

This paper reports a composite microfluidic system for performing protein crystallization trials in nanoliter aqueous droplets inside capillaries suitable for X-ray diffraction and the evaluation of the quality of the crystals directly by on-chip X-ray diffraction. Crystallization conditions can be screened with both microbatch and vapor-diffusion techniques by eliminating evaporation of solutions and by controlling diffusion of water between droplets.
Growing of high-quality crystals of proteins and other macromolecules plays an important role in structural biology. Crystallization conditions are usually identified by performing a large number of trials in which variable ratios of solutions of a protein, precipitants, and additives are pipetted together by hand (≈1 μL droplets) or with a robotic dispenser (≈100–10 nL droplets).[1] This process could be improved by a system that minimizes the amount of protein sample consumed, reduces labor, and is simple and economical enough to be implemented in individual laboratories. Micro-fluidics could serve as a platform for such a system because it allows sophisticated handling of small volumes (nL–pL) of reagents in a potentially simple, inexpensive format.[2] In addition, microfluidics may provide an opportunity to perform experiments that are difficult to do on larger scale—for example, Hansen et al. have crystallized proteins on a polydimethylsiloxane (PDMS) microfluidic device by free interface diffusion.[3] Previously, we screened crystallization conditions[4] on a PDMS microfluidic chip by using nanoliter droplets[5] formed in microfluidic channels.
In this work, we rely on the same droplet-based platform, but take three steps that advance this work beyond our previous report. 1) We implemented the microbatch technique, in which we completely eliminated problems of uncontrolled evaporation through PDMS. PDMS is water- and oil-permeable, and the crystallization of proteins in PDMS, at least in our hands, previously required careful control of humidity to limit evaporation. 2) We implemented a controlled vapor-diffusion technique, in which the rate of removal of water from the droplet containing the protein could be easily adjusted. 3) We developed a method for evaluating the quality of crystals by direct on-chip X-ray diffraction. This method eliminates the need for manual handling of crystals that was previously required for the extraction of crystals from the device and subsequent mounting. Manual handling was tedious and had the potential to damage the crystal.
We used composite PDMS/glass capillary microfluidic devices to eliminate the evaporation of solutions during crystallization trials (Figure 1a, b). Composite microfluidic devices are well known, especially in capillary electrophoresis.[6,7] Glass capillaries have been used to grow protein crystals by using the microbatch technique[8] and several diffusion techniques, such as equilibrium dialysis,[9] free interface diffusion,[10] and gel-acupuncture methods.[11,12] Crystallization of organic molecules has also been performed in glass capillaries.[13–15] We used the PDMS section of the device to generate droplets that contain protein, additives and precipitants in controlled ratios by using a procedure described previously.[4] Briefly, we flowed aqueous streams that of solutions of the protein, additives, and precipitants into a flowing stream of fluorinated oil, in which all the aqueous streams formed droplets surrounded and carried by the flow of oil (Figure 1a). We varied the composition of the droplets by varying the relative flow rates of the aqueous streams. The flow of oil transported droplets into a glass capillary attached to the outlet of the PDMS section. The flow was stopped when the capillary was filled with droplets of desired composition, and the capillary was disconnected, sealed with wax, incubated at 18°C, and monitored periodically. Crystals formed in the droplets (Figure 1c) under the conditions similar to those we found by using the previous system.[4] In contrast to the case in which droplets were incubated in the PDMS channels, droplets and crystals incubated in a sealed glass capillary were stable even in the absence of humidity control, and did not show signs of evaporation over six months. This long-term stability is crucial for protein crystallization on nanoliter scale, because the probability of a nucleation event decreases as the volume of the solution decreases.[16–18] For example, nucleation rate is proportional to droplet volume for homogeneous nucleation and to the surface area of the droplet for heterogeneous nucleation. We have observed that it takes a longer time to obtain crystals in nL droplets than μL droplets under microbatch conditions. The acceleration of nucleation while still using nanoliter volumes would be desirable for protein crystallization used in high-throughput structural determination.
Figure 1.
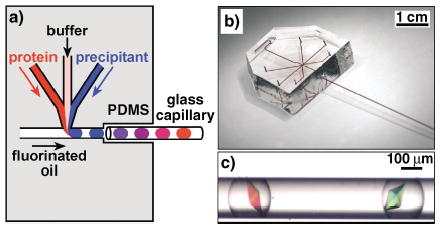
a) A schematic illustration of the method of forming droplets for protein crystallization trials in a PDMS/glass capillary composite microfluidic device. b) A photograph of the composite device. All inlets and channels were filled with [Fe(SCN)x](3−x)+ solution for clarity. c) A microphotograph of thaumatin crystals grown inside droplets in a capillary. Droplets were generated by the method illustrated in (a).
