due to filament life or optical alignment, all images to be used for analysis were digitized and stored for further analysis. The intensity of staining with the different antibodies was standardized to that of the integrated OD and determined by comparison with the integrated OD of the FG tumor, which was set at 100 [27].
For quantification of PCNA expression, the number of PCNA + cells was counted in four fields of 0.161-mm2 each at 100 x magnification at the areas of most intense PCNA expression.
Antibodies and Oligonucleotide Probes
All antibodies were purchased as listed: rabbit anti-bFGF (Sigma), rabbit anti-VEGF/VPF (Santa Cruz, Santa Cruz, CA), monoclonal mouse anti-MMP-9 AB3 (Oncogene Research, Uniondale, NY), rat anti-mouse CD31/PECAM-1 and peroxidase-conjugated rat anti-mouse IgG1 (Pharmingen, San Diego, CA), mouse anti-PCNA clone PC 10 (DAKO A/S, Copenhagen, Denmark), peroxidase-conjugated F(ab′)2 goat anti-rabbit IgG F(ab′)2 and peroxidase-conjugated goat anti-rat IgG [H + L] (Jackson Research Laboratories, West Grove, CA), peroxidase-conjugated rat anti-mouse IgG2a (Serotec, Harlan Bioproducts for Science, Inc., Indianapolis, IN), and peroxidase-conjugated F(ab′)2 anti-mouse IgG1 (Oncogene).
Specific antisense oligonucleotide DNA probes were designed complementary to the mRNA transcripts of metastasis and angiogenesis-related genes (MMP-2, MMP-9, E-cadherin, VEGF, bFGF, interleukin-8 [IL-8]) based on published reports of the cDNA sequence: VEGF/VPF (TGG′TGA′TGT′TGG′-ACT′CCT′CAG′TGG′GCU), 57.7% guanosine-cytosine (CG) content [28]; bFGF (CGG′GAA′GGC′GCC′GCT′GCC′GCC), 85.7% GC content [27]; IL-8 (CTC′CAC′AAC′CCT′CTG′CAC′CC), 65% GC content [29]; MMP-2 (TGG′GCT′ACG′GCG′CGG′CGG′CGT′GGC), 88.9% GC content [30]; MMP-9 (CCG′GTC′CAC′CTC′GCT′GGC′GCT′CCG′GU), 80.0% GC content [31]; E-cadherin (mixture) (TGG′AGC′GGG′CTG′GAG′TCT′GAA′CTG′), 62.5% CG content and (GAC′GCC′GGC′GGC′CCC′TTC′ACA′GTC′), 75% CG content [32]. The specificity of the oligonucleotide sequence was initially determined by Gene Bank European Molecular Biology Library database search with the use of Genetics Computer Group sequence analysis program (GCG, Madison, WI) based on the FastA algorithm [33]. These sequences showed 100% homology with the target gene and minimal homology with nonspecific mammalian gene sequences.
In Situ Hybridization
In situ hybridization (ISH) was carried out according to the microprobe manual staining system (Fisher Scientific Co, Pittsburgh, PA) [34,35]. Tissue sections (5 µm) of formalin-fixed, paraffin-embedded specimens were mounted on Silane-coated ProbeOn slides (Fisher Scientific). The slides were placed in the microprobe slide holder, dewaxed, and dehydrated with Autodewaxer and Autoalcohol (Research Genetics), followed by an enzymatic digestion with pepsin [36]. Hybridization of the biotinylated probe was carried out sequentially for 3 minutes at 100°C, 4 minutes at room temperature, and 45 minutes at 45°C. The samples were then washed 3 times with 2 x standard saline citrate (SSC) for 2 minutes at 45°C. The samples were incubated for 30 minutes in alkaline phosphatase-labeled avidin (DAKO, Carpineria, CA) at 45°C, briefly rinsed in Tris-buffered saline (50 mmol/L Tris-HCl [pH 7.6], 150 mmol/L NaCl), rinsed for 1 minute in alkaline phosphatase enhancer (Biomeda Corp, Foster City, CA), and finally incubated for 30 minutes with chromogen substrate FastRed (Research Genetics) at 45°C. Slides were washed several times with distilled water, counterstained with Gill's 3 hematoxylin (Sigma), and mounted with Universal Mount (Research Genetics). A positive reaction in this assay stained red. Control for endogenous alkaline phosphatase included treatment of the samples in the absence of the biotinylated probe and use of chromogen in the absence of any oligonucleotide probes. To check the specificity of the hybridization signal, the following controls were used: 1) RNase pretreatment of tissue sections; 2) a biotin-labeled sense probe; and 3) competition assay with unlabeled antisense probe [37]. A markedly decreased or absent signal was obtained after all of these treatments.
Stained sections were examined under a Zeiss photo-microscope (Zeiss) equipped with a 3-chip-charged coupled device color camera (model DXC-960 MD, Sony). The images were analyzed by using the Optimas Image Analysis software (version 5.2, Bothell, WA). The samples were not counterstained, so the OD was due solely to the product of the ISH reaction. For each section, we determined the OD in several 2 x 2 mm zones located at the center and periphery of the tumor. Three to five different fields in each 2 x 2 mm zone were quantified to derive an average value. The intensity of staining was standardized to that of the integrated OD of poly d(T0)20 and determined by comparison with the integrated OD of nonpathological pancreatic epithelial cells, which was set at 100.
In Vitro Growth Kinetics
Pancreatic cancer cells were harvested from 75% confluent monolayer cultures by a brief trypsinization with 0.1% trypsin and 0.1% EDTA. The cells were washed in cold HBSS, plated at a density of 1500 cells per 38-mm2 well, and then given supplemented DMEM containing 10% FBS. Cell growth was monitored after 8 hours and 1, 2, 3, and 4 days by using the MTT assay as described previously [38]. In vitro doubling times were calculated from the log phase growth curve [39].
In Vitro Migration Assay
A modified Boyden chamber assay was used to examine the in vitro motility of the different pancreatic cancer cells. Polycarbonate filters (pore size, 8 µm; diameter, 13 mm) were glued to inserts, which were placed in the wells of 24-well tissue culture plates. The lower part of the chamber was filled with fibronectin (100 ng/mL) as a chemoattractant. Cells (2 x 104) were suspended in 100-µl HBSS and placed in the upper part of the chamber. After 6 hours incubation at 37°C, the filters were carefully removed from the inserts, stained with hematoxylin = losin for 10 minutes, and mounted on microscopic slides. The number of stained cells was counted in 10 microscopic fields (each 0.161 mm2) at 100 x magnification. Migratory activity was quantified by the average number of cells per microscopic field.
Endothelial Cell Proliferation Assay
Primary human umbilical vein endothelial cells (HUVEC) were cultured in complete DMEM containing 15% FBS and 10 ng/mL bFGF. Cell culture flasks were coated with 4 mg/mL gelatin. Cells were plated in 96-well plates at a density of 3 x 103 cells per well. After 24 hours, the supernatant in each well was removed, and 200 µl of conditioned medium of the different pancreatic cancer cells was added in various concentrations (10% and 50% conditioned medium). Conditioned medium was prepared by culturing 5 x 105 pancreatic cancer cells in a 6 well plate in supplemented DMEM containing 10% FBS for 5 days. After 4 days of incubation, the growth of HUVECs in various concentrated conditioned media of the different pancreatic cancer cells was monitored by MTT assay and compared to the growth of HUVECs treated with supplemented DMEM containing 15% FBS and 10 ng/mL bFGF only.
