

Abstract
Excellent animal models are available of virus-induced and autoimmune heart disease that are remarkably similar to human disease. Developing good animal models for heart disease is crucial because cardiovascular disease is now the leading cause of death in the United States and is estimated to be the leading cause of death in the world by the year 2020. A significant proportion of heart disease in Western populations is associated with inflammation. Myocarditis, or inflammation of the heart muscle, is the major cause of sudden death in young adults. Although most individuals recover from acute myocarditis, genetically susceptible individuals may go on to develop chronic myocarditis and dilated cardiomyopathy (DCM) resulting in congestive heart failure. In this article, we describe a model of autoimmune myocarditis and DCM induced by inoculation with heart-passaged coxsackievirus B3 (CVB3). Intraperitoneal inoculation of susceptible mice with CVB3 induces acute cardiac inflammation from days 7 to 14 post infection (pi) that progresses to chronic myocarditis and DCM from day 28 to at least 56 pi. The model of CVB3-induced myocarditis presented here allows dissection of the contribution of viral infection and xenobiotics on immune dysregulation and inflammation in the heart. An improved understanding of the interaction between environmental exposures and the development of heart disease represents a clear challenge for immunotoxicologists.
Keywords: immunotoxicology, autoimmunity, myocarditis, coxsackievirus, animal model
1. Introduction
Cardiovascular disease is the number one killer of men and women in the United States and the second most common cause of death worldwide [1,2]. Although the true incidence of inflammation in the heart is unknown, it is estimated that 1 in 4 individuals in the United States have some form of inflammatory heart disease [1,3]. Acute myocarditis is a principal cause of heart failure in young adults and often progresses to chronic myocarditis, DCM and congestive heart failure requiring heart transplantation [3]. CVB3 infection is believed to be a principle etiologic agent in human myocarditis, and the same virus strain that infects humans (ignoring the species barrier) induces biphasic myocarditis in genetically susceptible strains of mice [3–6].
Currently, several animal models are available that closely resemble human myocarditis [7]. Murine models fall into two basic categories: acute cardiac inflammation and sudden death induced by direct viral damage (where nearly 70% of animals die at day 4 to 7 after infection) [8–11], or inflammation triggered by adjuvant and cardiac myosin or virus and cardiac myosin (heart-passaged virus) resulting in acute myocarditis with no deaths that progresses to chronic heart disease and DCM [5,6,12–15]. In this article, we describe a model of autoimmune myocarditis and DCM induced by inoculation with heart-passaged CVB3 [7,15].
In the model of CVB3-induced myocarditis presented here, intraperitoneal (ip) inoculation of BALB/c mice with heart-passaged CVB3 (Nancy strain), which contains virus and cardiac myosin, induces inflammatory heart disease that is remarkably similar to disease induced by inoculation with adjuvant and cardiac myosin (experimental autoimmune myocarditis or EAM) [14–17]. Acute CVB3-induced myocarditis develops in susceptible BALB/c mice from day 7 to 14 post infection (pi) and progresses to chronic myocarditis and DCM from day 35 to at least 56 pi (Fig. 1A) [6,15,18,19]. Resistant strains of mice (i.e. C57BL/6) do not develop the chronic phase of the disease (Fig. 1B). Furthermore, susceptible male mice develop significantly increased acute (Fig. 2A) and chronic (data not shown) myocarditis in response to CVB3 infection compared to female mice. Following infection with heart-passaged CVB3, mice quickly develop immunoglobulin (Ig)G autoantibodies specific for cardiac myosin, similar to those found in EAM and in patients with myocarditis and DCM [5,6,20,21]. Acute CVB3-induced myocarditis is characterized by a focal cellular infiltrate with little necrosis or fibrosis, comprised primarily of macrophages, neutrophils, CD4+ T cells, and CD8+ T cells with lower numbers of B cells, mast cells, natural killer cells, and dendritic cells (Fig. 2B) [19]. In this model, virus replicates at relatively low levels in the heart during acute myocarditis (102–104 PFU) compared with other CVB3-induced models (107–109 PFU) [8–11,18,19]. By day 14 pi, infectious virus is cleared from the heart and does not reactivate during chronic myocarditis [6,15]. In genetically susceptible mice a diffuse, lymphocytic infiltrate develops around day 35 pi with large areas of necrosis and fibrosis (Fig. 3A) as well as pericarditis (Fig. 3B) and DCM (Fig. 3D) [15]. Although viral genome can be detected in heart tissue during chronic myocarditis, persistent virus is found in both susceptible and resistant strains of mice [13,22].
