

Abstract
Duchenne muscular dystrophy is a relatively common disease that affects skeletal muscle, leading to progressive paralysis and death. There is currently no resolutive therapy. We have developed a treatment in which we combined the effects of nitric oxide with nonsteroidal antiinflammatory activity by using HCT 1026, a nitric oxide-releasing derivative of flurbiprofen. Here, we report the results of long-term (1-year) oral treatment with HCT 1026 of two murine models for limb girdle and Duchenne muscular dystrophies (α-sarcoglycan-null and mdx mice). In both models, HCT 1026 significantly ameliorated the morphological, biochemical, and functional phenotype in the absence of secondary effects, efficiently slowing down disease progression. HCT 1026 acted by reducing inflammation, preventing muscle damage, and preserving the number and function of satellite cells. HCT 1026 was significantly more effective than the corticosteroid prednisolone, which was analyzed in parallel. As an additional beneficial effect, HCT 1026 enhanced the therapeutic efficacy of arterially delivered donor stem cells, by increasing 4-fold their ability to migrate and reconstitute muscle fibers. The therapeutic strategy we propose is not selective for a subset of mutations; it provides ground for immediate clinical experimentation with HCT 1026 alone, which is approved for use in humans; and it sets the stage for combined therapies with donor or autologous, genetically corrected stem cells.
Keywords: HCT 1026, satellite cells, skeletal muscle, α-sarcoglycan-null mice, mdx mice
Muscular dystrophies are clinically and molecularly heterogeneous genetic diseases causing wasting of skeletal muscle, severe local inflammation, and at least initially muscle regeneration for which there is no resolutive therapy (1). In the most severe forms, such as Duchenne muscular dystrophy, muscle regeneration is progressively exhausted, leading the patient to complete paralysis and death, usually by respiratory and/or cardiac failure (1). The therapeutic protocols currently in use, based on corticosteroid administration, provide some delay in the progression of the disease, but they are associated with severe side effects (2). The alternative pharmacological strategies attempted so far did not yield favorable outcomes in clinical trials, and they have not entered into the clinical practice (3).
Studies in mouse models have explored possible therapeutic strategies, ranging from deacetylase inhibitors (4), inhibition of myostatin (5), insulin-like growth factor 1 overexpression (6) to skipping of the mutated exon (7) and cell therapy with mesoangioblast stem cells (8). Although encouraging, these approaches still lack approval for use in patients and data on long-term efficacy, safety, and tolerability; in addition, they are expensive and in some cases they target only subsets of patients. The development of stem cell therapies is also hampered by problems related with isolation, in vitro expansion, and efficient engraftment of the cells used (9).
To tackle these issues, we developed a treatment by combining the known beneficial effects of nitric oxide (NO) in muscle repair and regeneration (10–15) with nonsteroidal antiinflammatory activity. As a drug of choice, we selected HCT 1026, a derivative of flurbiprofen, one of the most potent nonsteroidal antiinflammatory drugs, that releases NO and does not have the severe side effects of corticosteroids (16, 17). Of importance for an immediate testing in the clinic is the fact that HCT 1026 is safe at the gastrointestinal level, and it has been approved for use in humans; it is effective on oral administration, and it is thus suited for long-term treatment. As a second important step in the development of an efficacious therapeutic protocol for muscular dystrophy, we combined this pharmacological treatment with intraarterial delivery of mesoangioblasts.
The effect of HCT 1026 was tested on two models of muscular dystrophy, the α-sarcoglycan (SG)-null and mdx mice, in a long-term (1-year) treatment. HCT 1026 dramatically slowed the progress of the disease, and it maintained muscle integrity and function through mechanisms involving inhibition of inflammation and preservation of satellite cell number and activity. HCT 1026 was significantly more potent than prednisolone, which was used as a reference corticosteroid, and it did not induce detectable side effects. Moreover, the drug treatment improved significantly the therapeutic potential of mesoangioblasts by increasing their homing to dystrophic muscles. These results open a window to an effective cure of muscular dystrophy.