Statistical Analysis
The unpaired Student t test was used to compare the in vitro and in vivo results. Survival analysis was computed by the Kaplan-Meier method and compared by the log-rank test [40]. The Mann-Whitney test was used to compare the level of gene expression among the different cell lines (univariate analyses). Significance was defined as a two-sided P < .05 [40].
Results
Selection of Highly Metastatic Variant Lines
In the first set of experiments, we injected cells from the FG line derived from COLO357 human pancreatic adeno-carcinoma cell line [21,22] into the spleen or pancreas of nude mice. Intrasplenic injection allows cells to reach the liver parenchyma within minutes after injection [13,14]. Liver colonization can occur rapidly and without necessity to overcome the barrier of the primary site microenvironment. The liver lesions thus formed are termed experimental metastases. In contrast, to produce liver lesions, human pancreatic cancer cells implanted into the pancreas must complete all the steps of the process. The lesions then are designated as spontaneous metastases. Experimental or spontaneous liver metastases were harvested, established in tissue culture, and designated as L3.4sl and L3.4pl, respectively. Cells harvested from these cultures were injected into the spleen or pancreas of another set of nude mice. Liver lesions were again isolated, and cells were established in culture. After three such selection cycles, lines L3.6sl and L3.6pl were established in culture (Figure 1). Cytogenetic analysis confirmed the human origin of the cells.
Figure 1.

In vivo selection of metastatic human pancreatic cancer cells. L3.3 cells (derived from the FG line) were injected into the spleen or the pancreas of nude mice. Experimental liver metastases (spleen injection) or spontaneous liver metastases (pancreas injection) were harvested, established in culture, and designated L3.4sl (spleen-liver) and L3.4pl (pancreas-liver). The cultures were harvested and injected into the spleen or pancreas of another set of nude mice. The cycles were repeated twice more to yield lines L3.6sl and L3.6pl.
To determine whether the isolated variants had increased metastatic potential, cells from the original FG line and from the L3.6sl, and L3.6pl lines were injected into the spleen and/or the pancreas of nude mice (n = 20). The mice were killed when moribund or by day 105 after injection. Tumorigenicity, production of metastases, and survival were determined (Table 1). In the first assay for experimental metastasis, we injected the cells into the spleen. In trasplenic injection of FG cells produced spleen tumors in 5 of 20 animals, experimental liver metastases in 4 of 20 mice, and a median survival time of 92 days. Injection of L3.6sl cells produced spleen tumors in 19 of 20 mice, experimental liver metastases in 10 of 19 mice (P < .01), and a median survival time of 71 days. For L3.6pl cells, 20 of 20 mice had spleen tumors and 13 of 20 had liver metastases (P < .01); the median survival time was 34 days (P < .01). The highest median number of liver metastases was 10 (3–75), found after intrasplenic injection of L3.6pl cells, suggesting that these cells have a higher capacity to colonize the liver.
Table 1.
Tumorigenicity and Production of Metastasis by Human Pancreatic Cancer Cells Subsequent to Implantation in Athymic Nude Mice.
| Site of Implantation* | Cell Line | Median Survival | Tumorigenicity | Liver Metastasis | Lymph Node | |
| (days) | Incidence† | Median (range) | Metastasis‡ | |||
| Spleen | FG | 92 | 5/20 | 4/20 | 0 (0–2) | 2/10 |
| L3.6sl | 71 | 19/20‖ | 10/19 | 3 (0–10) | 7/19§ | |
| L3.6pl | 34 | 20/20‖ | 13/20 | 10 (0–75) | 3/20 | |
| Pancreas | FG | 78 | 20/20 | 1/20 | 0 (0–1) | 3/20 |
| L3.6sl | 56 | 20/20 | 5/20 | 0 (0–3) | 12/20‖ | |
| L3.6pl | 36 | 20/20 | 10/20 | 1 (0–15) | 9/20§ | |
Nude mice were injected into the spleen, pancreas or vein with 1 x 106 viable cells. The mice were killed when moribund and necropsied. The presence of tumor lesions in the spleen, pancreas, liver, lymph nodes, or lungs was determined with the aid of a dissecting microscope.
Number of tumor-positive mice per number of injected mice.
Number of mice with macroscopically enlarged regional lymph node per number of injected mice.
P < .01 versus FG.
P < .001 versus FG.
In the second assay for spontaneous metastasis, we injected the cells into the pancreas. All three lines were 100% tumorigenic after intrapancreatic injection (Table 1). Liver metastases were produced in 1 of 20, 5 of 20, and 10 of 20 mice after injection of FG, L3.6sl, and L3.6pl cells, respectively. The median survival time was 84, 56, or 36 days after the intrapancreatic injection of FG, L3.6sl, and L3.6pl cells, respectively (Figure 2). These data show that selection of cells for experimental metastasis also increased their ability to form spontaneous metastasis. The selection of cells for spontaneous metastasis, however, yielded cells with the highest metastatic potential.
Figure 2.
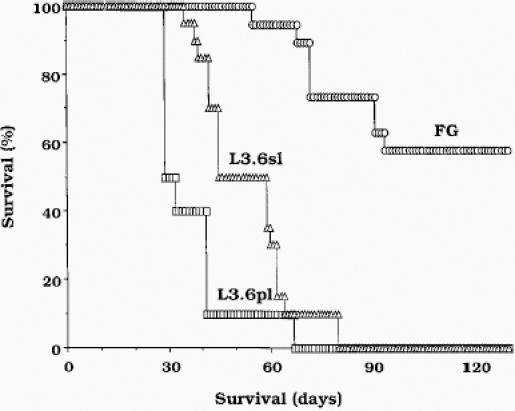
Percent cumulative survival. The pancreases of nude mice were injected with 1 x 106 viable FG, L3.6sl, or L3.6pl cells. Moribund mice were killed and necropsied. Survival analysis was computed by the Kaplan-Meier method, and the unpaired Student t test was used to compare the results among the three different cell lines. FG versus L3.6sl, P < .002; FG versus L3.6pl, P < .0001; L3.6sl versus L3.6pl, P < 0.02.
The intravenous (IV) injection of 1 x 106 FG cells produced a median of 75 experimental lung metastasis per mouse (range, 18–142), whereas the intravenous injection of L3.6sl cells produced a median number of 3 lung lesions. The IV injection did not yield a significant number of liver metastases.
Cell Growth in Vitro and in Vivo
The in vitro doubling time of FG, L3.6sl and L3.6pl was 25, 20, and 16 hours, respectively. The average number of PCNA + cells (per microscopic field) in the pancreatic tumors agreed with the in vitro results. The average number of PCNA + cells per microscopic field was 178 ± 42, 289 ± 48, and 399 ± 33 in 72- to 84-day-old pancreatic tumors of FG, 53- to 59-day-old pancreatic tumors of L3.6sl, and 35- to 37-day-old pancreatic tumors produced by L3.6pl cells (Figure 3; Table 2).
Figure 3.
