

SUMMARY
The ability of p53 to induce apoptosis plays an important role in tumor suppression. Here we describe a previously unknown post-translational modification of the DNA-binding domain of p53. This modification, acetylation of lysine 120, occurs rapidly after DNA damage and is catalyzed by the MYST family acetyltransferases hMOF and TIP60. Mutation of lysine 120 to arginine, as occurs in human cancer, debilitates K120 acetylation and diminishes p53-mediated apoptosis without affecting cell-cycle arrest. The K120R mutation selectively blocks the transcription of pro-apoptotic target genes such as BAX and PUMA while the non-apoptotic targets p21 and hMDM2 remain unaffected. Consistent with this, depletion of hMOF and/or TIP60 inhibits the ability of p53 to activate BAX and PUMA transcription. Furthermore, the acetyl-lysine 120 form of p53 specifically accumulates at pro-apoptotic target genes. These data suggest that K120 acetylation may help distinguish the cell cycle arrest and apoptotic functions of p53.
Keywords: p53, hMOF, TIP60, acetylation, apoptosis, BAX
INTRODUCTION
The tumor suppressor p53 protects mammalian cells from malignant transformation by inducing either cell cycle arrest or apoptosis when genotoxic insults are encountered. Not surprisingly, the p53 pathway is inactivated in the majority of human cancers (Levine, 1997; Vousden and Lu, 2002). This inactivation often takes the form of debilitating missense mutations in p53 itself. Alternatively, genes encoding other components of the p53 pathway can exhibit mutations (Vogelstein et al., 2000; Vousden and Lu, 2002). While cell cycle arrest depends on the ability of p53 to induce the transcription of target genes such as the CDK inhibitor p21/waf1 (el-Deiry et al., 1993), apoptosis depends on induction of a distinct class of target genes, including BAX, PUMA, NOXA, p53AIP1, PERP, and PIG3 (Vousden and Lu, 2002). The combined induction of these genes promotes mitochondrial outer membrane permeabilization and cytochrome c release. These events lead to the activation of caspases and ultimately apoptotic cell death (Green, 2000). What remains unclear is precisely how cells with genetic damage choose between cell cycle arrest and apoptosis, the two distinct biological responses initiated by p53 activation. Understanding the bifurcation of these pathways remains a clinically important goal in cancer biology as most chemotherapeutic strategies are aimed at triggering the apoptosis of tumor cells.
The human MYST family of acetyltransferases consists of five members, hMOF, TIP60, HBO1, MOZ and MORF. The highly related hMOF and TIP60 proteins have been implicated in regulating several critical steps in the DNA damage response (Gupta et al., 2005; Ikura et al., 2000; Kusch et al., 2004; Legube et al., 2002; Sun et al., 2005). For example, exposure of cells to DNA damage leads to rapid acetylation of ATM (ataxia telangiectasia-mutated). Loss of acetylation due to hMOF or TIP60 depletion, prevents ATM from phosphorylating downstream effectors, such as p53 and CHK2 (Sun et al., 2005). hMOF and TIP60 physically interact with ATM and p53 (Dou et al., 2005; Tyteca et al., 2006). In vitro, hMOF binds and synergizes with p53 to increase histone H4 lysine 16 acetylation and target gene transcription (Dou et al., 2005). Furthermore, depletion of TIP60 prevents p53 from inducing expression of p21(Berns et al., 2004; Legube et al., 2004).
Many acetyltransferases both bind and directly acetylate p53 (Gu and Roeder, 1997; Sakaguchi et al., 1998), thereby regulating many of the biological properties of p53, (e.g. DNA binding, stability and co-factor recruitment). Given that p53 is a substrate of these other acetyltransferases, the possibility that MYST family proteins directly acetylate p53 was examined. We report here that both hMOF and TIP60 acetylate p53 at lysine 120 (K120) within the DNA binding domain. Structural studies have suggested that K120 acetylation may provide p53 with a preference for certain target gene promoters over others (Kitayner et al., 2006). Rare mutations at K120 have been reported in human cancer, with some of these mutations altering the lysine to a highly conserved, but non-acetylatable arginine residue. Remarkably, the loss of acetylation potential selectively blocks the ability of p53 to induce the transcription of the pro-apoptotic target genes BAX and PUMA, while having no effect on the transcription of non-apoptotic targets. Furthermore, acetylation of K120 on endogenous p53 occurs rapidly after DNA damage, and the acetyl-K120 form of p53 selectively accumulates at pro-apoptotic target gene promoters. Thus, MYST-mediated acetylation of K120 within the DNA binding domain of p53 may provide a mechanism by which p53 can distinguish distinct target genes and may ultimately explain how it selectively triggers either cell cycle arrest or apoptosis.