Figure 1.
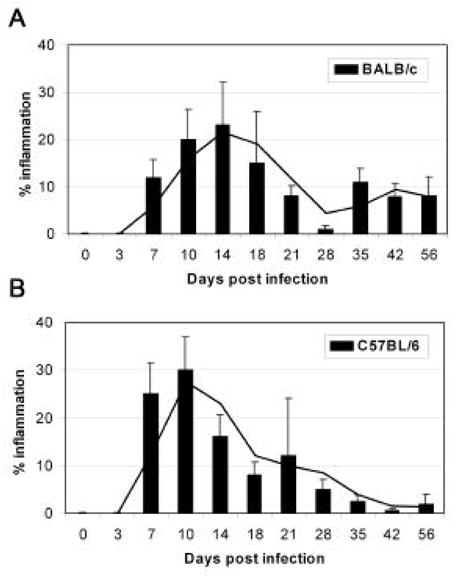
Progression to chronic autoimmune myocarditis following CVB3 infection of susceptible BALB/c mice. Intraperitoneal inoculation of BALB/c (A) or C57BL/6 (B) mice with a heart-passaged stock of CVB3 (Nancy strain) induces inflammation in the heart from day 7 to 21 after infection (acute myocarditis). During acute myocarditis, infectious virus can be detected in the heart of both mouse strains but does not correlate with the percent inflammation. Susceptible strains of mice (i.e. BALB/c) progress to a chronic phase of disease from day 35 to at least day 56 characterized by large areas of fibrosis, necrosis, pericarditis and DCM. The progression of myocarditis in female BALB/c and C57BL/6 mice is shown.
Figure 2.
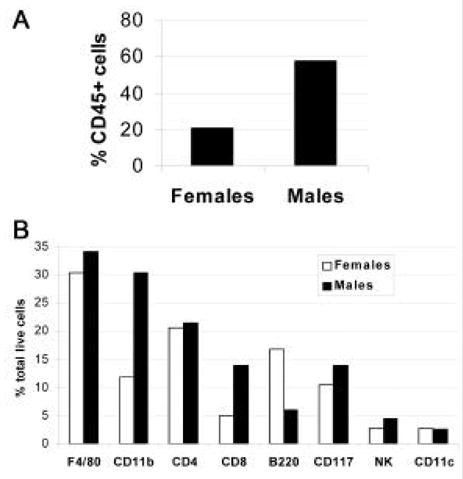
Male mice develop significantly increased inflammation in the heart during acute myocarditis. BALB/c mice were infected with CVB3 on day 0 and CD45+ immune cells isolated from the heart and analyzed by FACS on day 8 pi. Males have increased numbers of CD45+ immune cells in the heart compared to females (A). The infiltrate is composed of (B) macrophages (F4/80), macrophages and neutrophils (CD11b), CD4+ helper T cells, CD8+ cytolytic T cells, B cells (B220), mast cells (CD117), natural killer (NK) cells, and dendritic cells (CD11c).
Figure 3.
