

Abstract
The plasma concentration of soluble adhesion receptors is increased under pathological circumstances, but their function remains enigmatic. Soluble P-selectin (sP-sel) is shed from activated platelets and endothelial cells. Mice genetically engineered to express P-selectin without the cytoplasmic tail (ΔCT) constitutively show a 3- to 4-fold increase of sP-sel in plasma. We observed that the ΔCT mice formed fibrin very readily. In an ex vivo perfusion chamber, there was more fibrin deposited at the site of platelet thrombus formation than in wild type (WT), whereas no fibrin deposits were detected using P-selectin-deficient blood during the same interval. Similarly, in vivo, the hemorrhage produced by local Shwartzman reaction was smaller in the ΔCT mice than in WT. In contrast, we previously showed hemorrhage to be more prominent in P-selectin knock-out mice. Infusion of mouse P-sel-Ig chimera produced the same protective effect in WT mice as seen in the ΔCT mice, indicating that the effect was due to increased levels of sP-sel. Mice infused with P-sel-Ig showed significantly more fibrin deposited on the luminal face of the injured vessels than control mice. Plasma from ΔCT mice or mice infused with P-sel-Ig contained higher concentration of pro-coagulant microparticles and clotted one minute faster than WT. This pro-coagulant phenotype of ΔCT mice could be reversed by a 4-day treatment with PSGL-Ig, a P-selectin inhibitor. We propose that sP-sel should no longer be considered only as a marker of inflammation or platelet activation, but also as a direct inducer of pro-coagulant activity associated with vascular and thrombotic diseases.
P-selectin is a member of the selectin family localized in the membranes of α-granules of platelets and the Weibel-Palade bodies of endothelial cells (1). A soluble form of P-selectin can be found in the plasma as a circulating protein (2). In vivo, two main physiological roles are attributed to the integral membrane form of P-selectin. First, in inflammation, P-selectin is redistributed onto the surface of activated endothelial cells where it mediates the rolling of leukocytes (3). Second, in thrombosis, P-selectin expressed on activated platelets present in a thrombus supports the recruitment of leukocytes (4). Soluble P-selectin (sP-sel) of healthy individuals has been suggested to originate from the alternatively spliced form found in endothelial cells and platelets (5). Alternatively, elevated levels of sP-sel may reflect platelet activation (6) because P-selectin is proteolytically shed from the plasma membrane in vivo shortly after activation (7, 8). Therefore, plasma levels of sP-sel have been considered a useful tool to predict thrombotic consumptive platelet disorders (9–12), but they can also reflect endothelial cell activation (13, 14). Although the circulating form of P-selectin is potentially active because only the lectin and epidermal growth factor (EGF) domains are required to bind its receptor, P-selectin glycoprotein ligand-1 (PSGL-1) (15), the biological role of sP-sel and similarly that of other soluble adhesion receptors circulating in blood is not known (16).
Previous work in our laboratory has shown that P-selectin-deficient mice exhibit a slightly prolonged bleeding time, as well as an increased hemorrhagic response in a local Shwartzman reaction (17), suggesting that P-selectin could play a role in hemostasis. To further evaluate this possibility, we studied the hemostatic properties of mice genetically engineered to express P-selectin without the cytoplasmic domain (ΔCT mice) (18). In these mice, P-selectin is constitutively expressed on the surface of the endothelial cell and shed from the plasma membrane, leading to a 3- to 4-fold increase of sP-sel in plasma. We now report that the increased levels of sP-sel accelerate hemostasis in these mice. Similarly, wild-type (WT) animals infused with a P-selectin-Ig fusion protein (P-sel-Ig) chimera entered a pro-coagulant state.
Materials and Methods
Reagents.
Human IgG1 was from Sigma, and P-sel-Ig was from PharMingen. The P-sel-Ig is composed of N-terminal fragment of mouse P-selectin including the first two complement-binding domains fused to the Fc region (hinge, C1 and C2) of human IgG1 (19). PSGL-Ig (a generous gift from Genetics Institute, Cambridge, MA) is composed of the first 47 aa from the N-terminal end of mature human PSGL-1 fused to the Fc region of human IgG1 (20). The control protein (control-Ig, Genetics Institute) is a murine IgG2a produced in Chinese hamster ovary (CHO) cells. The protein has been mutated in the FcγRI and C′1q binding sites to inhibit Fc binding and complement directed cytolysis. The same sites were mutated in the human PSGL-Ig molecule.
Mice.
C57BL/6J/129Sv mice, WT and ΔCT, (18) were compared. C57BL/6J mice were used as recipient for the injection of P-sel-Ig, human IgG1, PSGL-Ig, and control-Ig. Animals were housed at the Center for Blood Research, Harvard Medical School. Experimental procedures were approved by the Animal Care and Use Committee of the Center for Blood Research.
Ex Vivo Perfusion Chamber.