RESULTS
MYST family acetyltransferases, hMOF and TIP60 acetylate p53 at lysine 120
To determine whether MYST family proteins can acetylate p53, p53 was ectopically expressed with TIP60, hMOF or HBO1 in the p53 null cell line H1299. The co-expression of TIP60 or hMOF robustly enhanced acetylation of p53 (Figure 1A). In contrast, no acetylation of p53 by HBO1 was observed. To determine whether MYST family members acetylate p53 at the same lysine residues targeted by other acetyltransferases, a mutant version of p53 was generated in which the lysines targeted by p300/CBP or PCAF/hGCN5 were mutated to arginines (p53-9K/R) (Ard et al., 2002). hMOF was still able to acetylate this mutant version of p53 (Figure 1B), suggesting that hMOF-mediated acetylation occurs at a distinct site within p53. Identical results were obtained for TIP60 (Supp. Figure 1A). Analysis of deletion mutants truncating the C-terminus of p53 demonstrated that hMOF primarily acetylates the central DNA binding domain of p53 (data not shown). In order to map the specific site(s) of acetylation within this domain, individual lysines were converted to arginines. As above, these mutants were analyzed for acetylation by co-expression with hMOF or TIP60. Conversion of lysines 101, 132, and 139 did not affect acetylation by hMOF (Figure 1C). However, mutating K120 to arginine abolished hMOF-mediated acetylation of p53, raising the possibility that K120 is the major site targeted by hMOF. Conversion of K120 to arginine also abolished TIP60-mediated acetylation of p53 (Supp. Figure 1B). The apparent loss of p53 acetylation upon mutation of K120 might result from structural changes in the protein caused by the mutation, that occlude the true acetylation site targeted by hMOF and TIP60. To assess this possibility, p53 was purified from H1299 cells after co-expression with either hMOF or TIP60. After purification, sites of hMOF-mediated acetylation were directly identified by μLC-MS/MS. Analysis of the hMOF sample revealed that of the eight unique tryptic or chymotryptic peptides spanning K120, six were acetylated at K120 (Figure 1D). Of the unique peptide spectra spanning the same residue from p53 expressed in the absence of hMOF, none were acetylated. μLC-MS/MS analysis of p53 acetylated by TIP60 also documented K120 as the site targeted (Supp. Figure 1C). Thus, K120 on p53 is a shared site of acetylation by the MYST family enzymes hMOF and TIP60.
Figure 1. hMOF and TIP60 enhance acetylation of p53.
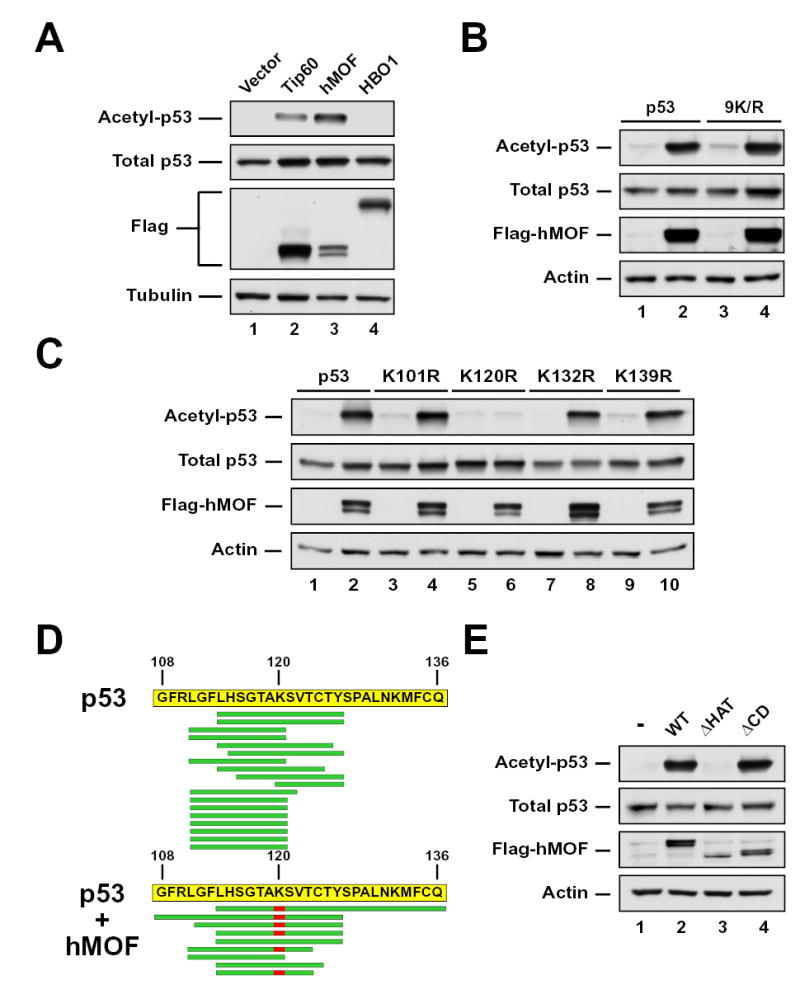
(A) H1299 cells were co-transfected with p53 and either vector, FLAG-TIP60, FLAG-hMOF, or FLAG-HBO1. Following 4 hours of treatment with 10mM sodium butyrate, cells were lysed and subjected to immunoprecipitation (IP) with a pan-acetyl-lysine (AcK) antibody. Precipitates were blotted with a p53 antibody to quantify p53 acetylation. Input lysates were also blotted with FLAG, p53, and tubulin antibodies. (B) H1299 cells were co-transfected with either wild type or 9K/R in the presence or absence