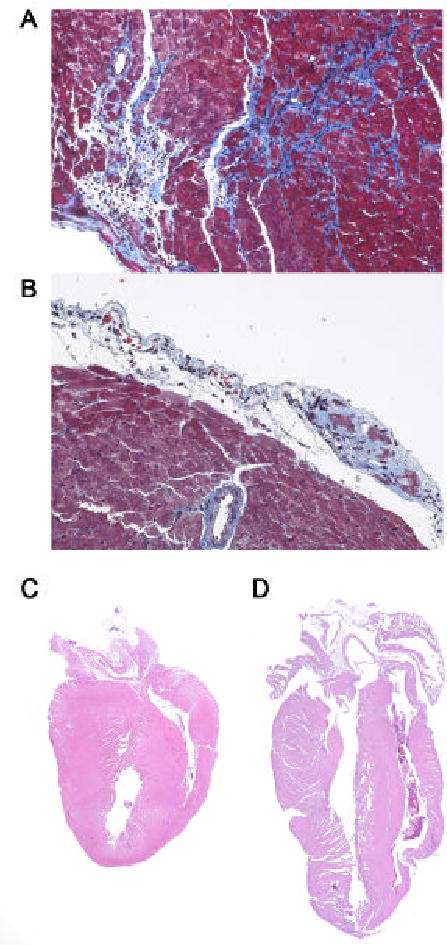
Chronic, autoimmune myocarditis following CVB3 infection is characterized by fibrosis, pericarditis and DCM. BALB/c mice were infected with CVB3 on day 0 and the heart examined at day 35 pi for fibrosis (collagen deposition stains bright blue) (A), pericarditis (B) and DCM (D). DCM is not present during acute myocarditis (at day 12 pi) (C). Original magnificantion, x64 (A and B) and x2.5 (C and D).
The development of autoimmune disease depends on a complex interaction between genetic, environmental, and endogenous factors [23–25]. Environmental agents, such as infections, diet, hormones and drugs, contribute significantly to the risk of developing and/ or dying from heart disease [2,14,26–28]. The incidence and severity of heart disease is higher among men, but the incidence increases in women as they age [1,29]. Young male BALB/c mice (6 to 8 weeks old) develop significantly greater acute and chronic myocarditis than female mice (Fig. 2), similar to humans [1,30]. Public health measures have dramatically reduced exposure to many infections and toxic chemicals. However, the effect of xenobiotics on the immune system and the development of inflammatory heart disease following infection has not been fully evaluated. Recent studies suggest that low dose exposure to immunotoxicants, such as mercury, may exacerbate inflammatory heart disease induced by viral infection or adjuvant and cardiac myosin [31]. A clear challenge for immunotoxicologists is to better understand the mechanisms involved in the progression of inflammatory heart disease. Only then can we develop more effective prevention and intervention strategies for these diseases.
2. Materials
2.1. Reagents and equipment
Vero cells (American Type Culture Collection, ATCC, Manassas, VA, USA)
Minimum essential medium (MEM), liquid and powder (Mediatech Inc., Herndon, VA, USA)
Heat-inactivated fetal bovine serum (FBS) (Invitrogen Life Technologies, Carlsbad, CA, USA)
Penicillin/ streptomycin, 5000 U (Pen/ Strep) (Mediatech Inc., Herndon, VA, USA)
Methyl cellulose, 4000 centipoises (Fischer Scientific, Pittsburgh, PA, USA)
Bleach (Clorox, USA)
Susceptible mice, age 6 to 8 weeks old (Jackson Laboratory, Bar Harbor, ME, USA)
10% phosphate-buffered formalin (Decal Corporation, Tallman, NY, USA)
PBS (Biofluids, Biosource Int., Camarillo, CA, USA)
Collagenase II, 1 mg/ mL (Sigma-Aldrich, St. Louis, MO, USA)
Protease XIV, 0.5 mg/ mL (Sigma–Aldrich, St. Louis, MO, USA)
CVB3, Nancy strain (ATCC, Manassas, VA, USA)
CD45 paramagnetic beads (30F11.1; Miltenyi Biotec, Auburn, CA, USA)
Paramagnetic beads (Miltenyi Biotec, Auburn, CA, USA)
FACS antibodies for cell surface markers (BD PharMingen or eBioScience, San Diego, CA, USA)
FACSCalibur flow cytometer (BD BioSciences, San Diego, CA, USA)
Cell Quest software (BD BioSciences, San Diego, CA, USA)
3. Methods
3.1. Tissue culture virus
Grow Vero cells until confluent at 37°C and 5% CO2 in MEM supplemented with Pen/ Strep and 10% FBS (10% MEM). Remove media and replace with MEM supplemented with Pen/ Strep and 2% FBS (2% MEM) (FBS inhibits viral entry). CVB3 is infectious to people as well as mice. A 20% bleach solution or UV light will kill the virus. Always wear a protective mask and replace gloves immediately after working with the virus to prevent spread. Wash all surfaces and utensils with 20% bleach and expose the hood to UV light for at least 15 min after working with the virus. Add 1 mL CVB3 (ATCC) to a confluent flask of Vero cells and incubate at 37°C and 5% CO2 until cells round up and detach from the flask (approximately 2 days). Carefully collect cells and supernatant from the flask, centrifuge at 795 g for 20 min and collect supernatant containing infectious virus. Aliquot supernatant and freeze at −80°C. The viral stock should last at least one year at −80°C. Add bleach to the flask and centrifuge tube to kill remaining virus and discard.