Vapor diffusion may be used to accelerate the nucleation of protein crystals. During protein crystallization by vapor diffusion, a droplet containing the crystallization mixture is placed in the vicinity of a reservoir that contains a high concentration of salt. Slow diffusion of water from the crystallization solution into the concentrated solution of salt is due to the difference in osmotic pressures of the two solutions. This diffusion leads to a decrease in volume of the crystallization solution, and therefore to a sufficient increase in the supersaturation of the crystallization solution for the nucleation of the protein crystal to occur.
To implement crystallization by vapor diffusion inside capillaries, we wished to generate droplets of the crystallization mixture alternating with droplets that contained a high concentration of salt. We hypothesized that if the oil separating the droplets were water-permeable, crystallization would occur as water diffused from droplets of low concentration to droplets of high concentration until osmotic pressures were equalized. We found that alternating droplets of two sets of aqueous solutions can be easily generated by flowing the two sets of aqueous solutions head-on into a flowing stream of water-immiscible oil (Figure 2a). In the experiment shown in Figure 2b two of the three aqueous streams from the upper set were labeled with a red dye, and two of the three aqueous streams from the lower set were labeled with a green dye. Flowing oil served as the barrier between the two sets of aqueous streams, thus preventing them from coming into contact (Figure 2b). We used fluorescent markers to confirm that there was no cross-contamination between the two sets of solutions.
Figure 2.
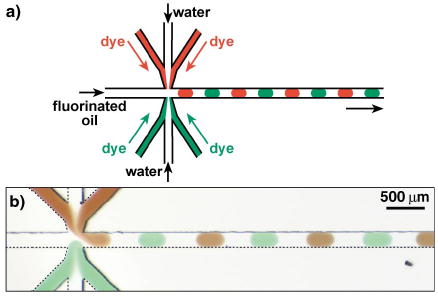
a) A schematic illustration and b) a microphotograph of the microfluidic method of forming alternating droplets in the microfluidic channel. Alternating droplets formed spontaneously when water and aqueous solutions of food dyes (red and green) were continuously flowed into a steady stream of fluorinated oil.
We used alternating droplets formed with 1.5M and 0.5M aqueous solutions of NaCl to measure the rate of transport of water through various oils by measuring the change in size of the droplets (Figure 3a). We found that perfluoroalkanes were impermeable to water, and over a period of several weeks we observed no transport of water between adjacent droplets separated by ≈100 μm of a perfluoroalkane. This result is consistent with our results in microbatch crystallization, in which we used perfluoroalkanes as the oils and did not observe any transport of water between adjacent droplets. We found that polytrifluoropropylmethylsiloxane homopolymer (FMS-121) was permeable to water. A similar oil has been used to absorb water from small droplets to drive the self-assembly of colloids.[19] To quantify the rate of transport of water through this oil in capillaries, we formed isolated pairs of droplets containing 1.5M and 0.5M aqueous solutions of NaCl. To aid visualization we colored 0.5M NaCl solution with 0.05M [Fe(SCN)x](3−x)+ in 0.3M KNO3. Each pair of droplets in a capillary was separated by ≈50 to ≈400 μm of oil. Capillaries were stored at 18°C and monitored. Initially, all droplets were ≈25 nL in volume, but the increase in volume of the droplets that contained 1.5M NaCl and the decrease in volume of the droplets that contained 0.5M NaCl became noticeable in a few hours (Figure 3b). The initial change of volume was linear with time, and linear changes of up to 30% were observed over several days (Figure 3b). The rate of transport decreased as osmotic pressure inside the droplets equalized. The observed rate of change in volume is similar to that used in traditional vapor diffusion methods, and can be tuned by controlling the distance between the droplets (Figure 3b). This distance can be controlled by changing the relative flow rates of the aqueous and fluorous phases.[20] The control of the evaporation rate of water is important for protein crystallization under vapor-diffusion conditions. An increase in the rate of evaporation of water results in shorter equilibration time, therefore the crystallization trials can be carried out more rapidly.[21] On the other hand, it has also been proposed that larger, fewer, and better quality crystals are more likely to be obtained by reducing the evaporation rate.[22] Both approaches could be appropriate depending on a specific protein, and both of them can be implemented in this microfluidic system.