of a plasmid expressing FLAG-hMOF. Acetylation was assessed by IP using the strategy described in (A). Total p53 expression was assessed by western blot of input lysates. (C) Expression vectors for mutants of p53 carrying individual lysine-to-arginine substitution mutations within the DNA-binding domain of p53 were transfected into H1299 cells with or without a plasmid expressing FLAG-hMOF. Acetylation was assessed by IP using the strategy described above. Total p53 expression was assessed by western blot of input lysates. (D) FLAG epitope-tagged p53 expressed in either the presence or the absence of hMOF was affinity purified from transiently transfected H1299 cells. The purified p53 proteins were subjected to μLC-MS/MS analysis to identify in vivo sites of hMOF-mediated acetylation. The sequence of the FLAG-epitope tagged p53 protein is displayed with green bars indicating the position of the tryptic or chymotryptic peptide spectra detected in the μLC-MS/MS analysis from the sample co-expressed with hMOF. Acetylated lysines are displayed in red. (E) H1299 cells were co-transfected with p53 and plasmids expressing FLAG-hMOF, FLAG-hMOF HAT (represents a 60-amino acid deletion within the HAT domain of hMOF), or FLAG-hMOF CD (represents a 66-amino acid deletion of the hMOF chromodomain). Following transfection cells were lysed and subjected to IP with a pan-acetyl-lysine antibody. To assess p53 acetylation pan-acetyl-precipitates were subjected to western blot with a p53 antibody (Top panel). Input lysates were also analyzed by western blot with the indicated antibodies (bottom three panels).
hMOF and TIP60 display a common domain structure, with an N-terminal chromodomain and a centrally located acetyltransferase domain. Deletion of amino acids 273–332 within the HAT domain of hMOF (hMOFΔHAT) eliminated its ability to acetylate p53 (Figure 1E). In contrast, deletion of the chromodomain failed to inhibit hMOF’s ability to acetylate p53.
Although the data presented suggests that p53 may be a direct substrate of hMOF and TIP60, it remained possible that these MYST proteins activate an intermediate enzyme to acetylate p53. To assess this possibility, in vitro acetylation reactions were performed using purified components. Recombinant forms of the p53 DNA binding domain (amino acids 94–312) and full-length hMOF were purified and incubated in vitro with 3H-acetyl-CoA. As a positive control, parallel reactions included a 19-amino acid (1–19) histone H4 peptide that spans K16, which has been previously shown to be acetylated by hMOF (Dou et al., 2005; Gupta et al., 2005; Taipale et al., 2005). As evident in Figure 2A, hMOF directly acetylates the DNA-binding domain of p53, in a dose-dependent manner. No hMOF-mediated acetylation was detected in samples lacking the p53 and H4 substrates. The direct in vitro acetylation of the p53 DNA binding domain by hMOF was eliminated by mutating K120 to arginine (Figure 2B), consistent with K120 being the major site of acetylation within this region.