3.2. Heart passage of the virus
Inoculate 6 week old susceptible mice (i.e. BALB/c) with 0.1 mL of tissue culture-derived virus ip. Three days later, sacrifice the mice and collect the hearts. Sera and organs from these mice contain infectious virus, so use appropriate care. Blot excess blood from the hearts and immediately add to cold 2% MEM (10% w/v). Homogenize hearts with an electric homogenizer and centrifuge at 795 g for 20 min. Collect supernatant containing infectious virus (and cardiac myosin) and aliquot and store at −80°C until used for inoculating mice. Uninfected hearts from the same mouse strain can be treated in the same manner and used as uninfected controls in experiments. The viral stock should last at least one year at −80°C. Add bleach to the centrifuge tube to kill the virus and discard.
3.3 Plaque assay
The plaque assay determines the level of infectious virus in a sample by the degree of killing observed in Vero cells. The standard protocol has been described in detail previously [18,32]. The level of infectious virus in tissue culture and heart-passaged CVB3 stocks is assessed by plaque assay. In brief, virus stocks (or homogenized tissue supernatants from experiments) are serially diluted in 2% MEM and added to Vero cells that are approximately 80% confluent. The virus is incubated in 24 well trays for one hour at 37°C and 5% CO2 to allow viral attachment, and then incubated for 3 days with methyl cellulose in 2% MEM to allow plaque formation. Wells are stained with 1% methylene blue in 10% formalin (which inactivates the virus) over night at room temperature. The next day trays are washed and plaques counted. Virus titers are expressed as the mean plaque forming units (PFU)/ mL or g of tissue ±SEM.
3.4 Infection of mice and assessment of myocarditis and DCM
Infect 6 to 8 week old mice (i.e. BALB/c, A/J) with 0.1 mL of 103 PFU of heart-passaged CVB3 diluted in sterile PBS ip on day 0. Hearts should be collected on day 7 to 12 pi for acute myocarditis and from day 35 to 56 pi for chronic myocarditis [15,18]. No deaths occur during the acute phase of myocarditis [15]. Mice inoculated ip with PBS or uninfected heart homogenate do not develop acute or chronic myocarditis [15]. Cut hearts longitudinally, fix in 10% phosphate-buffered formalin and embed in paraffin. Sections are cut 5 μm thick at various depths in the section and stained with hematoxylin and eosin (H&E) for assessment of inflammation in the heart or with Masson’s trichrome to detect collagen deposition (fibrosis). Myocarditis is assessed as the percentage of the heart section with inflammation compared to the overall size of the heart section, with the aid of a microscope eyepiece grid (Fig. 1) [18]. DCM is assessed by gross observation of H&E stained sections at low magnification for dilation of the chambers of the heart at days 35 to 56 pi (Fig. 3D) [15]. DCM is absent during the acute phase at days 7 to 12 pi (Fig. 3C).
3.5 Heart digestion and FACS analysis of the cellular infiltrate
Perfuse the heart with cold PBS at a constant flow of 14 mL/ min for 2 min [19]. Release immune cells from the myocardium by digestion with collagenase II (1 mg/ mL) and protease XIV (0.5 mg/ mL) in PBS for 7 min at 37ºC. Further dislodge immune cells from the tissue using razor blades. Individual cell suspensions from 7 to 10 mice/ group should be pooled by group and immune cells separated from heart cells using a magnetic column and anti-CD45 paramagnetic beads (30F11.1). Mast cells and macrophages should be separated using anti-CD117 and anti-FITC paramagnetic beads, respectively. Immune cells are stained with monoclonal antibodies diluted in 1% FBS in PBS to label cell surface markers (Fig. 2B). Cell fluorescence is measured using a FACSCalibur flow cytometer and data analyzed using Cell Quest software.