Results
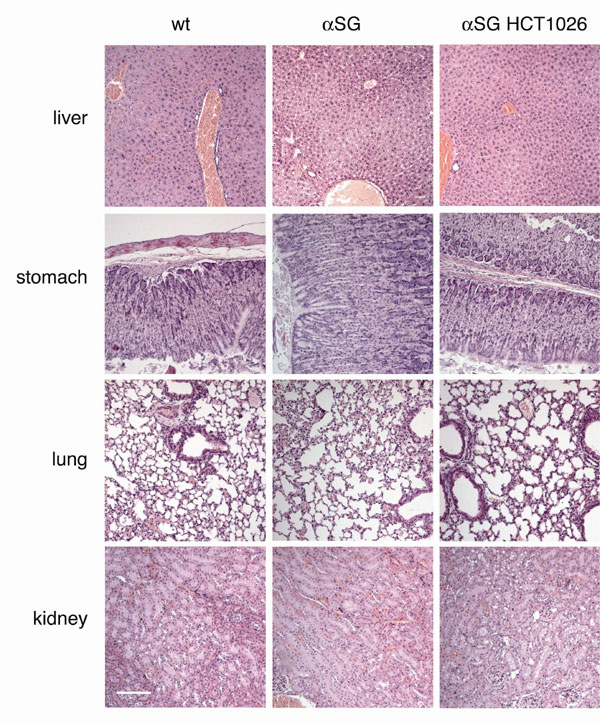
The first set of experiments was carried out in the α-SG-null mice, a mouse model of limb girdle muscular dystrophy with a severe phenotype (18). Groups of mice (18 animals per group) were treated for up to 12 months with HCT 1026 (30 mg/kg of body weight) or prednisolone (2 mg/kg), incorporated in the diet. Control groups receiving the same diet without any drug or the NO donor isosorbide dinitrate (ISDN; 30 mg/kg) were analyzed in parallel. Flurbiprofen was not used because of its known long-term toxicity (16, 17), whereas the daily dose of HCT 1026 we used produces antiinflammatory activity without signs of toxicity (19). Incorporation of the drug into the diet led to plasma levels of flurbiprofen (the active and detectable metabolite of HCT 1026) in the range of 16–20 μM after 30 days or 44 μM after 6 months (19). This plasma concentration is in the same order of magnitude of that found in healthy volunteers after treatment for 7 days with HCT 1026 at 100 and 200 mg orally (20). The dose of prednisolone was similar to that used in other studies in mice (21). We started the treatment 1 month after weaning, roughly at the onset of the disease symptoms, thus mimicking a treatment starting in pediatric patients. To evaluate efficacy, we relied on morphological, histochemical, and functional parameters. At the time of animal killing we carried out histological analyses of bone, liver, spleen, brain, kidney, stomach, lung, and heart, to check for any major sign of HCT 1026 toxicity. No relevant histological differences in these organs were observed between α-SG-null and wild-type (WT) mice, and no significant histological alterations were observed in the 12-month time window of drug treatment [see supporting information (SI) Fig. 5] (data not shown).
HCT 1026 Reduces Creatine Kinase Serum Levels in α-SG-Null Mice.
Creatine kinase, an enzyme released by damaged or necrotic fibers, is commonly used as a biomarker of the severity of muscular dystrophies. The serum levels of creatine kinase in untreated α-SG-null mice were higher than those of WT animals already within the first 2 months of life, and they increased progressively up to 6 months, after which a decline was observed (Fig. 1A), consistent with the dramatic reduction in muscular tissue known to occur at later stages of the disease (18) (see also Fig. 2). In the group receiving HCT 1026, creatine kinase levels were reduced significantly with respect to those observed in untreated animals (by 22.2%, 44.9%, and 75.7% at 3, 4, and 6 months, respectively; P < 0.05, n = 9). A reduction in creatine kinase levels was observed also in prednisolone-treated animals (by 41.0%, 42.2%, and 53.6% at 3, 4, and 6 months, respectively P < 0.05; n = 9) (Fig. 1A), whereas ISDN did not have significant effects (SI Table 1).
Fig. 1.

Effects of treatment of α-SG-null mice with HCT 1026 or prednisolone on parameters of muscle function. (A) Creatine kinase serum levels. wt, wild-type. (B and C) Locomotor performance measured on the running-wheel (B) and exhaustion treadmill (C) tests. (D) Mechanical analysis of intact soleus and extensor digitorum longus (EDL) muscle contractile activity, assessed by measuring tetanic force. Values shown are the results of experiments on nine animals per group ±SEM. Asterisks and crosses indicate statistical significance vs. untreated α-SG-null mice analyzed in parallel as control (NT) or prednisolone-treated animals, respectively (P < 0.05).
Fig. 2.

Effects of treatment of α-SG-null mice with HCT 1026 or prednisolone on morphological muscle parameters. (A–C) Tibialis anterior and diaphragm muscle cross-cryosections were stained with hematoxylin/eosin (H&E). (A) Numbers of necrotic (n) and centronucleated (c) fibers. Values shown are the results of experiments on nine animals per group ± SEM. Asterisks indicate statistical significance vs. untreated α-SG-null mice analyzed in parallel as control (NT) or prednisolone-treated animals, respectively (P < 0.05). wt, wild-type. (B) Histological images obtained after 6 months of treatment representative of three reproducible experiments. (C) Fiber membrane damage was evaluated after the treadmill exhaustion test in EDL muscles at the 12-month time point by assessing Evans blue dye uptake. Shown are histological images representative of three reproducible experiments. (D) Distribution of CSA values of 300 single muscle fibers of tibialis anterior muscles isolated from nine animals per group. (Scale bar, 400 μm.)