Glass capillary tubes (0.56 mm inner diameter) were coated with 1 mg/ml type III fibrillar collagen (Sigma) as described (21). Mice were anesthetized with 2.5% tribromoethanol (0.15 ml/10 g). Non-anticoagulated blood was collected from the vena cava by using a 25G butterfly needle, and perfused through the collagen-coated perfusion chamber via silastic tubing. A flow rate of 220 μl/min was established for 2 min by a syringe pump mounted distal to the chamber, resulting in a 212 s−1 shear rate. Immediately after the blood perfusion, the thrombotic deposits formed on the collagen surface were rinsed for 20 s with PBS and fixed in ice-cold 2.5% cacodylate-buffered glutaraldehyde (pH 7.4) at the same shear rate. The perfusion chamber was then removed and fixed in a freshly prepared fixative for 24 h at 4°C. En face visualization of the thrombotic deposits was performed under light microscopy after epon embedding.
Determination of sP-sel and Fibrinogen Levels in Plasma.
Determination of the level of sP-sel was performed by using a modified sandwich ELISA procedure previously described (18). Calculations of the amount of sP-sel in the serum samples were made by comparing the specific sP-sel values with a standard curve of titrated P-sel-Ig. The plasma level of fibrinogen was measured according to Sigma Diagnostics Procedure No. 886.
Local Shwartzman Reaction.
Twelve- to fourteen-week-old age-matched WT and ΔCT male mice were primed on day 0 by a s.c. injection of Escherichia coli LPS 055:B5 (Difco) at 100 μg/mouse in 0.1 ml of sterile PBS. Twenty-four hours later (day 1), recombinant murine TNF-α (Genzyme) at 0.3 μg/mouse was injected at the same site (17). On day 2, the hemorrhagic lesions were examined and scored on a scale of 0 to 4 without knowledge of genotypes. Hematoxylin-eosin-stained paraffin sections were prepared from the lesion site, and the degree of inflammatory cell infiltration as well as hemorrhage were scored microscopically, on a scale of 0 to 4 (17).
Immunohistology.
Paraffin sections from the Shwartzman lesion site were de-paraffinized, sequentially blocked with avidin D solution and biotin blocking solution (Vector Laboratories) and stained with a rabbit anti-human fibrinogen (1:1000 dilution; Dako), which crossreacts with mouse fibrin/fibrinogen. Sections were then sequentially treated with a biotinylated goat anti-rabbit antibody (Zymed), and an ABC mix solution (Vector Laboratories). Development was done by treating the sections with an AEC substrate kit for horseradish peroxidase (Vector Laboratories). Sections were counterstained with hematoxylin.
Plasma Clotting Time Assay.
One milliliter of blood was drawn from the retro-orbital venous plexus by using plain microhematocrit capillary tubes (VWR Scientific) and collected into polypropylene tubes (Eppendorf; Marsh Biochemical Products, Rochester, NY) containing 10% final volume of acid-citrate-dextrose (ACD; 38 mM citric acid/75 mM trisodium citrate/100 mM dextrose). Platelet-poor plasma (PPP) was prepared by centrifugation at 1,500 × g for 25 min. PPP was centrifuged once more for 2 min at 15,000 × g to remove contaminating cells from the plasma. Plasma clotting was induced under stirring conditions (800 rpm) at 37°C in an aggregometer (Sienco, Inc., Wheat Ridge, CO) by adding a volume of prewarmed 20 mM CaCl2 solution to an equal volume of plasma in a siliconized tube. The time (in seconds) needed to clot was determined.
Flow Cytofluorometry of Plasma Microparticles.
PPP was prepared as above. Three hundred microliters was obtained per mouse. PPP from three mice was pooled, diluted 1:3 with a Hepes buffer [10 mmol/L Hepes/5 mmol/L KCL/1 mmol/L MgCl2/136 mmol/L NaCl (pH 7.4)], and centrifuged for 1.5 h at 100,000 × g. The supernatant was removed, and the pelleted microparticles were resuspended in 120 μl of a 10 mM Hepes, 136 mM NaCl (pH 7.4) buffer. Flow cytometric analysis was performed on a Becton-Dickinson FACSCalibur with CELLQUEST software (Becton-Dickinson). The light scatters and fluorescent channels were set at logarithmic gain (forward scatter was E00 with a threshold of 12 and sideward scatter was 300). To count the total microparticle population, 30-μl aliquots were incubated for 15 min in the dark with 0.25 μg/ml calcein AM (Molecular Probes). The total number of events was counted for a set interval of 10 s. Samples were stained for 20 min at room temperature with a rat monoclonal anti-murine Mac-1 (CD11b/CD18, 10 μg/ml; Boehringer Mannheim) and with a sheep anti-rabbit tissue factor IgG (5 μg/ml; American Diagnostica, Greenwich, CT), which recognizes mouse tissue factor. A phycoerythrin-conjugated goat anti-rat-IgG (1:200; Immunotech-Coulter, Marseille) and FITC-conjugated rabbit anti-sheep IgG (1:1000; Zymed) were second antibodies. Rat IgG (Sigma) and FITC-conjugated sheep IgG (Caltag Laboratories, South San Francisco, CA) were control antibodies.