4. Concluding remarks
The model of CVB3-induced myocarditis presented here allows dissection of the contribution of viral infection and xenobiotics on immune dysregulation and the severity of inflammation in the heart. Because patients rarely die from acute viral myocarditis [3], autoimmune models of CVB3-induced myocarditis may more closely resemble disease as it occurs in human populations. Indeed, clinical observations closely resemble findings using this model. Little is known about the interaction of infections and chemicals or other toxic agents on the development of heart disease. An improved understanding of the mechanisms involved in the pathogenesis of heart disease is urgently needed to develop better approaches to therapy. Treatment of myocarditis and DCM remains problematic, and with the worldwide rise in inflammatory heart disease better treatment options are essential. Good animal models of autoimmune disease will provide answers to many mechanistic questions that simply cannot be investigated in patients. The model presented here may be used to evaluate the adverse effects of various exogenous agents on the cardiovascular and immune systems.
Acknowledgments
Funding for this research was provided by grants from the National Institutes of Health (HL67290, HL70729, AI51835 and ES03819).
References
- 1.American Heart Association. Heart Disease & Stroke Statistics- 2005 Update (2005) . www.americanheart.org.
- 2.Willerson JT, Ridker PM. Circulation. 2004;109(suppl II):II2–II10. doi: 10.1161/01.CIR.0000129535.04194.38. [DOI] [PubMed] [Google Scholar]
- 3.Dec GW. In: Myocarditis: From Bench to Bedside. Cooper LT Jr, editor. Humana Press; Totowa, New Jersey: 2003. pp. 157–281. [Google Scholar]
- 4.Yusuf S, Reddy S, Ounpuu S, Anand S. Circulation. 2001;104:2746–2753. doi: 10.1161/hc4601.099487. [DOI] [PubMed] [Google Scholar]
- 5.Rose NR, Wolfgram LJ, Herkowitz A, Beisel KW. Ann NY Acad Sci. 1986;475:146–156. doi: 10.1111/j.1749-6632.1986.tb20864.x. [DOI] [PubMed] [Google Scholar]
- 6.Fairweather D, Kaya Z, Shellam GR, Lawson CM, Rose NR. J Autoimm. 2001;16:175–186. doi: 10.1006/jaut.2000.0492. [DOI] [PubMed] [Google Scholar]
- 7.Fairweather D, Rose NR. Drug Discov Today: Dis Models. 2004;1:381–386. [Google Scholar]
- 8.Huber SA, Gauntt CJ, Sakkinen P. Adv Virus Res. 1998;51:35–80. doi: 10.1016/s0065-3527(08)60783-6. [DOI] [PubMed] [Google Scholar]
- 9.Horwitz MS, La Cava A, Fine C, Rodriguez E, Ilic A, Sarvetnick N. Nat Med. 2000;6:693–697. doi: 10.1038/76277. [DOI] [PubMed] [Google Scholar]
- 10.Fong IW. In: Infections and the Cardiovascular System: New perspectives. Fong IW, editor. Kluwer Academic: Plenum Publishers; New York: 2003. pp. 3–31. [Google Scholar]
- 11.Fuse K, Chan G, Liu Y, Gudgeon P, Husain M, Chen M, Yeh WC, Akira S, Liu PP. Circulation. 2005;112:2276–2285. doi: 10.1161/CIRCULATIONAHA.105.536433. [DOI] [PubMed] [Google Scholar]
- 12.Neu N, Rose NR, Beisel KW, Herskowitz A, Gurri-Glass G, Craig SW. J Immunol. 1987;139:3630–3636. [PubMed] [Google Scholar]
- 13.Fairweather D, Frisancho-Kiss S, Rose NR. Rev Med Virol. 2005;15:17–27. doi: 10.1002/rmv.445. [DOI] [PubMed] [Google Scholar]