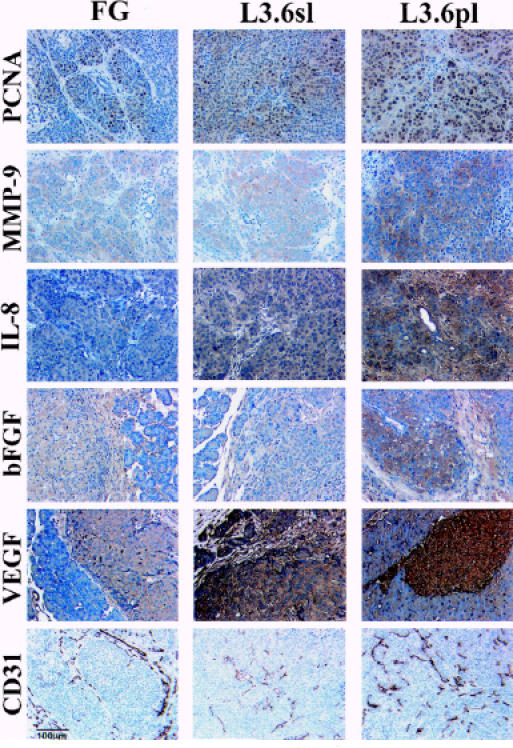
Representative IHC staining of FG, L3.6sl, and L3.6pl human pancreatic cells growing in the pancreas of nude mice. Tissue sections were stained for expression of PCNA (to show cell division), MMP-9, IL-8, bFGF, VEGF/VPF, and CD31 (to show endothelial cells).
Table 2.
Immunohistochemical Analysis of Pancreatic Cancers Produced by Orthotopic Implantation in Athymic Nude Mice.
| Cell Line* | PCNA† | CD-31‡ | VEGF/VPF§ | bFGF§ | IL-8§ | MMP-9§ |
| FG | 180 (152–228) | 7 (6–13) | 100 (97–108) | 100 (92–103) | 100 (97–105) | 100 (98–107) |
| L3.6sl | 262 (244–350) | 35 (33–57) | 133 (130–139) | 136 (131–147) | 141 (137–145) | 123 (118–124) |
| L3.6pl | 374 (366–391) | 73 (59–112) | 178 (168–185) | 176 (169–182) | 157 (155–165) | 145 (133–150) |
Tumor cells were injected into the pancreas of athymic nude mice. Tumors were harvested and processed for IHC as described in Materials and Methods.
Median (range) number of PCNA + cells was determined by counting positive cells in four fields of 0.161 mm2 each at 100 x magnification. FG v L3.6sl, P < .004; FG v L3.6pl, P < .002; L3.6sl v L3.6pl, P < 0.002.
Median (range) number of CD31-positive cells was determined by counting positive cells in four fields of 1.0 mm2 each at 40 x magnification at the area of most intense CD31 expression. FG v L3.6sl, P < .01; FG v L3.6pl, P < .006; L3.6sl v L3.6pl, P < .01.
Median (range) OD of VEGF/VPF, bFGF, and MMP-9 was determined by measurement of 100 cells per tumor at 200 x magnification with the Optimas Image Analysis software. The integrated OD was compared with the integrated OD in the FG tumors, which was set at 100. VEGF/VPF: FG v L3.6sl, P < .05; FG v L3.6pl, P < .01; L3.6sl v L3.6pl, P < 0.05. bFGF: FG v L3.6sl, P < .05; FG v L3.6pl, P < .01; L3.6sl v L3.6pl, P < .05. MMP-9: FG v L3.6al, P < .05; FG v L3.6pl, P < .02; L3.6sl v L3.6pl, P < .04.
Cell Motility
To determine whether the increased metastatic potential was associated with increased motility, we used a modified Boyden chamber assay with fibrinogen (100 ng/mL) as a chemoattractant. The motility of L3.6sl and L3.6pl cells differed significantly from that of the FG cells. Specifically, the number of FG, L36sl, and L3.6pl cells per 10 microscopic fields was 3 ± 1, 38 ± 5, and 44 ± 7 cells, respectively (P < .001).
Relative Expression of MMP-2/MMP-9 and E-cadherin
Detachment of malignant cells from the primary tumor and penetration of host stroma including basement membranes and extracellular matrix (ECM) is an essential part of the metastatic process [7]. The degradation of the ECM components surrounding malignant tumors is primarily mediated by MMPs or type IV collagenases [41–46]. To determine whether the metastatic potential of the variant cells correlated with their production of MMPs, we examined the expression of MMPs by ISH.
Pancreatic tumors derived from L3.6pl and L3.6sl cells had significantly higher MMP-9 and MMP-2 mRNA levels than tumors derived from FG (Figure 4, Table 3). The MMP-9 mRNA expression level was significantly higher in pancreatic tumors derived from L3.6pl cells (spontaneous metastasis) than in L3.6sl cells (experimental metastasis). The expression of E-cadherin, a transmembrane glycoprotein that is directly related to cell-to-cell cohesion, was high in tumors derived from FG and low in tumors derived from L3.6sl and L3.6pl cells. IHC staining of pancreatic tumors with anti-MMP-9 antibody revealed a significant increase in the expression of MMP-9 protein in tumors derived from metastatic L3.6sl and L3.6pl cells (Figure 3, Table 2).
Figure 4.
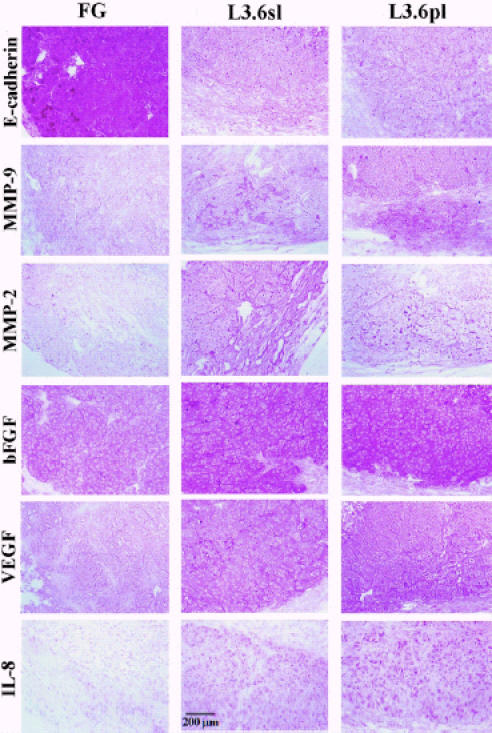
Representative ISH of FG, L3.6sl, and L3.6pl human pancreatic cells growing in the pancreas of nude mice. Metastatic cells (L3.6sl, L3.6pl) concurrently expressed high levels of MMP-2, MMP-9, bFGF, VEGF/VPF, IL-8, and low levels of E-cadherin. The low metastatic cells expressed high levels of E-cadherin.
Table 3.
In Situ Hybridization Analysis for Relative Expression of Metastasis-Regulating Genes by Human Prostate Cancer Cell Lines Growing in the Pancreas of Nude Mice.
| Cell Line | Median Expression Index (Range) | MMP/E-Cadherin Ratio | |||||
| VEGF/VPF | bFGF | IL-8 | MMP-2 | MMP-9 | E-cadherin | ||
| FG | 46 (127–147) | 166 (155–172) | 84 (75–940) | 119 (96–142) | 92 (83–100) | 73 (65–80) | 1.5 (1.4–1.5) |
| L3.6sl | 295 (163–353) | 263 (223–285) | 208 (210–215) | 201 (180–225) | 186 (167–195) | 55 (46–56) | 3.7 (3.3–4.0) |
| L3.6pl | 280 (233–315) | 240 (224–252) | 198 (149–210) | 200 (171–248) | 146 (152–150) | 48 (41–55) | 3.8 (3.3–3.9) |
| P value | |||||||
| FG v L3.6sl | .001 | .0006 | .001 | .0006 | .002 | .006 | .0006 |
| FG v L3.6pl | .001 | .0006 | .001 | .0006 | .001 | .006 | .0006 |
| L3.6sl v L3.6pl | NS | NS | .02 | NS | .001 | NS | NS |
Note: Tumor cells were injected into the pancreas of athymic nude mice. Tumors were harvested and processed for in situ hybridization analysis as described in Materials and Methods. Relative expression of specific mRNA was determined by examining at least 7 different fields/specimen. The expression of each gene in normal epithelium of mouse pancreas was set at the value 100.