HCT 1026 Ameliorates in Vivo and ex Vivo Muscle Function in α-SG-Null Mice.
Muscle function was evaluated by using the running-wheel and treadmill tests, which measure animal voluntary motility and muscle endurance, respectively. Untreated α-SG-null mice showed a significantly reduced motility in the running-wheel test (Fig. 1B) and a lower time of exhaustion in the treadmill test (Fig. 1C) compared with WT mice. The groups receiving HCT 1026 performed significantly better than untreated dystrophic mice in both tests at all time points. At 6 months, the group receiving prednisolone performed significantly worse than the group receiving HCT 1026 on the running wheel, and they performed at comparable levels on the treadmill. At 12 months, prednisolone-treated animals also performed worse on the treadmill than those receiving HCT 1026. Prednisolone-treated mice performed significantly better than the untreated α-SG-null mice only in the treadmill test. The group receiving ISDN behaved similarly to untreated α-SG-null mice (SI Table 1).
We determined whether the reduced creatine kinase serum levels and the increase in the in vivo performance induced by the treatments correlated with increased muscle strength. To this end, at 12 months we measured force production by soleus and EDL muscles. Muscles from α-SG-null mice developed less force during tetanic contraction compared with WT mice (Fig. 1D). Treatment with HCT 1026 reduced this difference significantly. Prednisolone was significantly less effective in preventing the loss of muscle force in soleus muscle, and it did not show any effect in EDL. At the end of the experiments, the integrity of the sarcolemma was investigated by evaluating the uptake of Evans blue dye, which stains severely damaged and dying fibers. EDL and soleus from α-SG-null mice, untreated or treated with prednisolone, showed similarly higher dye uptake compared with EDL from WT mice (Fig. 2C). No significant differences between these groups could be detected. By contrast, muscles from α-SG-null mice treated with HCT 1026 displayed a significantly lower dye uptake (Fig. 2C and data not shown).
HCT 1026 Enhances Regeneration and Protects Skeletal Muscle in α-SG-Null Mice from Damage.
Evaluation of H&E-stained sections was carried out on the tibialis anterior and diaphragm muscles. The muscle obtained from untreated α-SG-null mice revealed a progressive degeneration, with the appearance of necrotic and calcified fibers, which was accompanied by an increase in centronucleated, regenerating fibers early on, followed by a decrease, most likely because of depletion of satellite cells (22) (Fig. 2 A and B). The treatment with prednisolone only slightly ameliorated these parameters, whereas ISDN produced no significant effects (SI Table 1). By contrast, muscle architecture was nearly normal in the groups receiving HCT 1026, with a significantly lower number of necrotic fibers, even at the 12-month time point. Interestingly, the number of centronucleated fibers was higher in HCT 1026-treated animals, and it did not decline with time, indicating that exhaustion of the satellite pool did not occur, consistent with the creatine kinase serum level determinations and the observed reduction in muscle wasting. Further support to a beneficial effect on skeletal muscle by HCT 1026 came from the analyses of frequency histograms of the cross-sectional area (CSA) of the single fibers. In the untreated α-SG-null mice, fiber CSA was very heterogeneous compared with fiber CSA of WT mice, with values distributed between 250 and 3,000 μm2, the largest diameters being a sign of degeneration (pathological hypertrophy) (Fig. 2D). By contrast, the frequency histograms of HCT 1026-treated animals showed a significantly more homogeneous distribution, with CSA values not significantly different from those observed in the WT mice. Homogeneity in CSA values was observed also in prednisolone-treated animals, but only up to 6 months; after 12 months CSA value distribution was as heterogeneous as that of untreated α-SG-null mice.
HCT 1026 Increases Satellite Cell Differentiation and Resistance to Apoptosis, and It Preserves Their Number in Vivo.
The results obtained so far suggest that the mechanism of action of HCT 1026 involves specific beneficial effects on satellite cells. We therefore assessed the action of the drug on satellite cell differentiation, proliferation, and resistance to apoptogenic cues found in the cytotoxic environment of the dystrophic muscle. The results obtained by both in vitro and in vivo experiments clearly show that treatment with HCT 1026 significantly increased satellite cell differentiation and survival, leading to an increased satellite cell population (SI Results and SI Fig. 6). The efficacy of HCT 1026 on these parameters was still significant after 12 months of treatment.
HCT 1026 and Prednisolone Exert an Antiinflammatory Action on Skeletal Muscle of α-SG-Null Mice.