Figure 3.
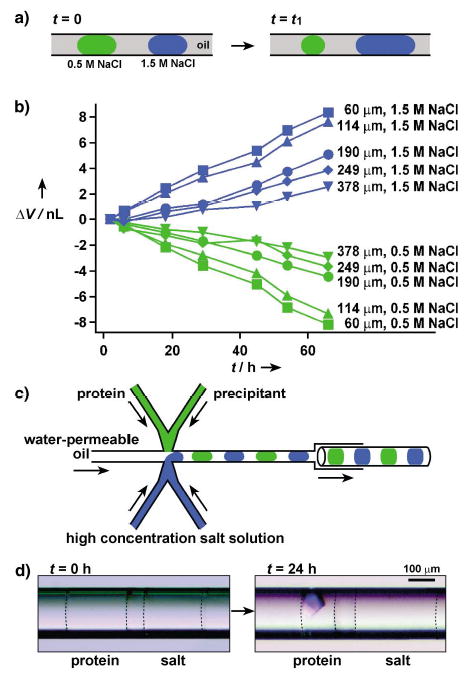
Protein crystallization by vapor diffusion in capillaries: a) A schematic illustration of the principle of vapor diffusion in a capillary. Water diffused through oil from droplets that contained a low concentration of salt to droplets that contained a high concentration of salt. b) A plot of change in the volume of droplets that were initially 25 nL (ΔV) as a function of time (t). Legends on the right side of each curve show the distance between the droplets. The concentrations of NaCl in the droplets are also indicated. c) A schematic illustration of the PDMS/glass composite device for setting up pairs of droplet for protein crystallization under vapor-diffusion conditions in capillaries. d) Two microphotographs that show a pair of droplets immediately after (left) and 24 h after (right) the setup. The droplet of the protein solution yielded a crystal after it had lost about 50% of its water content. Dashed lines were added to show the interface between the aqueous droplets and oil.
To use this system for the crystallization of proteins by vapor diffusion, one may envision flowing the crystallization solutions through one set of channels, and flowing solutions containing high concentrations of salts through the other set of channels (Figure 3c). For simplicity, Figure 3c shows only two channels for each set of solutions, but we have performed these experiments with up to four channels for each set; the device shown in Figure 1b has three channels for each set of solutions. To screen the crystallization conditions, all concentrations were varied by changing the flow rates of the corresponding aqueous streams.[4]
To demonstrate crystallization of a protein by vapor diffusion in capillaries, we generated a sequence of droplets that contained lysozyme and precipitants in the first set of droplets, and contained a high-concentration of salt in the second set of droplets (Figure 3c). After 24 hours of incubation, ≈50% of the water in the droplets of lysozyme solution had diffused into salt droplets, thus inducing crystallization (Figure 3d). More than half of the droplets of lysozyme solution yielded crystals after two days. To demonstrate that crystallization is due to the transport of water, a control experiment was performed. The capillary was filled with droplets of lysozyme solution, all at identical concentrations equal to those used in the vapor-diffusion experiment, so that no water transport between droplets could occur. None of the droplets yielded crystals after an incubation period of one month.
The evaluation of the quality of the crystals is an important part of the screening process. The ultimate test of crystal quality is X-ray diffraction. Crystals obtained from 10–20 nL droplets had dimensions of about 50–200 μm on each side and were large enough for synchrotron diffraction experiments. We have been able to extract crystals grown inside droplets in capillaries simply by cutting the capillary and gently flowing or wicking the liquid our together with the crystal. More importantly, we could obtain diffraction patterns from crystals grown in these thin-walled capillaries without manipulation of the crystals, by subjecting the capillaries with crystals directly to the synchrotron X-ray beam (Figure 4). We have observed scattering patterns from the glass and the mother liquor present around the crystal, but this scattering did not obscure the diffraction of crystals. Thaumatin crystals diffracted to ≈1.8 Å resolution (Figure 4, diffraction to 2.0 Å is shown). A tetragonal space group with correct unit-cell dimensions could be obtained from a single diffraction image. The diffraction image shown in Figure 4 was refined to unit-cell dimensions a = 58.45 Å, b = 58.45 Å, c = 149.02 Å and α = β = γ = 90° by using 567 unique reflections; and the literature values are a = 58.60 Å, b = 58.60 Å, c = 151.80 Å and α = β = γ = 90°.[23] Lysozyme crystals diffracted to ≈1.8 Å resolution. The unit-cell dimensions of the crystal derived from the diffraction were a = 78.99 Å, b = 78.99 Å, c = 38.40 Å and α = β = γ = 90°. The reported values are a = 79.10 Å, b = 79.10 Å, c = 37.90 Å and α = β = γ = 90°.[24]
Figure 4.