For analysis of microparticles in plasma of ΔCT mice treated with PSGL-Ig, 200 μl of blood was collected by retro-orbital puncture on day 0. PPP was obtained, and 40 μl was diluted in 260 μl PBS and immediately analyzed for microparticle number by FACS. Mice were then infused i.v. (days 0 and 2) with 10 mg/kg PSGL-Ig or control-Ig. On day 4, 200 μl of blood was collected from the other eye, and the number of microparticles was determined.
Tissue Factor Activity in PPP.
PPP was prepared from pooled plasma of WT and ΔCT mice. A first centrifugation step at 12,000 × g was performed to remove contaminating cells. The supernatant was then diluted in 20 mM Hepes, 1 mM EDTA (pH 7.2) solution and ultracentrifuged at 200,000 × g for 1.5 h. The pelleted microparticles were resuspended (1/2 of the initial volume) as described above. Determination of tissue factor activity of the microparticles solution was evaluated by its ability to promote the activation of factor X (150 nM) by factor VIIa (5 nM) in the presence of 1 mM CaCl2. A chromogenic substrate of factor Xa, Spectrozyme fXa, was added (0.3 mM). The linear changes in absorbance at 405 nM were recorded by using a plate reader equipped with kinetics software (DYNEX Technologies, Chantilly, VA). The changes directly correlate with the concentration of factor Xa generated in the assay.
Results
Excessive Fibrin Deposition on Platelet Thrombi Formed in a Flow Chamber Perfused with ΔCT Blood.
We compared the thrombotic process in WT, P-selectin-deficient (P−/−), and ΔCT mice by perfusing non-anticoagulated blood through glass capillaries coated with fibrillar collagen type III. Platelet deposits were observed in all capillaries with no significant differences in size, but, surprisingly, we observed striking differences in the amount of deposited fibrin (Table 1, Fig. 1). Perfusion of WT blood showed microscopically visible depositions of short fibrin tails in only 4/11 cases. We did not observe any fibrin tails when blood from P−/− mice was perfused under identical conditions. On the other hand, long fibrin tails distal to the thrombi were found in eight of nine chambers perfused with blood from ΔCT mice. It should be noted that, when fibrin was detected, it always originated from the adhering platelets (Fig. 1), confirming that the collagen surface is not pro-coagulant by itself (22). Because the activated platelets from WT and ΔCT mice have similar P-selectin expression on the plasma membrane (18), we hypothesized that the difference in fibrin formation at the platelet thrombi most likely reflected a function of sP-sel in plasma. As described previously (18), plasma of ΔCT mice contains elevated levels of a 100-kDa P-selectin fragment. We determined by comparison to known concentrations of P-sel-Ig that the concentration of sP-sel is approximately 1 μg/ml in ΔCT mice and 0.3 μg/ml in WT mice. We also checked that there were no significant differences in fibrinogen levels between WT and ΔCT mice (367 ± 24 and 344 ± 14 mg/dl, respectively, for each group, n = 13).
Table 1.
Fibrin deposition in an ex vivo perfusion chamber
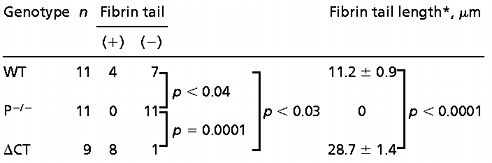 |
Differences between groups presenting fibrin or not were evaluated using the Log-rank test.
Values were expressed as the mean ± SEM of the length of the fibrin tail when it was detected. Student's t test was used to analyze differences in the length of the fibrin tail between groups.
Figure 1.

Platelet and fibrin deposition in an ex vivo flow chamber. Representative photomicrographs are shown of collagen-coated capillaries exposed for 2 min to flow of non-anticoagulated blood from WT, P−/−, and ΔCT mice at shear rate of 212 s−1. White arrowheads point to platelet-rich thrombi formed on the collagen surface. Note the presence of long fibrin tails distal to the thrombi in the ΔCT perfusion chamber (black arrows). After a longer perfusion, some fibrin could be detected also with the P−/− blood. Blood flow was from left to right. Bar = 20 μm.
sP-sel Reduces Hemorrhage and Affects Fibrin Deposition in a Local Shwartzman Reaction.