Gelatin zymography of tumor lysates revealed 5-fold and 3.5-fold increases in activity of the 92-kDa MMP-9 in pancreatic tumors (or culture supernatants) derived from L3.6sl and L3.6pl cells, respectively, as compared with FG cells. MMP-2 activity was indistinguishable among the 3 different tumors. Additionally, a 3-fold increase of MMP-9 activity was determined in lysates of pancreatic tumors from L3.6pl versus L3.6sl cells (Figure 5).
Figure 5.
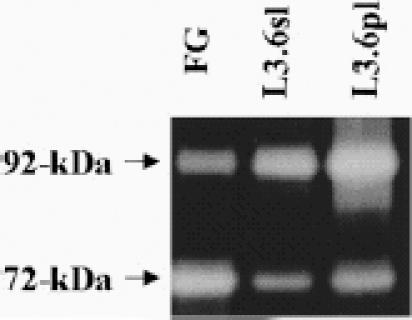
Gelatin zymography. Lysates from FG L3.6sl and L3.6pl pancreatic tumors were prepared. Protein (1 µg) was loaded on a 7.5% polyacrylamide slab gel-impregnated with 1 mg/mL gelatin under nonreducing conditions.
Expression of VEGF, bFGF, IL-8, and Quantification of Microvessel Density
In situ hybridization of tissue sections demonstrated a significant difference in VEGF/VPF, bFGF, and IL-8 mRNA expression level among pancreatic tumors derived from FG, L3.6sl, and L3.6pl cells (see Figure 4). IL-8 mRNA expression level was significantly higher in pancreatic tumors derived from L3.6pl than in L3.6sl or FG cells (see Figure 4). Similar results were obtained in vitro by Northern blot analysis of cultured cells (data not shown).
IHC staining with specific antibodies demonstrated increased protein expression of VEGF/VPF, bFGF, or IL-8 in L3.6sl and L3.6pl tumors (see Figure 3, Table 2) (FG v L3.6sl/L3.6pl, P < .01, L3.6sl v L3.6pl, P < .03). These in vivo results are supported by Western blot analysis of tissue culture lysates for bFGF as well as ELISA assay of culture supernatants for IL-8 and VEGF/VPF (data not shown).
To determine whether the proangiogenic molecules produced by the pancreatic tumor cells were biologically active, we incubated HUVEC with culture supernatants of the three cell lines and determined HUVEC proliferation by the MTT assay [38,39]. Conditioned media of FG, L3.6sl, and L3.6pl cells stimulated proliferation of HUVECs (P < .02). To further investigate angiogenesis in the orthotopic tumors, we quantified MVD after staining frozen sections of tumors with CD31/PECAM-1. In pancreatic tumors derived from FG, L3.6sl, and L3.6pl, the average number of CD31/PECAM-1 positive vessels (per 1-mm2 field at 40 x magnification) was 3 ± 1, 44 ± 11, and 89 ± 25, respectively (P < .001).
Discussion
Metastasis is a highly selective process that favors the survival and growth of unique subpopulations of metastatic cells that preexist within heterogeneous malignant neoplasms [47,48]. The isolation of clonal populations of cells that differ in their metastatic capacity has supported the hypothesis of metastatic heterogeneity [7–10,48]. Depending on the neoplasm, the selection process is gradual, requiring several cycles of in vivo selection [12,16,19]. This cyclic procedure was originally used to isolate the B16-F10 metastatic cell line from the wild-type B16 murine melanoma cell line [47] and, subsequently, human renal cell carcinoma [13], colon carcinoma [11,12], transitional cell carcinoma [15,16], and prostate carcinoma [18,19]. We implanted pancreatic cancer cells into the spleen (experimental liver metastasis) or pancreas (spontaneous liver metastasis) and harvested metastatic lesions. The recovered cells were expanded in culture to repeat the process. After several cycles, the behavior of the cells was compared with that of the parental cells. The metastatic potential of the pancreatic cancer cells in nude mice was determined by two assays. The first involved the implantation of the cells into the pancreas and a count of spontaneous lymph nodes and liver metastasis. The second measured to ability of the pancreatic cancer cells to grow in the liver parenchyma subsequent to intrasplenic implantation (experimental metastasis). In general, there was good agreement in the results of the two assays.
Earlier data from our laboratory established that the implantation site of metastatic human tumor cells determines whether they will produce distant metastasis. Specifically, the growth of tumors in orthotopic organs facilitates production of metastasis, whereas growth in ectopic organs does not [7–10]. Indeed, human pancreatic cancer cells implanted into the pancreas (orthotopic) or subcutis (ectopic) of severe combined immunodeficient mice produced tumors that differed in growth rate and status of differentiation [49]. These and other biologic differences may be due to differential interaction of tumor cells with organ-specific factors [50].
The intrasplenic injection of human pancreatic carcinoma cells into nude mice produced experimental metastases in the liver and in the lung [51,52]. Cells injected into the spleen reach the liver parenchyma within minutes [11]. Thus, the intrasplenic implantation of cells measures their ability to grow in the liver parenchyma but fails to measure cell detachment from the primary pancreatic tumor followed by invasion of host stroma [11,12]. This limitation stimulated the development of an orthotopic model for human pancreatic cancer that used tumor fragments, sutured onto the pancreas of nude mice [22,51,52]. The use of tumor fragments avoids the issue of neoplastic heterogeneity [53] and, thus, the orthotopic inoculation of human pancreatic cancer cell suspensions is mandatory for reproducible studies of the biology of pancreatic cancer metastasis. In this study, we undertook a series of intrapancreatic and intrasplenic implantation experiments with cell suspensions of a human pancreatic cancer cell line to select and isolate cell lines with higher tumorigenicity and increased metastatic potential. Intrasplenic and intrapancreatic implantation demonstrated a significant increase in the incidence of primary tumors (pancreas, spleen) and liver metastasis, as well as in median number of liver metastases for L3.6sl and L3.6pl cells over FG cells. Moreover, the incidence and median number of liver metastases were higher after either intrapancreatic or intrasplenic injection of L3.6pl as compared with L3.6sl cells, suggesting that the repeated pancreas-to-liver selection procedure isolated cells with higher potential for metastasis. More than a century ago, Paget [54] proposed that the outcome of metastasis depended on the interaction of specific tumor cells with a favorable organ environment. The present data support the ‘seed and soil’ hypothesis [54]. After IV injection of the more heterogeneous FG cells, a significantly greater differential favoring lung over liver metastases was produced than after the IV injection of pancreatic cancer cells selected for their ability to produce liver metastases.
The increased metastatic potential of the L3.6pl and L3.6sl variants correlated with expression of several metastasis-regulating genes [55]. First, E-cadherin is a cell surface glycoprotein involved in calcium-dependent homotypic cell-to-cell adhesion [56]. E-cadherin is localized at the epithelial junction complex and is responsible for the organization, maintenance, and morphogenesis of epithelial tissues [57–60]. Reduced levels of E-cadherin are associated with a decrease in cellular/tissue differentiation and increased grade in different epithelial neoplasms [61–65], and transfection of E-cadherinencoding cDNA into invasive cancer cells has been shown to inhibit motility and invasion [66].