Inflammation plays a role in the pathogenesis of muscular dystrophy because an abundant inflammatory infiltrate dispersed mostly in the fibrous/connective tissue of dystrophic muscle progressively replaces myofibers, causing reduction of muscle strength and worsening the disease (23). Azan–Mallory staining revealed frequent foci of fibrosis in muscles of untreated α-SG-null mice (Fig. 3A and B), with an infiltrate composed mainly of CD11+ macrophages among fibers, which were revealed by laminin staining (Fig. 3D). We examined also the expression of several proinflammatory cytokines, and we established that in dystrophic muscle TGF-β, macrophage inflammatory protein-1α (MIP-1α), and monocyte chemoattractant protein-1 (MCP-1) are increased (Fig. 3C). We found that treatment with HCT 1026 significantly reduced the number of inflammatory infiltrates, the amount of fibrotic tissue, and the concentrations of TGF-β, MIP-1α, and MCP-1, indicating that inhibition of inflammation plays a role in the action of HCT 1026 (Fig. 3). Prednisolone reduced the inflammatory infiltrates and cytokine expression; however, it did not reduce the formation of fibrous tissue. These effects, together with the limited effect on long-term muscle regeneration and protection from damage (Fig. 2), may contribute an explanation of why prednisolone does not significantly ameliorate muscle function.
Fig. 3.

Effects of treatment of α-SG-null mice with HCT 1026 or prednisolone on muscle inflammation. (A) Number of inflammatory infiltrates measured on Azan–Mallory-stained serial muscle sections of tibialis anterior (TA) and diaphragm (DIA) muscles. (B) TA muscles were isolated after 6 months of treatment and rapidly homogenized. The concentrations of the indicated cytokines were measured on muscle homogenates with appropriate antibodies. Values shown in A and B are the results of nine experiments ±SEM. Asterisks indicate statistical significance vs. untreated α-SG-null mice analyzed in parallel as control (NT) (P < 0.05). wt, wild-type. (C) Staining of TA muscle serial sections with Azan–Mallory reveals an accumulation of extracellular scar tissue (blue). (D) Presence of macrophages, revealed by staining of TA sections with the anti-CD11 and anti-laminin antibodies. (C and D) Histological images representative of nine reproducible experiments. [Scale bar (D), 400 μm.]
HCT 1026 Ameliorates the Dystrophic Phenotype of the mdx Mouse.
mdx mice (nine animals per group) were treated with HCT 1026 (30 mg/kg) incorporated in the diet, or they received plain diet (controls). Creatine kinase serum levels, motility on the running-wheel and treadmill tests, and muscle morphology were assessed after 6 months. The results clearly show that the long-term treatment with HCT 1026 exerts beneficial effects also in these animals. Data reported in SI Table 2 and SI Fig. 7 show that HCT 1026 significantly reduced creatine kinase levels, improved motility tests, reduced damage to the muscles, and increased centronucleated fibers. Also, in the mdx mouse we found no signs of toxicity with HCT 1026 in a variety of tissues and organs (data not shown).
HCT 1026 Enhances the Therapeutic Efficacy of Mesoangioblasts by Increasing Their Ability to Migrate and Engraft to Dystrophic Muscles.
The treatment with HCT 1026 does not correct the genetic defect underlying muscular dystrophy. To address this issue we combined the pharmacological treatment with delivery of mesoangioblasts. Mesoangioblasts were transduced with a lentiviral vector expressing GFP (8) to trace them into the various tissues, and they were used either untreated or after a 12-h ex vivo exposure to stromal cell-derived factor 1 (SDF-1; 50 ng/ml), a treatment that increases their homing to muscle (24). At variance with a previous report in which cells were injected in α-SG-null mice three times (8), here we performed a single injection to evaluate best the changes in therapeutic efficacy induced by drug treatment. Migration of mesoangioblasts was assessed by injecting them at a dose of 5 × 105 cells per animal in α-SG-null mice untreated or treated for 3 months with HCT 1026. Gastrocnemius and filter organs (spleen, liver, lung, and kidney, where mesoangioblasts that flow through the circulation may be trapped) were recovered 6 h after the injection, and the percentage of the cells migrated in them was evaluated by real-time PCR with specific primers for GFP. Mesoangioblast migration in gastrocnemius on the side of injection (right side) was significantly enhanced in HCT 1026-treated animals (Fig. 4A). A synergic effect of HCT 1026 with pretreatment of mesoangioblasts with SDF-1 before their injection was also observed. The percentage of cells retained by the filter organs was reduced in all conditions favoring mesoangioblast migration to the muscle.
Fig. 4.