The local Shwartzman reaction is a hemorrhagic and necrotic lesion that can be induced by endotoxin and cytokines and is attributed to a multitude of interactions between platelets, neutrophils, and endothelium (23). P−/− mice display increased hemorrhage in this model (17). To evaluate a potential role for sP-sel in hemostasis, local Shwartzman reaction was studied in ΔCT mice, as well as WT mice of similar genetic background. Macroscopic evaluation of the injection sites revealed that, in contrast with P−/− mice, the average size of the hemorrhagic lesion in ΔCT mice was reduced to about 50% of WT (Table 2). This reduction in hemorrhage was also observed by microscopic examination of paraffin sections (Table 2). To determine whether sP-sel was the cause of this protective effect, we studied the local Shwartzman reaction in C57/BL6J WT mice injected with 1.2 μg/g body weight of either P-sel-Ig or control IgG1 1 h before the challenge dose of tumor necrosis factor-α (TNFα) was administered. Infusion of P-sel-Ig significantly reduced the size of hemorrhagic lesions measured both macroscopically and evaluated microscopically (Table 2). Immunohistological staining for fibrin at the lesion site revealed that the P-sel-Ig-treated mice had more vessels in the lesion with fibrin deposited on the luminal surface of the vessel wall than did IgG1-infused mice (Fig. 2 A and B). In animals treated with IgG1, the fibrin was preferentially deposited outside the injured vessel (Fig. 2 A and B). This suggested that the reduction in the hemorrhage was probably due to an increased or more rapid deposition of fibrin on the damaged vessel. The degree of inflammatory cell infiltration was the same in WT and ΔCT animals. A slight reduction in infiltration was observed in the P-sel-Ig-treated group compared with the IgG1-treated group (Table 2). This reduction might be explained by the occupancy of PSGL-1 receptors by the injected P-sel-Ig chimera, which could affect the leukocyte rolling. The infused P-sel-Ig concentration in plasma could be higher than the level of sP-sel found in the plasma of ΔCT mice.
Table 2.
Severity grading of the local Shwartzman reaction
| WT ΔCT | WT + IgG1 WT + P-sel-Ig | |||
|---|---|---|---|---|
| Macroscopic grading of lesion size | 3.1 ± 0.2 1.5 ± 0.3 | 3 ± 0.2 1.1 ± 0.3 | ||
| P = 0.001 | P < 0.0001 | |||
| Microscopic grading | ||||
| hemorrhage | 3.5 ± 0.2 2.1 ± 0.3 | 2.5 ± 0.3 0.8 ± 0.2 | ||
| P = 0.003 | P < 0.0001 | |||
| Infiltrate | 2.6 ± 0.3 2.6 ± 0.3 | 2.7 ± 0.1 2 ± 0.1 | ||
| P = 0.4 | P = 0.0002 |
Macroscopic or microscopic grading of the lesion was on a scale of 0 to 4 and was conducted without knowledge of the mouse genotype or the infused substance. Values are expressed as the mean ± SEM. n = 7–10; P values were calculated using Mann–Whitney test.
Figure 2.

Effect of sP-sel on fibrin distribution at the Shwartzman reaction lesion site. (A) Micrographs showing fibrin deposition (F) in paraffin section from the Shwartzman lesion site. Tissue sections from WT mice treated with IgG1 or P-sel-Ig were immunostained with antibodies to fibrinogen. Note the presence of hemorrhage (H) in the section from the IgG1-treated animals, and a diffuse staining for fibrin inside and outside the vessel. A strong fibrin staining (F) is found on the luminal face of the vessel wall in the P-sel-Ig-treated mice, without detectable fibrin deposition in the surrounding tissue. White arrowheads point to the vessel wall. Bar = 40 μm. (B) Evaluation of fibrin localization relative to the vessel wall of all blood vessels observed in paraffin sections at the lesion site. “Leakage” annotates blood vessels that presented fibrin staining outside the vessel wall. Blood vessels that had fibrin staining only at the luminal face of the vessel wall were classified as “luminal ring.” The frequency of vessels showing leakage was 3-fold higher in WT mice treated with IgG1 than with P-sel-Ig (*, P < 0.0001). In contrast, the luminal ring was more common in the P-sel-Ig-treated group than in the IgG1 controls (†, P < 0.005). The counting was performed on samples from 7 mice injected with P-sel-Ig and from 10 mice injected with control IgG1. A minimum of 100 vessels were observed per lesion site.
Shorter Plasma Clotting Time in ΔCT Mice and in Mice Infused with P-sel-Ig.
To evaluate whether the pro-coagulant activity seen in ΔCT mice or mice infused with P-sel-Ig is intrinsic to plasma of these mice, we have measured the plasma clotting time. Plasma clotting time of ΔCT mice (214 ± 7 s) was significantly shorter than that of WT mice (276 ± 8 s) (Fig. 3A). Adding P-sel-Ig to the plasma of WT mice just before the clotting assay did not change the clotting time (not shown), showing that sP-sel does not directly affect coagulation. On the other hand, injection of P-sel-Ig into WT mice 22 h before collecting the blood samples reduced the clotting time by 1 min, i.e., to 218 ± 14 s, a time similar to ΔCT plasma (Fig. 3B). This time was also significantly shorter than that obtained with the plasma from the IgG1-treated mice (369 ± 21 s). Our results indicate that the presence of high levels of sP-sel in mice increases the coagulability of their plasma. Interestingly, when plasmas of WT and ΔCT mice were ultracentrifuged at 200,000 × g for 1 h, clotting times of both supernatants were dramatically increased and were no longer different (ΔCT, 1064 ± 79 s, n = 4; WT, 1033 ± 141 s, n = 3; P = 0.84). This indicated that the pro-coagulant activity found in ΔCT mice was probably associated with the microparticles that were removed by ultracentrifugation.
Figure 3.