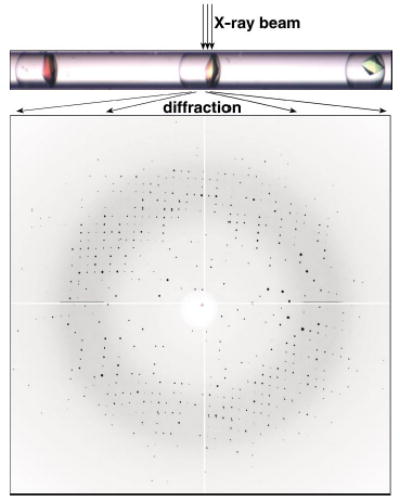
The capillary containing protein crystals (top) can be directly subjected to X-ray diffraction. A diffraction pattern (bottom) of thaumatin crystal from on-chip X-ray diffraction that shows the reflections on the edge of the image at a resolution of 2.0 Å.
Exposure of a crystal to synchrotron radiation at room temperature is known to damage the crystal. We observed deterioration of the diffraction patterns for these simple proteins after 20–50 exposures, each exposure lasting five seconds. From one crystal at room temperature, it would be difficult to obtain a complete set of data required for structural determination, especially for less-stable proteins. Most protein crystals should survive at least a single short exposure to the beam (or two exposures along orthogonal axes) required to obtain a single diffraction frame. As is, this technique would be especially valuable for evaluating and comparing the intrinsic quality of crystals obtained under different crystallization conditions, quality that has not deteriorated through handling and freezing. Freezing of crystals, either extracted from the capillary or directly inside the capillary, should increase their stability sufficiently for structural determination, and we are currently exploring both of these approaches by using a range of cryoprotectants.
In conclusion, we have demonstrated a versatile microfluidic system for screening protein-crystallization conditions in droplets inside capillaries. The system is compatible with both microbatch and vapor-diffusion methods, both of which have advantages and disadvantages.[25] The quality of crystals obtained in this system can be evaluated directly by X-ray diffraction. Several steps may be taken to improve this system further. We could not obtain capillaries longer than 10 cm, thus limiting us to ≈100 experiments per capillary. The cost of capillaries (≈$2 each) was a less significant limitation. A source of inexpensive long glass or plastic capillaries suitable for X-ray diffraction would make this system even more useful. Fluorocarbons used as carrier fluids in this system can dissolve large amounts of oxygen, and care must be taken to degas the fluids when working with air-sensitive proteins. Once the fluids are deoxygenated, they may be saturated with other gases. Because the capillaries are sealed after the experiment, this method maybe useful for long-term crystal growth of proteins[26,27] that require a controlled environment. An important application of the vapor-diffusion method is to concentrate very low volumes of solutions of proteins. This method may be useful for the crystallization of proteins that cannot be obtained at high concentrations, and may also find applications for concentrating solutions of other samples available in small quantities. Overall, we believe this microfluidic system will be useful for fundamental studies of crystallization phenomena, and will find applications for crystallizing targets ranging from small organic and inorganic molecules to large biological macromolecules.