Invasion of the host stroma and degradation of the blood vessel basement membrane are necessary for metastasis [41–46,67]. The levels of collagenase type IV in human and rodent neoplasms directly correlate with invasion and metastasis, and specific inhibitors of matrix metalloproteinases have been shown to inhibit tumor cell invasion [41–45]. We analyzed the tumors produced by FG cells (low metastatic) by ISH, IHC, and zymography of tumor tissue lysates and medium conditioned by the respective cultures. The metastatic variants expressed higher levels of MMP-9 as compared with the low metastatic FG cells. In contrast, the E-cadherin expression was significantly higher in the FG tumors than in the metastatic tumors. Thus, a decrease in the expression of E-cadherin and an increase in collagenase type IV activity was associated with enhanced tumor invasion and metastasis [37,67–69].
Increased motility is conducive to tumor cell invasion and metastasis [70,71]. Invasive pancreatic cancer cells can produce a dissociation factor that increases cell motility [72]. Similar to our previous study with metastatic human prostate cancer [19], we determined the relative motility of the FG, L3.6sl, and L3.6pl cells by using a modified Boyden chamber coated with fibronectin. This assay revealed that motility in vitro directly correlated with production of metastasis.
The progressive growth and metastasis of neoplasms is dependent on angiogenesis, the extent of which is determined by the balance between proangiogenic and antiangiogenic molecules secreted by both tumor and host cells [73–75]. Among the major molecules identified is bFGF, which induces the proliferation, migration, proteolytic activity, and differentiation of endothelial cells [73,76,77]. VEGF/VPF induces the proliferation of endothelial cells, increases the cells' vascular permeability, and induces the production of plasminogen activator by these cells [78–81]. IL-8, a chemoattractant cytokine produced by a variety of tissues and blood cells, attracts and activates neutrophils in inflammatory regions and is angiogenic [82,83]. The levels of MMP-2 and MMP-9 have been associated with active neo-vascularization because these MMPs can degrade the ECM, an essential step in the process of angiogenesis [84].
The expression of angiogenic molecules differed significantly among the 3 pancreatic cell lines. Both in vitro cultured cells and in vivo tumors derived from L3.6pl and L3.6sl cells expressed higher levels of VEGF/VPF, bFGF, IL-8, and MMP-9 than did the low metastatic FG cells or tumors. The increased expression level of the angiogenic molecules directly correlated with MVD of the tumor lesions as measured by immunostaining of endothelial cells with antibodies against CD31/PECAM-1. Finally, the biological activity of the angiogenic molecules was proven by their ability to stimulate growth of HUVEC. In good agreement with the IHC data, the conditioned media of all 3 pancreatic tumor cell variants stimulated the growth of HUVECs; however, the conditioned media of L3.6sl and L3.6pl cells stimulated the growth of HUVECs significantly more than the conditioned medium of FG cells.
Increased levels of the epidermal growth factor receptor and its ligand, transforming growth factor, have been associated with increased malignancy of human bladder, breast, and colon cancers [85–88]. The expression level for these genes, however, did not vary among the pancreatic cell lines. The c-met oncogene, which encodes the receptor for hepatocyte growth factor, has been shown to promote cell motility and invasiveness [89–92]. We did not find discernible differences in expression of c-met among the three lines.
In summary, we report the development of a reproducible orthotopic model to study the biology of human pancreatic cancer metastasis. We also succeeded in isolating variant lines with enhanced metastatic properties as demonstrated by a battery of assays. The availability of these unique cells and the in vivo model should facilitate in-depth studies to understand the interaction of metastatic pancreatic cancer cells with host factors and the design of biologically based new therapies.
Acknowledgements
The FG and L3.3 human pancreatic cancer cell lines were obtained from Dr. Lance M. Tibbetts (Veteran's Administration Medical Center, and Department of Surgery, Medicine, and Pathology, Roger Williams Medical Center and Brown University, Providence, RI).
The authors thank Donna Reynolds for technical assistance, Walter Pagel for editorial comments, and Lola López for expert assistance in the preparation of the manuscript.
Abbreviations
- matrix metalloproteinase-2
(MMP-2)
- bFGF
basic fibroblast growth factor
- DAB
diaminobenzidine
- FBS
fetal bovine serum
- FG
fast growing line
- HBSS
Hank's balanced salt solution
- HUVEC
human umbilical vein endothelial cells
- IHC
immunohistochemistry
- ISH
in situ hybridization
- MMP
matrix metalloproteinase
- OD
optical density
- PCNA
proliferating cell nuclear antigen
- VEGF/VPF
vascular endothelial growth factor/vascular permeabilit factory
Footnotes
This work was supported in part by Cancer Center Support Core Grant CA16672 and grant R35-CA42107 from the National Cancer Institute, National Institutes of Health, CaP CURE, and the Lise-Meitner-Stipendium, German Research Association, Nordrhein-Westfalen, Germany.
References
- 1.Moore M. Clinical experience with gemcitabine in pancreatic cancer. Oncology. 1997;11:1615–1622. [PubMed] [Google Scholar]
- 2.Wanebo HJ, Vezeridis MP. Pancreatic carcinoma in perspective. A continuing challenge. Cancer. 1996;78(Suppl):580–591. doi: 10.1002/(SICI)1097-0142(19960801)78:3<580::AID-CNCR38>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 3.Moore M. Activity of gemcitabine in patients with advanced pancreatic cancer. Cancer. 1996;78(Suppl):633–638. doi: 10.1002/(SICI)1097-0142(19960801)78:3<633::AID-CNCR44>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 4.Rothenberg ML, Abbruzzese JL, Portenoy RK, Robertson JM, Wanebo HJ. A rationale for expanding the endpoints for clinical trials in advanced pancreatic cancer. Cancer. 1996;78(Suppl):627–632. doi: 10.1002/(SICI)1097-0142(19960801)78:3<627::AID-CNCR43>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 5.Bramhall SR. The use of molecular technology in the differentiation of pancreatic cancer and pancreatitis. Int J Pancreatol. 1998;23:83–100. doi: 10.1385/IJGC:23:2:83. [DOI] [PubMed] [Google Scholar]
- 6.Breslin TM, Janjan NA, Lee JE, Pisters PWT, Wolff RA, Abbruzzese JL, Evans DB. Neoadjuvant chemoradiation for adenocarcinoma of the pancreas. Front Biosci. 1998;3:E193–E203. doi: 10.2741/a377. [DOI] [PubMed] [Google Scholar]