sP-sel and plasma clotting time. Plasma were incubated at 37°C with an equal volume of a 20 mM CaCl2 solution and stirred at 800 rpm in an aggregometer cuvette to measure the clotting time. Increased levels of sP-sel found in plasma of either ΔCT mice (A) or WT mice treated with 1.2 μg/g body weight P-sel-Ig (B) significantly shortened the plasma clotting time.
Elevated Levels of sP-sel Induce an Increase in Pro-Coagulant Microparticles.
To investigate further the possibility that the observed pro-coagulant activity is associated with microparticles, we measured the number of microparticles present in the plasma of WT and ΔCT mice by FACS analysis (Table 3). Plasma from ΔCT mice presented a 2-fold increase in the total number of microparticles compared with WT. As observed in the plasma clotting time assay, infusion of P-sel-Ig in WT mice induced a phenotype similar to that of the ΔCT mice, increasing the number of microparticles by 4-fold compared with the IgG1-treated mice (Table 3).
Table 3.
Number of microparticles in plasma
| Mouse | n | Mean | |
|---|---|---|---|
| WT | 4 | 5,910 ± 597 | |
| P < 0.004 | |||
| ΔCT | 4 | 11,288 ± 965 | |
| WT + IgG1 | 8 | 3,742 ± 1,233 | |
| P < 0.003 | |||
| WT + P-sel-Ig | 11 | 14,556 ± 2,455 |
Microparticles were collected by ultracentrifugation from PPP, and quantified by FACS analysis. High levels of sP-sel found in ΔCT mice or WT mice perfused with P-sel-Ig caused a significant increase in the number of microparticles when compared with respective controls.
We identified a population of Mac-1 (αMβ2 integrin) positive microparticles in WT mice, which showed their leukocyte origin (Fig. 4). This population was increased in ΔCT mice, and, interestingly, 13 + 3.5% of all microparticles (n = 3) stained for both Mac-1 and tissue factor (TF) (Fig. 4).
Figure 4.

Detection of Mac-1 and TF antigens at the surface of microparticles from WT and ΔCT mice by flow cytometry. Labeling of microparticles was performed with a rat monoclonal anti-murine macrophage Mac-1 antibody revealed by a phycoerythrin-conjugated goat anti-rat IgG, and a sheep anti-rabbit TF IgG revealed by a FITC-conjugated rabbit anti-sheep IgG. Control staining of microparticles from ΔCT mice was performed by using rat IgG and FITC-conjugated sheep IgG. A prominent population of doubly labeled microparticles is seen in samples from ΔCT mice. Figures are representative of three different experiments. TF-induced Xa activity was determined on the corresponding fraction of microparticles. The activity in samples from WT mice was 14.7 + 1.3 mOD/min, n = 5, and was significantly increased in ΔCT samples, 28.9 + 4.1 mOD/min, n = 5, P = 0.011.
PSGL-Ig Reduces the Pro-Coagulant Activity of ΔCT Mice.
We further evaluated whether infusion of a soluble form of the P-selectin receptor PSGL-1 (PSGL-Ig) would prevent the generation of the pro-coagulant state in ΔCT mice. We found that a 4-day treatment with PSGL-Ig significantly reduced the amount of microparticles and increased the plasma clotting time of ΔCT mice. Control-Ig had no such effect (Fig. 5 A and B). The clotting time of the PSGL-Ig-treated ΔCT was slightly longer than that of WT mice of similar age, indicating that the excess of microparticles observed in ΔCT mice was indeed due to sP-sel action.
Figure 5.

PSGL-Ig infusion decreases the number of microparticles and prolongs the clotting time in plasma of ΔCT mice. (A) Number of microparticles present in 40 μl of ΔCT plasma, before (open bars) and after (filled bars) two infusions of PSGL-Ig and control-Ig as described in Materials and Methods. *, P < 0.05, determined by paired t test. (B) Clotting time of plasma obtained at the end of the experiment was significantly longer in mice treated with PSGL-Ig (filled bar) than in control-Ig-treated group (striated bar), P < 0.05.
Discussion
The presence of soluble adhesion receptors in plasma has been widely reported, and it is often increased in pathological or stress conditions (24, 25). The physiological importance of these soluble receptors is not known. Until now, only speculations existed about the function of sP-sel. It was proposed that it may have an anti-inflammatory function by preventing leukocyte-vessel wall interactions because sP-sel can bind PSGL-1 on leukocytes (26, 27). This hypothesis is also supported by the fact that sP-sel binding to PSGL-1 activates leukocytes (28), and activated leukocytes may have a reduced ability to adhere to endothelium in vivo (29). It was also proposed that sP-sel could limit the extent of thrombosis by preventing additional leukocyte recruitment. On the other hand, sP-sel was shown to induce tissue factor expression on monocytes in vitro (30), indicating that it could have such an activity in vivo. Interestingly, infusion of anti-P-selectin antibodies in vivo, reduced fibrin deposition on dacron graft implanted within an arteriovenous shunt in baboons (4).