HCT 1026 increases the therapeutic efficacy of mesoangioblasts. α-SG-null mice, either untreated or exposed to a 3-month therapy with HCT 1026, were injected into the right femoral artery with GFP-expressing D16 mesoangioblasts treated or not with SDF-1. (A) Mesoangioblast migration to right (R) and left (L) gastrocnemius muscles (Gs) and liver (Li), lung (Lu), kidney (Ki), and spleen (Sp) evaluated by real-time PCR with specific primers for GFP on five animals per group. (B–E) Engrafting of mesoangioblasts to muscles. (B) Engrafting to the right and left quadriceps (Qd), Gs, and soleus (So) muscles evaluated by real-time PCR with specific primers for α-SG on three animals per group. (C) Western blot analysis of α-SG expression in Qd compared with that of GAPDH. The results shown are from one of three reproducible experiments. (D) Immunostaining of α-SG expressed in Qd fibers that were revealed in serial sections by staining with laminin; DAPI was used to identify nuclei. The results shown are from one of five reproducible experiments. (E) Specific tension of single muscle fibers (n = 101) of Gs from three animals per group. (F and G) Serum creatine kinase levels (F) and animal performance on the exhaustion treadmill tests (G), carried out as described in Fig. 1 on five animals per group. (E–G) Untreated indicates the values observed in α-SG-null mice which were neither treated with HCT 1026 nor injected with mesoangioblasts. (A, B, E–G) Bars represent SEM. Asterisks indicate statistical significance vs. α-SG-null mice injected with untreated mesoangioblasts analyzed in parallel as control (P < 0.05). wt, wild-type. [Scale bar (D), 200 μM.]
To test whether an increased migration would lead also to an increased correction of the dystrophic phenotype, mice treated with or without HCT 1026 were killed 2 months after mesoangioblast injection. The efficacy of the treatment was assessed by measuring α-SG mRNA expression by RT-PCR and α-SG protein levels by Western blotting and immunofluorescence. Mesoangioblasts engrafted and reconstituted muscle fibers in the quadriceps, gastrocnemius, and soleus muscles on the side of injection (Fig. 4 B–D). This effect was doubled in HCT 1026-treated animals and also by preexposure of mesoangioblasts to SDF-1. The combination of these two treatments increased 4-fold the percentage of fibers expressing a-SG protein. Of importance, both treatments increased engrafting in the contralateral muscles also. The ability of HCT 1026 to increase mesoangioblast-mediated fiber reconstitution was accompanied by increased single fiber-specific tension (Fig. 4E), reduced muscle damage (judged by reduced creatine kinase levels; Fig. 4F), and enhanced mouse performance on the treadmill (Fig. 4G), which were significant vs. those observed in animals receiving mesoangioblasts in the absence of HCT 1026 treatment, and they were further increased by SDF-1 pretreatment of mesoangioblasts. Thus, HCT 1026 greatly increases mesoangioblast engrafting, and it may synergize with other treatments, leading to a significantly more effective cell therapy.
Discussion
Two decades after the identification of the molecular defect responsible for Duchenne muscular dystrophy, there are still no effective cures for the disease. The failure of all previous pharmacological treatments has left corticosteroids as the only available drug treatment. Therapy with corticosteroids, despite current attempts to ameliorate the protocols of administration, is still of limited clinical benefits, and it is accompanied by severe side effects (21).
The treatment we tested here meets several criteria for an effective therapy, including the ability to block or at least significantly delay the progress of the disease, produce little or no side toxicity, be cost-effective, and, eventually, resolve the underlying genetic defect. In particular, the administration of HCT 1026 was sufficient on its own to delay significantly and persistently the progression of muscular dystrophy in two different models relevant to muscular dystrophy in humans. The drug was effective in correcting biochemical and morphological alterations and in limiting inflammation. Most importantly, the drug increased muscle strength and significantly increased the ability of animals to perform on different motility tests. This functional amelioration was persistent after 12 months of treatment, when disease in untreated animal was severe, clearly demonstrating the efficacy of the treatment in slowing disease progression. Long-term beneficial effects have not been reported for any of the experimental treatments of muscular dystrophy investigated so far.
The beneficial effects of NO on muscle function are well known, and the mechanisms of its action are well understood. In particular, NO is generated by skeletal muscle to stimulate key actions in muscle repair, including activation of satellite cells and release of myotrophic factors (10–12). In addition, NO stimulates vasodilation, and thus increases the supply of oxygen and glucose uptake, and it triggers mitochondrial biogenesis, all of which contribute to preserve muscle from damage during contraction (13, 14, 25, 26). Indeed, amelioration of the dystrophic phenotype had been observed in neuronal NO-synthase transgenic mice (15). These studies stimulated investigations about the therapeutic potential of treatments based on administration of l-arginine, a metabolic precursor of NO, or molsidomine, a NO donor, to increase NO release (26, 27). Although some amelioration of muscle morphology was observed, and in one study creatine kinase levels were reduced (26), those treatments did not yield recovery of muscle function, and they did not improve animal motility tests. Moreover, these experiments were short-term investigations. We found no amelioration of morphofunctional parameters in α-SG-null mice after a 6-month treatment with ISDN.