- 7.Fidler IJ. Critical factors in the biology of human cancer metastasis: Twenty-eighth GHA Clowes Memorial Award Lecture. Cancer Res. 1990;50:6130–6138. [PubMed] [Google Scholar]
- 8.Fidler IJ. Modulation of the organ microenvironment for the treatment of cancer metastases. J Natl Cancer Inst. 1995;87:1588–1592. doi: 10.1093/jnci/87.21.1588. (commentary) [DOI] [PubMed] [Google Scholar]
- 9.Aukerman SL, Price JE, Fidler IJ. Different deficiencies in the prevention of tumorigenic-low-metastatic murine K-1735 melanoma cells from producing metastases. J Natl Cancer Inst. 1986;77:915–924. [PubMed] [Google Scholar]
- 10.Fidler IJ. Orthotopic implantation of human colon carcinomas into nude mice provides a valuable model for the biology and therapy of cancer metastasis. Cancer Metastasis Rev. 1991;10:229–243. doi: 10.1007/BF00050794. [DOI] [PubMed] [Google Scholar]
- 11.Fidler IJ. Rationale and methods for the use of nude mice to study the biology and therapy of human cancer metastasis. Cancer Metastasis Rev. 1986;29:29–49. doi: 10.1007/BF00049529. [DOI] [PubMed] [Google Scholar]
- 12.Giavazzi R, Campbell DE, Jessup JM, Cleary K, Fidler IJ. Metastatic behavior of tumor cells isolated from primary and metastatic human colorectal carcinomas implanted into different sites in nude mice. Cancer Res. 1986;46:1928–1933. [PubMed] [Google Scholar]
- 13.Morikawa K, Walker SM, Jessup JM, Fidler IJ. In vivo selection of highly metastatic cells from surgical specimens of different primary human colorectal carcinomas implanted into nude mice. Cancer Res. 1988;48:1943–1948. [PubMed] [Google Scholar]
- 14.Naito S, von Eschenbach AC, Giavazzi R, Fidler IJ. Growth and metastasis of tumor cells isolated from a renal cell carcinoma implanted into different organs of nude mice. Cancer Res. 1986;46:4109–4115. [PubMed] [Google Scholar]
- 15.Price JE, Polyzos A, Zhang RD, Daniels LM. Tumorigenicity and metastasis of human breast carcinoma lines in nude mice. Cancer Res. 1990;50:717–721. [PubMed] [Google Scholar]
- 16.Ahlering TE, Dubeau L, Jones PA. A new in vivo model to study invasion and metastasis of human bladder carcinoma. Cancer Res. 1985;47:6660–6665. [PubMed] [Google Scholar]
- 17.Dinney CPN, Fishbeck R, Singh RK, Eve B, Pathak S, Brown N, Xie B, Fan D, Bucana CD, Fidler IJ, Killion JJ. Isolation and characterization of metastatic variants from human transitional cell carcinoma passaged by orthotopic implantation in athymic nude mice. J Urol. 1995;154:1532–1538. [PubMed] [Google Scholar]
- 18.McLemore TL, Liu MC, Blacker PC, Gregg M, Alley MC, Abbott BJ, Shoemaker RH, Bohlman ME, Litterst CC, Hubbard WC, Brennan RH, McMahon JB, Fine DL, Eggleston JC, Mayo JG, Boyd MR. Novel intrapulmonary model for orthotopic propagation of human lung cancers in athymic nude mice. Cancer Res. 1987;47:5132–5140. [PubMed] [Google Scholar]
- 19.Stephenson RA, Dinney CPN, Gohji K, Ordonez NG, Killion JJ, Fidler IJ. Metastatic model for human prostate cancer using orthotopic implantation in nude mice. J Natl Cancer Inst. 1992;84:951–957. doi: 10.1093/jnci/84.12.951. [DOI] [PubMed] [Google Scholar]
- 20.Pettaway CA, Pathak S, Greene G, Ramirez E, Wilson MR, Killion JJ, Fidler IJ. Selection of highly metastatic variants of different human prostatic carcinomas utilizing orthotopic implantation in nude mice. Clin Cancer Res. 1996;2:1627–1636. [PubMed] [Google Scholar]
- 21.Morgan RT, Woods LK, Moore GE, Quinn LA, McGavran L, Gordon SG. Human cell line (COLO375) of metastatic pancreatic adenocarcinoma. Int J Cancer. 1980;25:591–598. doi: 10.1002/ijc.2910250507. [DOI] [PubMed] [Google Scholar]
- 22.Vezeridis MP, Tzanakakis GN, Meitner PA, Doremus CM, Tibbetts LM. In vivo selection of a highly metastatic cell line of a human pancreatic carcinoma in the nude mouse. Cancer. 1992;69:2060–2063. doi: 10.1002/1097-0142(19920415)69:8<2060::aid-cncr2820690810>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 23.Shi SR, Key ME, Kalra KL. Antigen retrieval in formalin-fixed, paraffin-embedded tissues: an enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. J Histochem Cytochem. 1991;6:741–748. doi: 10.1177/39.6.1709656. [DOI] [PubMed] [Google Scholar]
- 24.Vecchi A, Garlanda C, Lampugnani MG, Resnati M, Matteucci C, Stoppacciaro A, Mantovani A. Monoclonal antibodies specific for endothelial cells of mouse blood vessels. Their application in the identification of adult and embryonic endothelium. Eur J Cell Biol. 1994;63:247–254. [PubMed] [Google Scholar]
- 25.Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis—correlation in invasive breast carcinoma. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi Y, Kitadai Y, Bucana CD, Cleary KR, Ellis LM. Expression of vascular endothelial growth factor and its receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer Res. 1995;55:3964–3968. [PubMed] [Google Scholar]
- 27.Yoneda J, Kuniyasu H, Crispens MA, Price JE, Bucana CD, Fidler IJ. Expression of angiogenesis-related genes and progression of human ovarian carcinomas in nude mice. J Natl Cancer Inst. 1998;90:447–454. doi: 10.1093/jnci/90.6.447. [DOI] [PubMed] [Google Scholar]
- 28.Berse B, Brown L, Van de Water L, Dvorak H, Senger DR. Vascular permeability factor (vascular endothelial cell growth factor) is expressed differentially in normal tissues, macrophages, and tumors. Mol Biol Cell. 1992;3:211–220. doi: 10.1091/mbc.3.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsushima K, Morishita K, Yoshimura T, Lavu S, Kobayashi Y, Lew W, Appella E, Kung HF, Leonard EJ, Oppenheim JJ. Molecular cloning of a human monocyte-derived neutrophil chemotactic factor (MDNCF) and the induction of mDNCF mRNA by interleukin 1 and tumor necrosis factor. J Exp Med. 1988;167:1883–1893. doi: 10.1084/jem.167.6.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collier IE, Wilhelm SM, Eisen AZ, Marmer BL, Grant GA, Seltzer JL, Kronberger A, He C, Bauer EA, Goldberg GI. H-ras oncogene-transformed human bronchial epithelial cells (TBE-1) secrete a single metalloproteinase capable of degrading basement membrane collagen. J Biol Chem. 1988;263:6579–6587. [PubMed] [Google Scholar]
- 31.Greene GF, Kitadai Y, Pettaway CA, von Eschenbach AC, Bucana CD, Fidler IJ. Correlation of metastasis-related gene expression with metastatic potential of human prostate carcinoma cells implanted in nude mice using in situ messenger RNA hybridization technique. Am J Pathol. 1997;150:1571–1582. [PMC free article] [PubMed] [Google Scholar]
- 32.Bussenmakers MJG, van Bokhoven A, Mees SGM, Kemler R, Schalken JA. Molecular cloning and characterization of human E-cadherin cDNA. Mol Biol Rep. 1993;17:123–128. doi: 10.1007/BF00996219. [DOI] [PubMed] [Google Scholar]