In this study, we found that sP-sel constitutes an endogenous activator of the coagulation process through the generation of circulating microparticles in plasma. Low levels of microparticles derived from platelets, leukocytes, and endothelial cells have been found in plasma of healthy individuals (31–34), and elevated numbers of microparticles circulate in the blood of patients with acute coronary syndromes and meningitis (35, 36). There are several explanations for a role of microparticles in modulating the coagulation process in vivo. First, microparticles carry pro-coagulant activity notably through the expression of prothrombinase activity on their membrane (37). Second, monocyte-derived microparticles were shown to activate endothelial cells in vitro, leading to expression of TF (32). In our study, we have found that elevated amount of sP-sel in ΔCT mice enhanced the generation of leukocyte-derived microparticles (Mac-1+), some of which also expressed TF (Fig. 4). Functional TF activity in microparticles in whole blood from normal individuals has been recently reported (38, 33). In certain clinical conditions, such as meningococcal sepsis, the amount of pro-coagulant microparticles originating from either platelets or granulocytes is increased 10- to 100-fold (36). In ΔCT mice, only 13% of microparticles expressed tissue factor, which may explain why these mice do not show symptoms of disseminated intravascular coagulation [the differential cell count (18) and the fibrinogen level were normal].
In the perfusion chamber model, it appeared that the pro-coagulant microparticles were preferentially recruited to thrombi, probably via a Mac-1/fibrinogen interaction (39), or via a CD15/P-selectin interaction (40). An increased concentration of leukocyte-derived microparticles, especially when bound to platelets, would further accelerate the coagulation process on activated platelets (41, 42), via the TF/FVIIa pathway, as reported by Giesen and colleagues (33). A second line of evidence for an increased pro-coagulant activity at the site of injury comes from the local Shwartzman reaction model. An intense staining for fibrin/fibrinogen was found frequently on the luminal surface of the vessels in the lesions of WT mice infused with P-sel-Ig but not in control animals. This may be explained again by the recruitment of microparticles to activated platelets as described above.
Thus, we have found a role for sP-sel in plasma in promoting coagulation that is different from its usual role as surface receptor involved in leukocyte recruitment and transmigration (43). High levels of sP-sel observed in thrombotic consumptive platelet disorders such as disseminated intravascular coagulation and thrombotic thrombocytopenic purpura could be in part responsible for the generalized hypercoagulable state associated with these diseases (9). In addition, sP-sel could contribute to the formation of fibrin tails, which are often associated with thrombi formed under high shear rates (44). Moreover, elevated levels of sP-sel might be involved in the recurrent thrombotic process of angina (45) and in restenosis (46). Our findings support and provide an explanation for the promising results of antithrombotic therapies directed against P-selectin in various thrombotic models such as stroke (47), or deep vein thrombosis (48). A reduction in numbers of circulating pro-coagulant microparticles by PSGL-Ig could explain its efficacy in accelerating thrombolysis (20) and reducing venous thrombosis in vivo (49). On the other hand, sP-sel could be also considered as an adjunctive treatment for patients with congenital bleeding disorders such as hemophilia, for example, to enhance hemostasis before a surgical procedure. We would like to propose that sP-sel in plasma should no longer be viewed as a simple marker of platelet or endothelial activation, but rather as a direct inducer of a pro-coagulant state. It is possible that other adhesion receptors may have new yet unsuspected functions when present in their soluble form.
Acknowledgments
We thank Drs. Robert Schaub, Vincent Ling, Richard Zollner, and Jennifer Thomas for providing the PSGL-Ig and the control-Ig; Dr. Yale Nemerson for helpful advice; Maria Economopoulos for mouse husbandry; and Lesley Cowan for help with the preparation of the manuscript. This work was supported by National Institutes of Health Grants PO1 HL56949 and RO1 HL53756 (to D.D.W.).