- 14.Fairweather D, Rose NR. Lupus. 2005;14:646–651. doi: 10.1191/0961203305lu2192oa. [DOI] [PubMed] [Google Scholar]
- 15.Fairweather D, Frisancho-Kiss S, Yusung SA, Barrett MA, Gatewood SJL, Davis SE, Njoku DB, Rose NR. Am J Pathol. 2004;165:1883–1894. doi: 10.1016/s0002-9440(10)63241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Afanasyeva M, Georgakopoulos D, Fairweather D, Caturegli P, Kass DA, Rose NR. 2004 Circulation. 2004;110:2910–2917. doi: 10.1161/01.CIR.0000147538.92263.3A. [DOI] [PubMed] [Google Scholar]
- 17.Afanasyeva M, Georgakopoulos D, Belardi DF, Bedja D, Fairweather D, Wang Y, Kaya Z, Gabrielson KL, Rodriguez ER, Caturegli P, Kass DA. NR Rose Proc Natl Acad Sci USA. 2005;102:180–185. doi: 10.1073/pnas.0408241102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fairweather D, Yusung S, Frisancho(-Kiss) S, Barrett M, Gatewood S, Steele R, Rose NR. J Immunol. 2003;170:4731–4737. doi: 10.4049/jimmunol.170.9.4731. [DOI] [PubMed] [Google Scholar]
- 19.Fairweather D, Frisancho-Kiss S, Yusung SA, Barrett MA, Davis SE, Steele RA, Gatewood SJL, Rose NR. J Immunol. 2005;174:261–269. doi: 10.4049/jimmunol.174.1.261. [DOI] [PubMed] [Google Scholar]
- 20.Neu N, Beisel KW, Traystman MD, Rose NR. SW Craig J Immunol. 1987;138:2488–2492. [PubMed] [Google Scholar]
- 21.Lauer B, Schannwell M, Kuhl U, Strauer BE. HP Schultheiss J Am Coll Cardiol. 2000;35:11–18. doi: 10.1016/s0735-1097(99)00485-4. [DOI] [PubMed] [Google Scholar]
- 22.Lenzo JC, Fairweather D, Cull V, Shellam GR, Lawson CM. J Mol Cell Cardiol. 2002;34:629–640. doi: 10.1006/jmcc.2002.2003. [DOI] [PubMed] [Google Scholar]
- 23.Rose NR. Sem Liver Dis. 2002;22:387–394. doi: 10.1055/s-2002-35708. [DOI] [PubMed] [Google Scholar]
- 24.Davidson A, Diamond B. N Engl J Med. 2001;345:340–350. doi: 10.1056/NEJM200108023450506. [DOI] [PubMed] [Google Scholar]
- 25.Brix TH, Kyvik KO, Christensen K, Hegedus L. J Clin Endocrinol Metab. 2001;86:930–934. doi: 10.1210/jcem.86.2.7242. [DOI] [PubMed] [Google Scholar]
- 26.Fairweather D, Rose NR. Emerg Inf Dis. 2004;10:2005–2011. doi: 10.3201/eid1011.040367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brook RD, Franklin B, Cascio W, Hong Y, Howard G, Lipsett M, Luepker R, Mittleman M, Samet J, Smith SC, Jr, Tager I. Circulation. 2004;109:2655–2671. doi: 10.1161/01.CIR.0000128587.30041.C8. [DOI] [PubMed] [Google Scholar]
- 28.Doria A, Sarzi-Puttini P. Lupus. 2005;14:643–645. doi: 10.1191/0961203305lu2191ed. [DOI] [PubMed] [Google Scholar]
- 29.Liu PY, Death AK, Handelsman DJ. Endocrine Rev. 2003;24:313–340. doi: 10.1210/er.2003-0005. [DOI] [PubMed] [Google Scholar]
- 30.Huber SA, Pfaeffle B. J Virol. 1994;68:5126–5132. doi: 10.1128/jvi.68.8.5126-5132.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silbergeld EK, Silva IA, Nyland JF. Toxicol Appl Pharm. 2005;207:S282–S292. doi: 10.1016/j.taap.2004.11.035. [DOI] [PubMed] [Google Scholar]
- 32.Cihakova D, Sharma R, Fairweather D, Afanasyeva M, Rose NR. Methods Mol Med. 2004;102:175–194. doi: 10.1385/1-59259-805-6:175. [DOI] [PubMed] [Google Scholar]