- 33.Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bucana CD, Radinsky R, Dong Z, Sanchez R, Brigati DJ, Fidler IJ. A rapid colorimetric in situ mRNA hybridization technique using hyperbiotinylated oligonucleotide probes for analysis of mdr1 in mouse colon carcinoma cells. J Histochem Cytochem. 1993;41:499–506. doi: 10.1177/41.4.8095509. [DOI] [PubMed] [Google Scholar]
- 35.Radinsky R, Bucana CD, Ellis LM, Sanchez R, Cleary KR, Brigati DJ, Fidler IJ. A rapid colorimetric in situ messenger RNA hybridization technique for analysis of epidermal growth factor receptor in paraffin-embedded surgical specimens of human colon carcinomas. Cancer Res. 1993;53:937–943. [PubMed] [Google Scholar]
- 36.Park CS, Brigati DJ. Automated molecular pathology: One hour in situ DNA hybridization. J Histotechnol. 1991;14:219–229. [Google Scholar]
- 37.Kitadai Y, Ellis LM, Takahashi Y, Bucana CD, Anzai h, Tahara T, Fidler IJ. Multiparametric in situ mRNA hybridization analysis to detect metastasis-related genes in surgical specimens of human colon carcinoma. Clin Cancer Res. 1995;1:1095–1102. [PubMed] [Google Scholar]
- 38.Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988;48:589–602. [PubMed] [Google Scholar]
- 39.Fan D, Bucana CD, O'Brian CA, Zwelling LA, Seid CJ, Fidler IJ. Enhancement of murine cell sensitivity to Adriamycin by presentation of the drug in phosphatidylcholine-phosphatidylserine liposomes. Cancer Res. 1990;50:3619–3626. [PubMed] [Google Scholar]
- 40.Hosmer DW, Lemeshow S. Applied Logistic Regression. New York: John Wiley & Sons; 1980. [Google Scholar]
- 41.D'Errico A, Garbisa S, Liotta LA, Castronovo V, Stetler-Stevenson WG, Grigioni WF. Augmentation of type IV collagenase, laminin receptor, and Ki67 proliferation antigen associated with human colon, gastric, and breast carcinoma progression. Mod Pathol. 1991;4:239–246. [PubMed] [Google Scholar]
- 42.Matrisian LM, Bowden GT. Stromelysin/transin and tumor progression. Semin Cancer Biol. 1990;1:107–115. [PubMed] [Google Scholar]
- 43.Aznavoorian S, Murphy AN, Stetler-Stevenson WG, Liotta LA. Molecular aspects of tumor cell invasion and metastasis. Cancer. 1993;71:1368–1383. doi: 10.1002/1097-0142(19930215)71:4<1368::aid-cncr2820710432>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 44.Albini A, Melchiori A, Santi L, Liotta LA, Brown PD, Stetler-Stevenson WG. Tumour cell invasion inhibited by TIMP-2. J Natl Cancer Inst. 1991;83:775–779. doi: 10.1093/jnci/83.11.775. [DOI] [PubMed] [Google Scholar]
- 45.Xie B, Bucana CD, Fidler IJ. Density-dependent induction of 92-kd type IV collagenase activity in cultures of A431 human epidermoid carcinoma cells. Am J Pathol. 1994;144:1058–1067. [PMC free article] [PubMed] [Google Scholar]
- 46.DeClerck YA, Yean T-D, Chan D, Shimada H, Langley KE. Inhibition of tumor invasion of smooth muscle cell layers by recombinant human metalloproteinase inhibitor. Cancer Res. 1991;51:2151–2157. [PubMed] [Google Scholar]
- 47.Fidler IJ. Selection of successive tumor lines for metastasis. Nature. 1973;242:148–149. doi: 10.1038/newbio242148a0. [DOI] [PubMed] [Google Scholar]
- 48.Fidler IJ, Kripke ML. Metastasis results from preexisting variant cell within a malignant tumor. Science. 1977;19:893–895. doi: 10.1126/science.887927. [DOI] [PubMed] [Google Scholar]
- 49.Mohammed RM, Al-Katib A, Pettit GR, Vaitkevicius VK, Joshi U, Adsay V, Majumdar APN, Sarkar FH. An orthotopic model of human pancreatic cancer in severe combined immunodeficient mice: potential application for preclinical studies. Clin Cancer Res. 1998;4:887–894. [PubMed] [Google Scholar]
- 50.Fukumura D, Yuan F, Monsky WL, Chen Y, Jain RK. Effect of host microenvironment on the microcirculation of human colon adenocarcinoma. Am J Pathol. 1997;151:679–688. [PMC free article] [PubMed] [Google Scholar]
- 51.Vezeridis MP, Doremus CM, Tibbetts LM, Tzanakakis G, Jackson BT. Invasion and metastasis following orthotopic transplantation of human pancreatic cancer in the nude mouse. J Surg Oncol. 1989;40:261–265. doi: 10.1002/jso.2930400412. [DOI] [PubMed] [Google Scholar]
- 52.Vezeridis MP. Models of pancreatic cancer invasion and metastasis. Int J Pancreatol. 1994;16:237–240. [Google Scholar]
- 53.Fidler IJ, Hart IR. Biological diversity in metastatic neoplasms: Origins and implications. Science. 1982;217:998–1003. doi: 10.1126/science.7112116. [DOI] [PubMed] [Google Scholar]
- 54.Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;1:571–573. [PubMed] [Google Scholar]
- 55.Fidler IJ, Radinsky R. The search for genes that suppress cancer metastasis (editorial) J Natl Cancer Inst. 1996;88:1700–1703. doi: 10.1093/jnci/88.23.1700. [DOI] [PubMed] [Google Scholar]
- 56.Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;251:1451–1455. doi: 10.1126/science.2006419. [DOI] [PubMed] [Google Scholar]
- 57.Dorudi S, Sheffield JP, Poulsom R, Northover JMA, Hart IR. E-cadherin expression in colorectal cancer. An immunocytochemical and in situ hybridization study. Am J Pathol. 1993;142:1–986. [PMC free article] [PubMed] [Google Scholar]
- 58.Bohm M, Totzeck B, Birchmeier W, Wieland I. Differences of E-cadherin expression levels and patterns in primary and metastatic human lung cancer. Clin Exp Metastasis. 1994;12:55–62. doi: 10.1007/BF01784334. [DOI] [PubMed] [Google Scholar]
- 59.Frixen UH, Behrens J, Sachs M, Eberle G, Voss B, Warda A, Loechner D, Birchmeier W. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol. 1991;113:173–185. doi: 10.1083/jcb.113.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shimoyama Y, Hirohashi S, Hirano S, Noguchi M, Shimosato Y, Takeichi M, Abe O. Cadherin cell adhesion molecules in human epithelial tissues and carcinomas. Cancer Res. 1989;49:2128–2133. [PubMed] [Google Scholar]
- 61.Mayer B, Johnson JP, Leitl F, Jauch KW, Heiss MM, Schildberg FW, Birchmeier W, Funke I. E-cadherin expression in primary and metastatic gastric cancer: downregulation correlates with cellular dedifferentiation and glandular disintegration. Cancer Res. 1993;53:1690–1695. [PubMed] [Google Scholar]
- 62.Gamallo C, Palacios J Suarez A, Pizamo A, Navarro P, Quintanilla M, Cana A. Correlation of E-cadherin expression with differentiation grade and histological type in breast carcinoma. Am J Pathol. 1993;142:987–993. [PMC free article] [PubMed] [Google Scholar]
- 63.Schipper JH, Frixen UH, Behrens J, Unger A, Jahnke K, Birchmeier W. E-cadherin expression in squamous carcinomas of the head and neck: Inverse correlation with tumor dedifferentiation and lymph node metastasis. Cancer Res. 1991;51:6328–6337. [PubMed] [Google Scholar]
- 64.Shimoyama Y, Hirohashi S. Expression of E- and P-cadherin in gastric carcinomas. Cancer Res. 1991;51:2185–2192. [PubMed] [Google Scholar]