The detectable and persistent recovery of muscle function observed in our study is most likely because HCT 1026 combines the multiple beneficial effects of NO outlined above with the potent antiinflammatory action of flurbiprofen, yielding a drug endowed with new properties. We found that important mechanisms concur to determine the excellent therapeutic potential of HCT 1026. The drug inhibited significantly inflammation in the dystrophic muscle by reducing the generation of proinflammatory cytokines and reducing infiltration of proinflammatory cells. In addition, HCT 1026 had a specific action in preserving satellite cell number and function by protecting them from the proapoptotic environment of the dystrophic muscle and by increasing their ability to differentiate. This action explains why in HCT 1026-treated animals, the regenerating ability of muscle, assessed as an increased number of centronucleated, reduced number of necrotic fibers and normal CSA, was preserved. The beneficial effect of the treatment we propose was tested against prednisolone, one of the widely used corticosteroids for treatment of Duchenne muscular dystrophy. We found that HCT 1026 was more effective than the steroid in ameliorating morphological, biochemical, and functional parameters and that these actions occurred in the absence of toxic side effects. In particular, we did not observe gastric damage, which in HCT 1026 is minimized by the NO-donating moiety (16). Moreover, the limited beneficial effects of the steroid decreased with time, becoming insignificant at later stages of the disease.
Previous studies in the mdx mouse showing that corticosteroids either alone or combined with other treatments exert beneficial effects were designed only as short-term therapeutic protocols (2–3 months on average) (25, 28). In humans, however, treatments have to be delivered long-term, a situation in which the side effects of corticosteroid administration still represent a serious problem (21). In contrast, HCT 1026 has a profile of safety that holds promise for its future use in patients affected by Duchenne muscular dystrophy. Because of the absence of corticosteroid-related toxicity, HCT 1026 appears a more promising therapy than the combination of l-arginine and deflazacort recently proposed (25). In addition, l-arginine and deflazacort were tested only up to 3 months, and l-arginine may fail to produce long-term NO release because of the dramatic decrease of NO synthase observed in dystrophic muscles (13). The second important advantage of the treatment with HCT 1026 is that it significantly enhanced the engrafting to muscle of intraarterially delivered mesoangioblasts, a type of stem cells shown to be effective in correcting the genetic defect of the muscle (8). This enhancement resulted in a significant increase in their therapeutic efficacy as shown by morphological and biochemical parameters and by in vivo motility assay on the treadmill. The effect was further enhanced by in vitro exposure of donor cells to SDF-1, a molecule able to increase homing of mesoangioblasts (24), implying that HCT 1026 may be used in combined therapeutic strategies to yield synergic beneficial effects. Of importance, the increased homing of mesoangioblasts to the muscle was accompanied by a concomitant reduction of their number in the relevant filter organs. This number is large, and it may cause damage to these organs (9). An increased efficiency of stem cell delivery will reduce either the number of cells needed for a single injection or the number of injections, leading to an optimization of cell therapy.
Compared with the current experimental therapies, the treatment we propose has clear advantages. Despite recent encouraging results, therapy with stem cells alone cannot reverse the preexisting pathological changes, and it still suffers from inefficient engraftment (9). This inefficiency may originate from a limited homing of these cells to muscle and a reduced ability to resist to the cytotoxic environment existing in the damaged muscle (23, 29). These problems are reduced by the combination of mesoangioblasts with HCT 1026.
Within the current scenario of preclinical and early clinical studies, HCT 1026 shows the distinct advantages of being economically affordable (unlike humanized anti-myostatin antibodies), nonimmunogenic (like adeno-associated vectors), suitable for all mutations (unlike oligonucleotides for exon skipping), and with a tested profile of safety. We conclude that the therapeutic strategy presented in this work represents a significant advance and sets the stage for immediate trials in patients.
Materials and Methods
Animal Treatments.
Mice were treated with HCT 1026 and prednisolone compounded in the chow following the European Community guidelines, and with the approval of the Institutional Ethical Committee. Mesoangioblast D16-GFP cells, pretreated with or without SDF-1, were delivery by intraarterial injection through the right femoral artery (8).
Determination of Creatine Kinase Serum Levels.
Serum creatine kinase concentrations were measured from mouse blood samples by using the indirect colorimetric assay (Roche Diagnostics, Basel, Switzerland), following standard procedures.
Functional Tests.
For a description of the functional tests, see SI Materials and Methods.