Abbreviations
- sP-sel
soluble P-selectin
- P-sel-Ig
P-selectin-Ig fusion protein
- TF
tissue factor
- WT
wild-type
- P−/−
P-selectin-deficient
- ΔCT
mice that express P-selectin lacking the cytoplasmic domain
- PPP
platelet-poor plasma
- PSGL
P-selectin glycoprotein ligand
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.250475997.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.250475997
References
- 1.Wagner D D. Thromb Haemost. 1993;70:105–110. [PubMed] [Google Scholar]
- 2.Dunlop L C, Skinner M P, Bendall L J, Favaloro E J, Castaldi P A, Gorman J J, Gamble J R, Vadas M A, Berndt M C. J Exp Med. 1992;175:1147–1150. doi: 10.1084/jem.175.4.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mayadas T N, Johnson R C, Rayburn H, Hynes R O, Wagner D D. Cell. 1993;74:541–554. doi: 10.1016/0092-8674(93)80055-j. [DOI] [PubMed] [Google Scholar]
- 4.Palabrica T, Lobb R, Furie B C, Aronovitz M, Benjamin C, Hsu Y M, Sajer S A, Furie B. Nature (London) 1992;359:848–851. doi: 10.1038/359848a0. [DOI] [PubMed] [Google Scholar]
- 5.Johnston G I, Bliss G A, Newman P J, McEver R P. J Biol Chem. 1990;265:21381–21385. [PubMed] [Google Scholar]
- 6.Fijnheer R, Frijns C J, Korteweg J, Rommes H, Peters J H, Sixma J J, Nieuwenhuis H K. Thromb Haemost. 1997;77:1081–1085. [PubMed] [Google Scholar]
- 7.Berger G, Hartwell D W, Wagner D D. Blood. 1998;92:4446–4452. [PubMed] [Google Scholar]
- 8.Michelson A D, Barnard M R, Hechtman H B, MacGregor H, Connolly R J, Loscalzo J, Valeri C R. Proc Natl Acad Sci USA. 1996;93:11877–11882. doi: 10.1073/pnas.93.21.11877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chong B H, Murray B, Berndt M C, Dunlop L C, Brighton T, Chesterman C N. Blood. 1994;83:1535–1541. [PubMed] [Google Scholar]
- 10.Wu G, Li F, Li P, Ruan C. Haemostasis. 1993;23:121–128. doi: 10.1159/000216864. [DOI] [PubMed] [Google Scholar]
- 11.Blann A D, Dobrotova M, Kubisz P, McCollum C N. Thromb Haemost. 1995;74:626–630. [PubMed] [Google Scholar]
- 12.Smith A, Quarmby J W, Collins M, Lockhart S M, Burnand K G. Thromb Haemost. 1999;82:1593–1599. [PubMed] [Google Scholar]
- 13.Frijns C J, Kappelle L J, van Gijn J, Nieuwenhuis H K, Sixma J J, Fijnheer R. Stroke. 1997;11:2214–2218. doi: 10.1161/01.str.28.11.2214. [DOI] [PubMed] [Google Scholar]
- 14.Verhaar M C, Beutler J J, Gaillard C A, Koomans H A, Fijnheer R, Rabelink TJ. J Hypertens. 1998;16:45–50. doi: 10.1097/00004872-199816010-00008. [DOI] [PubMed] [Google Scholar]
- 15.Mehta P, Patel K D, Laue T M, Erickson H P, McEver R P. Blood. 1997;90:2381–2389. [PubMed] [Google Scholar]
- 16.Blann A D, Lip G Y. J Clin Endocrinol Metab. 2000;85:1745–1747. doi: 10.1210/jcem.85.5.6594. [DOI] [PubMed] [Google Scholar]
- 17.Subramaniam M, Frenette P S, Saffaripour S, Johnson R C, Hynes R O, Wagner D D. Blood. 1996;87:1238–1242. [PubMed] [Google Scholar]
- 18.Hartwell D W, Mayadas T N, Berger G, Frenette P S, Rayburn H, Hynes R O, Wagner D D. J Cell Biol. 1998;143:1129–1141. doi: 10.1083/jcb.143.4.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hahne M, Jager U, Isenmann S, Hallmann R, Vestweber D. J Cell Biol. 1993;121:655–664. doi: 10.1083/jcb.121.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar A, Villani M P, Patel U K, Keith J C, Jr, Schaub R G. Circulation. 1999;99:1363–1369. doi: 10.1161/01.cir.99.10.1363. [DOI] [PubMed] [Google Scholar]
- 21.André P, Arbeille B, Drouet V, Hainaud P, Bal dit Sollier C, Caen J P, Drouet L O. Arterioscler Thromb Vasc Biol. 1996;16:56–63. doi: 10.1161/01.atv.16.1.56. [DOI] [PubMed] [Google Scholar]
- 22.Sakariassen K S, Joss R, Muggli R, Kuhn H, Tschopp T B, Sage H, Baumgartner H R. Arteriosclerosis. 1990;10:276–284. doi: 10.1161/01.atv.10.2.276. [DOI] [PubMed] [Google Scholar]
- 23.Brozna J P. Semin Thromb Haemost. 1990;16:326–332. doi: 10.1055/s-2007-1002685. [DOI] [PubMed] [Google Scholar]
- 24.Gearing A J, Newman W. Immunol Today. 1993;14:506–512. doi: 10.1016/0167-5699(93)90267-O. [DOI] [PubMed] [Google Scholar]
- 25.Blann A D, McCollum C N, Steiner M, Jayson M I. Immunol Today. 1995;16:251–252. doi: 10.1016/0167-5699(95)80169-3. [DOI] [PubMed] [Google Scholar]