Figure 2. hMOF directly acetylates p53 in vitro.
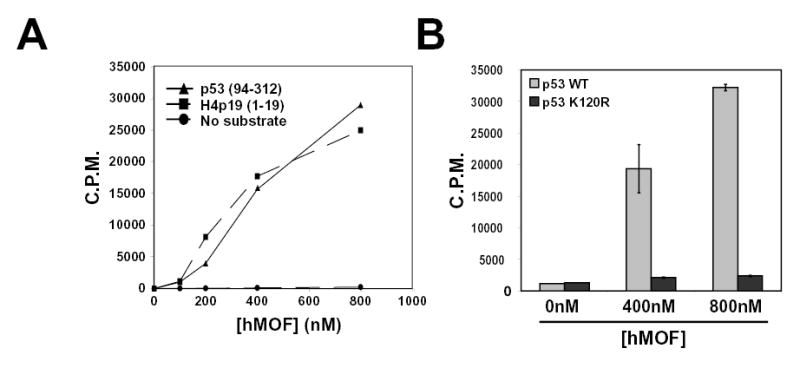
(A) Increasing amounts of recombinant full-length hMOF and radioactive acetyl Co-A were incubated with and without recombinant human p53 (amino acid residues 94 to 312). After incubation, liquid scintillation was used to assess enzymatic activity. Increasing amounts of full-length hMOF were incubated with a histone H4 peptide (S1-R19) as a positive control. (B) Both wild type and K120R versions of recombinant human p53 (described above) were purified from bacterial extracts. Subsequently each p53 protein was incubated separately with varying concentrations of hMOF. After incubation, liquid scintillation counting was used to detect the acetylation of wild-type and mutant p53.
K120 acetylation increases rapidly after genotoxic stress
In order to assess whether acetylation of p53 occurs at K120 in response to DNA damage, an antisera specific for acetyl-K120 was generated. The specificity of this reagent for the acetylated form of p53 was documented by experiments in which pre-incubation of the antisera with an acetylated form of the K120 peptide blocked reactivity with hMOF acetylated p53 (Figure 3A). Pre-incubation with a non-acetylated form of the K120 peptide did not block reactivity. The specificity of the antisera was further confirmed by its inability to recognize p53 when K120 was mutated to arginine (Figure 3B and Supp. Figure 2). This acetyl-K120 specific antibody was utilized to assess the induction of K120 acetylation on endogenous p53 following treatment of U2OS cells with UV light, γ-irradiation or the chemotherapeutic agents adriamycin and camptothecin. All four treatments resulted in increased levels of endogenous p53. Strikingly, all four treatments also resulted in the robust acetylation of p53 at K120 (Figure 3C). While some stimuli resulted in K120 acetylation that peaked simultaneously with the increase in total p53 (adriamycin and camptothecin), other stimuli resulted in induction of K120 acetylation that peaked earlier than total p53 levels (γ-irradiation and UV). The induction of K120 acetylation on endogenous p53 was also observed in response to DNA damage in the LNCaP prostate cancer cell line and MCF7 breast cancer cell line (Supp. Figure 2).
Figure 3. Acetylation of lysine 120 catalyzed by hMOF and TIP60 increases in response to genotoxic stress.