Supplementary Material
Footnotes
This work was supported by the NIH (R01 EB001903), and by Beckman Young Investigator Program, and was performed at the MRSEC microfluidic facility funded by NSF. L.S.R. is supported by the Burroughs Wellcome Fund Cross-Disciplinary Training Program in Biophysical Dynamics. We thank Adam Lyon for preliminary experiments. We thank Phoebe Rice, Keith Moffatt, Andrzej Joachimiak, Chuan He, and Erica Duguid for valuable discussions and help with protein crystallography. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38. Use of the BioCARS Sector 14 was supported by the National Institutes of Health, National Center for Research Resources, under grant number RR07707.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Rupp B. Acc Chem Res. 2003;36:173. doi: 10.1021/ar020021t. [DOI] [PubMed] [Google Scholar]
- 2.von d Woerd M, Ferree D, Pusey M. J Struct Biol. 2003;142:180. doi: 10.1016/s1047-8477(03)00049-2. [DOI] [PubMed] [Google Scholar]
- 3.Hansen CL, Skordalakes E, Berger JM, Quake SR. Proc Natl Acad Sci USA. 2002;99:16531. doi: 10.1073/pnas.262485199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng B, Roach LS, Ismagilov RF. J Am Chem Soc. 2003;125:11170. doi: 10.1021/ja037166v. [DOI] [PubMed] [Google Scholar]
- 5.Song H, Tice JD, Ismagilov RF. Angew Chem. 2003:115–792. doi: 10.1002/anie.200390203. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2003;42:768. [Google Scholar]
- 6.Bings NH, Wang C, Skinner CD, Colyer CL, Thibault P, Harrison DJ. Anal Chem. 1999;71:3292. doi: 10.1021/ac981419z. [DOI] [PubMed] [Google Scholar]
- 7.Figeys D, Aebersold R. Anal Chem. 1998;70:3721. doi: 10.1021/ac980502j. [DOI] [PubMed] [Google Scholar]
- 8.Penkova A, Chayen N, Saridakis E, Nanev C. Acta Crystallogr Sect D. 2002;58:1606. doi: 10.1107/s0907444902014166. [DOI] [PubMed] [Google Scholar]
- 9.Weber BH, Goodkin PE. Arch Biochem Biophys. 1970;141:489. doi: 10.1016/0003-9861(70)90166-9. [DOI] [PubMed] [Google Scholar]
- 10.Salemne FR. Arch Biochem Biophys. 1972;151:533. doi: 10.1016/0003-9861(72)90530-9. [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Ruiz JM, Moreno A. Acta Crystallogr Sect D. 1994;50:484. doi: 10.1107/S0907444993014350. [DOI] [PubMed] [Google Scholar]
- 12.López-Jaramillo FJ, García-Ruiz JM, Otálora JAGaF. J Appl Crystallogr. 2001;34:365. [Google Scholar]
- 13.Hilden JL, Reyes CE, Kelm MJ, Tan JS, Stowell JG, Morris KR. Cryst Growth Des. 2003;3:921. [Google Scholar]
- 14.Yokoyama C, Tamura Y, Nishiyama Y. J Cryst Growth. 1998;191:827. [Google Scholar]
- 15.Ukita T, Ishikawa K, Takezoe H, Fukuda A, Murayama H, Muta K-i. J Cryst Growth. 1994;143:66. [Google Scholar]
- 16.Garcia-Ruiz JM. J Struct Biol. 2003;142:22. doi: 10.1016/s1047-8477(03)00052-2. [DOI] [PubMed] [Google Scholar]
- 17.Bodenstaff ER, Hoedemaeker FJ, Kuil ME, Vrind HPMd, Abrahams JP. Acta Crystallogr Sect D. 2002;58:1901. doi: 10.1107/s0907444902016608. [DOI] [PubMed] [Google Scholar]
- 18.Turnbull D. J Appl Phys. 1950;21:1022. [Google Scholar]
- 19.Yi GR, Thorsen T, Manoharan VN, Hwang MJ, Jeon SJ, Pine DJ, Quake SR, Yang SM. Adv Mater. 2003;15:1300. [Google Scholar]
- 20.Tice JD, Song H, Lyon AD, Ismagilov RF. Langmuir. 2003;19:9127. [Google Scholar]
- 21.Santarsiero BD, Yegian DT, Lee CC, Spraggon G, Gu J, Scheibe D, Uber DC, Cornell EW, Nordmeyer RA, Kolbe WF, Jin J, Jones AL, Jaklevic JM, Schultz PG, Stevens RC. J Appl Crystallogr. 2002;35:278. [Google Scholar]
- 22.Li G, Xiang Y, Zhang Y, Wang DC. J Appl Crystallogr. 2001;34:388. [Google Scholar]
- 23.Ko TP, Day J, Greenwood A, McPherson A. Acta Crystal-logr Sect D. 1994;50:813. doi: 10.1107/S0907444994005512. [DOI] [PubMed] [Google Scholar]
- 24.Diamond R, Phillips DC, Blake CCF, North ACT. J Mol Biol. 1974;82:371. doi: 10.1016/0022-2836(74)90598-1. [DOI] [PubMed] [Google Scholar]
- 25.Chayen NE. Acta Crystallogr Sect D. 1998;54:8. doi: 10.1107/s0907444997005374. [DOI] [PubMed] [Google Scholar]
- 26.Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, Drennan CL. Science. 2004;303:76. doi: 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Einsle O, Tezcan FA, Andrade SLA, Schmid B, Yoshida M, Howard JB, Rees DC. Science. 2002;297:1696. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.