Mechanics of Isolated Muscles and Fibers.
Mechanical analysis of intact muscles was carried out as described in ref. 30. Tetanic force was normalized to the estimated CSA assuming cylindrical shape of the muscle and a density of 1.06 mg/mm3 (CSA corresponds to the wet weight divided by its length). Normalized tetanic force is expressed as kN/m2. Absolute (Po) and specific force (Po/CSA) of single muscle fibers were measured by using skinned preparations (31).
Histology Immunohistochemistry and Morphometry.
Muscles were dissected and frozen in liquid N2-cooled isopentane. Fiber membrane damage was evaluated Evans blue dye staining. Serial muscle sections were stained in H&E, by using the Azan–Mallory technique, or immunostained as described in ref. 8. Morphometrical analyses to evaluate distribution of fiber CSA were carried out on 300 fibers per muscle, on H&E-stained sections of tibialis anterior, by using Image 1.63 software (Scion Corporation, Frederick, MD).
Cytokine Analysis.
TGF-β and MIP-1α concentrations were determined on equal amounts of protein lysates from tibialis anterior muscles, by using the Quantikine ELISA kits (R&D Systems, Minneapolis, MN). MCP-1 concentration was assessed by flow cytometry by using the cytometric bead array (BD Bioscience, San Jose, CA).
RNAs and Protein Measurements.
Real-time quantitative PCR analysis was carried out on cDNA from tissue samples by using an Mx3000P real-time PCR detection system (Stratagene, La Jolla, CA). Each cDNA sample was amplified in duplicate by using the SYBR Green Supermix (Bio-Rad, Hercules, CA) for GFP (primers GFP: forward, AAGTTCATCTGCACCACCG; reverse, TCCTTGAAGAAGATGGTGCG). Protein samples from homogenized tissue were analyzed by Western blotting by using mAbs anti-α-SG (Novocastra, Newcastle upon Tyne, U.K.) and GAPDH (Biogenesis, Kingston, NH) (8).
Statistical Analyses.
Values were expressed as means ±SEM. Statistical significance was assessed by one-way ANOVA followed by the Student–Newman–Keul test. A probability of <5% (P < 0.05) was considered statistically significant.
Supplementary Material
Acknowledgments
We thank K. Campbell for the α-SG-null mice, M. Sampaolesi for advice, and C. Rinaldi for technical help. This work was supported by Telethon Grant GGP05007 (to E.C.), Association Française contre les Myopathies Grant 12048 (to E.C.), The European Community [Euro Stem Cell (to G.C.)], Associazione Parent Project Italia (to G.C.), the Italian Association of Cancer Research Grant 1016 (to E.C.), and the Ministry of Health [Ricerca Corrente (to E.C.)].
Abbreviations
- CSA
cross-sectional area
- EDL
extensor digitorum longus
- ISDN
isosorbide dinitrate
- MCP-1
monocyte chemoattractant protein 1
- MIP-1α
macrophage inflammatory protein 1α
- NO
nitric oxide
- SDF-1
stromal cell-derived factor 1
- SG
sarcoglycan.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0608277104/DC1.
References
- 1.Emery AE. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- 2.Manzur AY, Kuntzer T, Pike M, Swan A. Cochrane Database Syst Rev. 2004 doi: 10.1002/14651858.CD003725.pub2. CD003725. [DOI] [PubMed] [Google Scholar]
- 3.Skuk D, Vilquin JT, Tremblay JP. Curr Opin Neurol. 2002;15:563–569. doi: 10.1097/00019052-200210000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Minetti GC, Colussi C, Adami R, Serra C, Mozzetta C, Parente V, Fortuni S, Straino S, Sampaolesi M, Di Padova M, et al. Nat Med. 2006;12:1147–1150. doi: 10.1038/nm1479. [DOI] [PubMed] [Google Scholar]
- 5.Bogdanovich S, Krag TO, Barton ER, Morris LD, Whittemore LA, Ahima RS, Khurana TS. Nature. 2002;420:418–421. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- 6.Barton ER, Morris L, Musaro A, Rosenthal N, Sweeney HL. J Cell Biol. 2002;157:137–148. doi: 10.1083/jcb.200108071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu QL, Mann CJ, Lou F, Bou-Gharios G, Morris GE, Xue SA, Fletcher S, Partridge TA, Wilton SD. Nat Med. 2003;9:1009–1014. doi: 10.1038/nm897. [DOI] [PubMed] [Google Scholar]