- 65.Umbas R, Schalken JA, Alders TW, Carter BS, Karthaus HF, Schaafsma HE, Debruyne FM, Isaacs WB. Expression of the cellular adhesion molecule E-cadherin is reduced or absent in high-grade prostate cancer. Cancer Res. 1992;52:5104–5109. [PubMed] [Google Scholar]
- 66.Vleminckx K, Vakaet L, Jr, Mareel M, Fiers W, Van Roy F. Genetic manipulation of E-cadherin expression by epithelial tumor cell reveals an invasion suppressor role. Cell. 1991;66:107–119. doi: 10.1016/0092-8674(91)90143-m. [DOI] [PubMed] [Google Scholar]
- 67.Kitadai Y, Ellis LM, Tucker SL, Green GF, Bucana CD, Cleary KR, Takahashi Y, Tahara E, Fidler IJ. Multiparametric in situ mRNA hybridization analysis to predict disease recurrence in patients with colon carcinoma. Am J Pathol. 1996;149:1541–1551. [PMC free article] [PubMed] [Google Scholar]
- 68.Anzai H, Kitadai Y, Bucana CD, Sanchez R, Omoto R, Fidler IJ. Intratumoral heterogeneity and inverse correlation between expression of E-cadherin and collagenase type IV in human gastric carcinomas. Differentiation. 1996;60:119–127. doi: 10.1046/j.1432-0436.1996.6020119.x. [DOI] [PubMed] [Google Scholar]
- 69.Anzai H, Kitadai Y, Bucana CD, Sanchez R, Omoto R, Fidler IJ. Expression of metastasis-related genes in surgical specimens of human gastric cancer can predict disease recurrence. Eur J Cancer. 1998;34:558–575. doi: 10.1016/s0959-8049(97)10075-2. [DOI] [PubMed] [Google Scholar]
- 70.Partin A, Schoemiger TS, Mohler TL, Coffey DS. Fourier analysis of cell motility: Correlation of motility with metastatic potential. Proc Natl Acad Sci USA. 1989;86:1254–1258. doi: 10.1073/pnas.86.4.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Silletti S, Paku S, Raz A. Autocrine motility factor and the extracellular matrix. II. Degradation or remodeling of substratum components directs the motile response of tumor cells. Int J Cancer. 1998;76:129–135. doi: 10.1002/(sici)1097-0215(19980330)76:1<129::aid-ijc20>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 72.Kurizaki T, Egami H, Hirota M, Akagi J, Ohmachi H, Yamamoto S, Ogawa M. Characterization of cancer cell dissociation factor in a highly invasive pancreatic cancer cell line. Cancer. 1995;75(Suppl):1554–1561. doi: 10.1002/1097-0142(19950315)75:6+<1554::aid-cncr2820751528>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 73.Folkman J. Angiogenesis in cancer, vascular, rheumatoid, and other diseases. Nat Med. 1985;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 74.Singh RK, Gutman M, Bucana CD, Sanchez R, Llansa N, Fidler IJ. Interferons alpha and beta downregulate the expression of basic fibroblast growth factor in human carcinomas. Proc Natl Acad Sci USA. 1995;92:4562–4566. doi: 10.1073/pnas.92.10.4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fidler IJ, Ellis LM. The implications of angiogenesis for the biology and therapy of cancer metastasis. Cell. 1994;79:185–188. doi: 10.1016/0092-8674(94)90187-2. [DOI] [PubMed] [Google Scholar]
- 76.Shing Y, Folkman J, Haudenschild C, Lund D, Crum R, Klagsbrun M. Angiogenesis is stimulated by a tumor-derived endothelial cell growth factor. Cell Biochem. 1985;29:275–287. doi: 10.1002/jcb.240290402. [DOI] [PubMed] [Google Scholar]
- 77.Montesano R, Vassalli JD, Baird A, Guillemin R, Orci L. Basic fibroblast growth factor induces angiogenesis in vitro. Proc Natl Acad Sci USA. 1986;83:7297–7301. doi: 10.1073/pnas.83.19.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–985. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- 79.Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol. 1995;146:1029–1039. [PMC free article] [PubMed] [Google Scholar]
- 80.Ferrara N, Houck KA, Jakeman LB, Winer J, Leung DW. The vascular endothelial growth factor family of polypeptides. J Cell Biochem. 1991;47:211–218. doi: 10.1002/jcb.240470305. [DOI] [PubMed] [Google Scholar]
- 81.Conn G, Soderman D, Schaeffer MT, Wile M, Hatcher VB, Thomas KA. Purification of a glycoprotein vascular endothelial cell mitogen from a rat glioma-derived cell line. Proc Natl Acad Sci USA. 1990;87:1323–1327. doi: 10.1073/pnas.87.4.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Strieter RM, Kunkel SL, Elner VM, Martonyi CL, Koch EA, Polverini PJ. Interleukin-8. A corneal factor that induces neovascularizaton. Am J Pathol. 1992;141:1279–1284. [PMC free article] [PubMed] [Google Scholar]
- 83.Koch AE, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, Strieter RM. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science. 258:1798–1801. doi: 10.1126/science.1281554. [DOI] [PubMed] [Google Scholar]
- 84.Zetter BR. Cell motility in angiogenesis and tumor metastasis. Cancer Invest. 1990;8:669–671. doi: 10.3109/07357909009018942. [DOI] [PubMed] [Google Scholar]
- 85.Neal DE, Bennett MK, Hall RR. Epidermal growth-factor receptors in human bladder cancer: comparison of invasive and superficial tumors. Lancet. 1985;1:366–368. doi: 10.1016/s0140-6736(85)91386-8. [DOI] [PubMed] [Google Scholar]
- 86.Mendelsohn J. The epidermal growth factor receptor as a target for therapy with antireceptor monoclonal antibodies. Semin Cancer Biol. 1990;1:339–344. [PubMed] [Google Scholar]
- 87.Nicholson S, Richard J, Sainsbury C. Epidermal growth factor: results of a 6-year follow-up study on operable breast cancer with emphasis on the node negative subgroup. Br J Cancer. 1991;63:146–150. doi: 10.1038/bjc.1991.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Radinsky R, Risin S, Fan D, Dong Z, Bielenberg D, Bucana CD, Fidler IJ. Level and function of epidermal growth factor receptor predict the metastatic potential of human colon carcinoma cells. Clin Cancer Res. 1995;1:19–31. [PubMed] [Google Scholar]
- 89.Tsugawa K, Yonemura Y, Hirono Y, Fushida S, Kaji M, Miwa K, Miyazaki I, Yamamoto H. Amplification of the c-met, c-erbB-2, and epidermal growth factor receptor gene in human gastric cancers: correlation to clinical features. Oncology. 1998;55:475–481. doi: 10.1159/000011898. [DOI] [PubMed] [Google Scholar]
- 90.Otsuka T, Takayama H, Sharp R, Celli G, LaRochelle WJ, Bottaro DP Ellmore N, Vieira W, Ownes JW, Anver M, Merlino G. C-met autocrine activation induces development of malignant melanoma and acquisition of metastatic phenotype. Cancer Res. 1998;58:5157–5167. [PubMed] [Google Scholar]
- 91.Di Renzo MF, Olivero M, Giacomini A, Porte H, Chastre E, Mirossay L, Nordlinger B, Bretti S, Bottardi S, Giordano S, Plebani M, Gespach C, Comoglio PM. Overexpression and amplification of the Met/HGF receptor gene during the progression of colorectal cancer. Clin Cancer Res. 1995;1:147–154. [PubMed] [Google Scholar]
- 92.Perugini RA, McDade TP, Vittimberga FJ, Jr, Callery MP. The molecular and cellular biology of pancreatic cancer. Crit Rev Eukaryot Gene Expr. 1998;8:377–393. doi: 10.1615/critreveukargeneexpr.v8.i3-4.70. [DOI] [PubMed] [Google Scholar]