- 26.Gamble J R, Skinner M P, Berndt M C, Vadas M A. Science. 1990;249:414–417. doi: 10.1126/science.1696029. [DOI] [PubMed] [Google Scholar]
- 27.Wong C S, Gamble J R, Skinner M P, Lucas C M, Berndt M C, Vadas M A. Proc Natl Acad Sci USA. 1991;88:2397–2401. doi: 10.1073/pnas.88.6.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evangelista V, Manarini S, Sideri R, Rotondo S, Martelli N, Piccoli A, Totani L, Piccardoni P, Vestweber D, de Gaetano G, Cerletti C. Blood. 1999;93:876–885. [PubMed] [Google Scholar]
- 29.Von Andrian U H, Hansell P, Chambers J D, Berger E M, Torres Filho I, Butcher E C, Arfors K E. Am J Physiol. 1992;263:H1034–H1044. doi: 10.1152/ajpheart.1992.263.4.H1034. [DOI] [PubMed] [Google Scholar]
- 30.Celi A, Pellegrini G, Lorenzet R, De Blasi A, Ready N, Furie B C, Furie B. Proc Natl Acad Sci USA. 1994;91:8767–8771. doi: 10.1073/pnas.91.19.8767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abrams C S, Ellison N, Budzynski A Z, Shattil S J. Blood. 1990;75:128–138. [PubMed] [Google Scholar]
- 32.Mesri M, Altieri D C. J Biol Chem. 1999;274:23111–23118. doi: 10.1074/jbc.274.33.23111. [DOI] [PubMed] [Google Scholar]
- 33.Giesen P L, Rauch U, Bohrmann B, Kling D, Roque M, Fallon J T, Badimon J J, Himber J, Riederer M A, Nemerson Y. Proc Natl Acad Sci USA. 1999;96:2311–2315. doi: 10.1073/pnas.96.5.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Combes V, Simon A C, Grau G E, Arnoux D, Camoin L, Sabatier F, Mutin M, Sanmarco M, Sampol J, Dignat-George F. J Clin Invest. 1999;104:93–102. doi: 10.1172/JCI4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mallat Z, Benamer H, Hugel B, Benessiano J, Steg P G, Freyssinet J M, Tedgui A. Circulation. 2000;101:841–843. doi: 10.1161/01.cir.101.8.841. [DOI] [PubMed] [Google Scholar]
- 36.Nieuwland R, Berckmans R J, McGregor S, Boing A N, Romijn F P, Westendorp R G, Hack C E, Sturk A. Blood. 2000;95:930–935. [PubMed] [Google Scholar]
- 37.Satta N, Toti F, Feugeas O, Bohbot A, Dachary-Prigent J, Eschwege V, Hedman H, Freyssinet J M. J Immunol. 1994;153:3245–3255. [PubMed] [Google Scholar]
- 38.Key N S, Slungaard A, Dandelet L, Nelson S C, Moertel C, Styles L A, Kuypers F A, Bach R R. Blood. 1998;91:4216–4223. [PubMed] [Google Scholar]
- 39.Altieri D C, Bader R, Mannucci P M, Edgington T S. J Cell Biol. 1988;107:1893–1900. doi: 10.1083/jcb.107.5.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rauch U, Bonderman D, Bohrmann B, Badimon J J, Himber J, Riederer M A, Nemerson Y. Blood. 2000;96:170–175. [PubMed] [Google Scholar]
- 41.Swords N A, Tracy P B, Mann K G. Arterioscler Thromb. 1993;13:1613–1622. doi: 10.1161/01.atv.13.11.1613. [DOI] [PubMed] [Google Scholar]
- 42.Kirchhofer D, Tschopp T B, Steiner B, Baumgartner H R. Blood. 1995;86:3815–3822. [PubMed] [Google Scholar]
- 43.Vestweber D, Blanks J E. Physiol Rev. 1999;79:181–213. doi: 10.1152/physrev.1999.79.1.181. [DOI] [PubMed] [Google Scholar]
- 44.Barstad R M, Kierulf P, Sakariassen K S. Thromb Haemost. 1996;75:685–692. [PubMed] [Google Scholar]
- 45.Ikeda H, Takajo Y, Ichiki K, Ueno T, Maki S, Noda T, Sugi K, Imaizumi T. Circulation. 1995;92:1693–1696. doi: 10.1161/01.cir.92.7.1693. [DOI] [PubMed] [Google Scholar]
- 46.Tsakiris D A, Tschopl M, Jager K, Haefeli W E, Wolf F, Marbet G A. Atherosclerosis. 1999;142:193–200. doi: 10.1016/s0021-9150(98)00175-0. [DOI] [PubMed] [Google Scholar]
- 47.Connolly E S, Jr, Winfree C J, Prestigiacomo C J, Kim S C, Choudhri T F, Hoh B L, Naka Y, Solomon R A, Pinsky D J. Circ Res. 1997;81:304–310. doi: 10.1161/01.res.81.3.304. [DOI] [PubMed] [Google Scholar]
- 48.Downing L J, Wakefield T W, Strieter R M, Prince M R, Londy F J, Fowlkes J B, Hulin M S, Kadell A M, Wilke C A, Brown, et al. J Vasc Surg. 1997;25:816–828. doi: 10.1016/s0741-5214(97)70211-8. [DOI] [PubMed] [Google Scholar]
- 49.Wakefield T W, Strieter R M, Schaub R, Myers D D, Prince M R, Wrobleski S K, Londy F J, Kadell A M, Brown S L, Henke P K, Greenfield L J. J Vasc Surg. 2000;31:309–324. doi: 10.1016/s0741-5214(00)90162-9. [DOI] [PubMed] [Google Scholar]