- 8.Sampaolesi M, Torrente Y, Innocenzi A, Tonlorenzi R, D'Antona G, Pellegrino MA, Barresi R, Bresolin N, De Angelis MG, Campbell KP, et al. Science. 2003;301:487–492. doi: 10.1126/science.1082254. [DOI] [PubMed] [Google Scholar]
- 9.Cossu G, Sampaolesi M. Trends Mol Med. 2004;10:516–520. doi: 10.1016/j.molmed.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 10.Kaliman P, Canicio J, Testar X, Palacin M, Zorzano A. J Biol Chem. 1999;274:17437–17444. doi: 10.1074/jbc.274.25.17437. [DOI] [PubMed] [Google Scholar]
- 11.Pisconti A, Brunelli S, Di Padova M, De Palma C, Deponti D, Baesso S, Sartorelli V, Cossu G, Clementi E. J Cell Biol. 2006;172:233–244. doi: 10.1083/jcb.200507083. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Tatsumi R, Hattori A, Ikeuchi Y, Anderson JE, Allen RE. Mol Biol Cell. 2002;13:2909–2918. doi: 10.1091/mbc.E02-01-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stamler JS, Meissner G. Physiol Rev. 2001;81:209–237. doi: 10.1152/physrev.2001.81.1.209. [DOI] [PubMed] [Google Scholar]
- 14.Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, Pisconti A, Brunelli S, Cardile A, Francolini M, et al. Proc Natl Acad Sci USA. 2004;101:16507–16512. doi: 10.1073/pnas.0405432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wehling M, Spencer MJ, Tidball JG. J Cell Biol. 2001;155:123–131. doi: 10.1083/jcb.200105110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wallace JL, Reuter B, Cicala C, McKnight W, Grisham MB, Cirino G. Gastroenterology. 1994;107:173–179. doi: 10.1016/0016-5085(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 17.Scatena R. Curr Opin Investig Drugs. 2004;5:551–556. [PubMed] [Google Scholar]
- 18.Duclos F, Straub V, Moore SA, Venzke DP, Hrstka RF, Crosbie RH, Durbeej M, Lebakken CS, Ettinger AJ, van der Meulen J, et al. J Cell Biol. 1998;142:1461–1471. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Groen T, Kadish I. Brain Res Brain Res Rev. 2005;48:370–378. doi: 10.1016/j.brainresrev.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 20.Zacharowski P, Zacharowski K, Donnellan C, Johnston A, Vojnovic I, Forte P, Del Soldato P, Benjamin N, O'Byrne S. Clin Pharmacol Ther. 2004;76:350–358. doi: 10.1016/j.clpt.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 21.Dubowitz V. Lancet Neurol. 2005;4:264. doi: 10.1016/S1474-4422(05)70050-8. [DOI] [PubMed] [Google Scholar]
- 22.Jejurikar SS, Kuzon WM., Jr Apoptosis. 2003;8:573–578. doi: 10.1023/A:1026127307457. [DOI] [PubMed] [Google Scholar]
- 23.Engvall E, Wewer UM. FASEB J. 2003;17:1579–1584. doi: 10.1096/fj.02-1215rev. [DOI] [PubMed] [Google Scholar]
- 24.Galvez BG, Sampaolesi M, Brunelli S, Covarello D, Gavina M, Rossi B, Costantin G, Torrente Y, Cossu G. J Cell Biol. 2006;174:231–243. doi: 10.1083/jcb.200512085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Archer JD, Vargas CC, Anderson JE. FASEB J. 2006;20:738–740. doi: 10.1096/fj.05-4821fje. [DOI] [PubMed] [Google Scholar]
- 26.Voisin V, Sebrie C, Matecki S, Yu H, Gillet B, Ramonatxo M, Israel M, De la Porte S. Neurobiol Dis. 2005;20:123–130. doi: 10.1016/j.nbd.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 27.Barton ER, Morris L, Kawana M, Bish LT, Toursel T. Muscle Nerve. 2005;32:751–760. doi: 10.1002/mus.20425. [DOI] [PubMed] [Google Scholar]
- 28.Chakkalakal JV, Thompson J, Parks RJ, Jasmin BJ. FASEB J. 2005;19:880–891. doi: 10.1096/fj.04-1956rev. [DOI] [PubMed] [Google Scholar]
- 29.Tews DS, Goebel HH. J Neuropathol Exp Neurol. 1996;55:342–347. doi: 10.1097/00005072-199603000-00009. [DOI] [PubMed] [Google Scholar]
- 30.Rossi R, Bottinelli R, Sorrentino V, Reggiani C. Am J Physiol. 2001;281:C585–C594. doi: 10.1152/ajpcell.2001.281.2.C585. [DOI] [PubMed] [Google Scholar]
- 31.D'Antona G, Pellegrino MA, Adami R, Rossi R, Carlizzi CN, Canepari M, Saltin B, Bottinelli R. J Physiol (London) 2003;552:499–511. doi: 10.1113/jphysiol.2003.046276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}