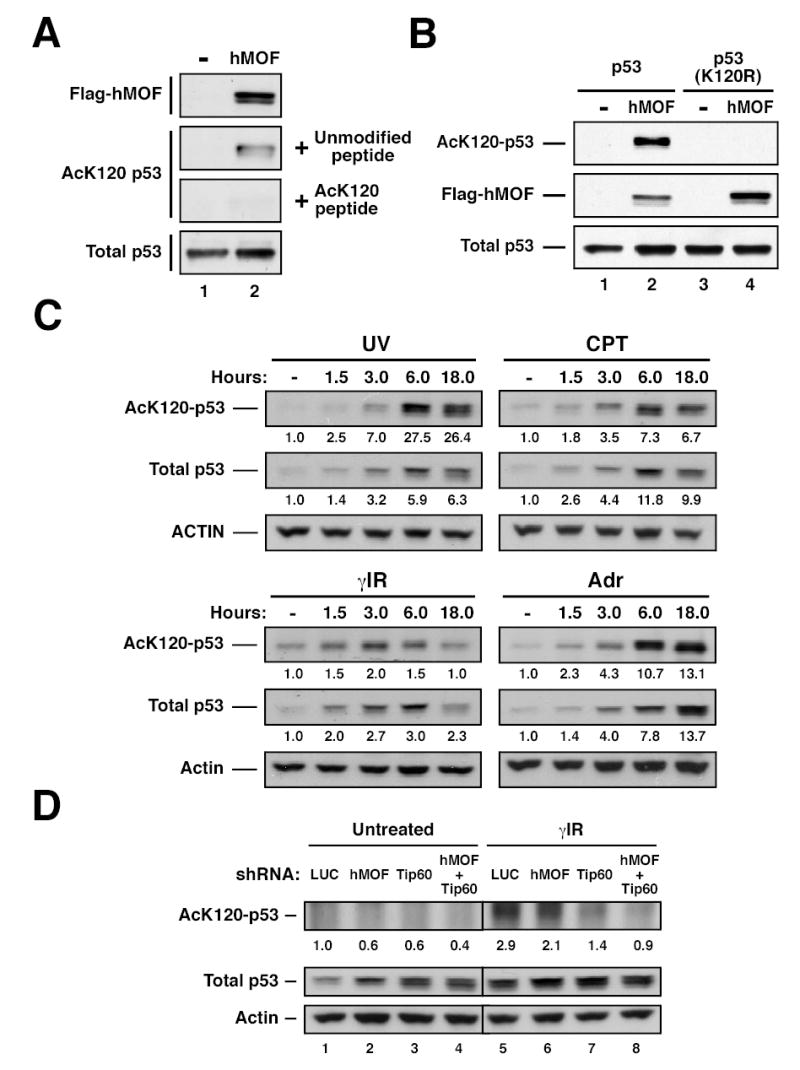
(A) H1299 cells were transfected with p53 in the presence or absence of hMOF. After transfection, cells were treated with 10mM sodium butyrate for 2–4 hours. Following treatment, the cell lysate was divided into two aliquots. One aliquot was subjected to immunoprecipitation (IP) with AcK120 antibody pre-blocked with 250μM modified peptide. The other aliquot was subjected to IP with AcK120 antibody pre-blocked with 25μM of unmodified peptide. Precipitates were then western blotted with a p53 antibody to quantify lysine-120 acetylation. Western blots were performed on the input lysates to ensure that equal amounts of p53 were used for each IP. (B) H1299 cells were transfected with wild-type p53 in the presence or absence of hMOF. In parallel, H1299 cells were co-transfected with p53-(K120R) and either vector control or hMOF. After transfection, cells were treated with 10mM sodium butyrate for 2–4 hours. Following treatment, cells were lysed and subjected to IP with an AcK120 antibody. Precipitates were western blotted with a p53 antibody to quantify lysine-120 acetylation. Input lysates were also subjected to western blot as described above. (C) U2OS cells were treated with 25 J/m2 UV, 5μM camptothecin (CPT), 10 Gy γ-irradiation (γIR), or 0.5 μg/ml adriamycin (Adr) and then harvested at the indicated times. Ninety minutes prior to each time point, cells were treated with 10mM Sodium Butyrate. K120 acetylation was assessed as described above. (D) U2OS cells were infected with recombinant lentiviruses that express shRNA molecules targeted to the corresponding genes. Seventy-two hours following infection, cells from each shRNA condition were treated with 10 Gy γ–Irradiation or left untreated. Two hours after irradiation, cells were treated with 10 mM Sodium Butyrate for 1 hour. Cells were then harvested and subjected to immunoprecipitation and subsequent western blot as described above.
Depletion of MYST family members diminishes K120 acetylation
To determine the contribution of hMOF and TIP60 to K120 acetylation on endogenous p53, U2OS cells were infected with lentiviruses encoding shRNAs targeting either hMOF or TIP60. (The hMOF and TIP60 shRNA constructs reduce protein levels by 70% and 80%, respectively (see. Figure 6)). γ-irradiation of control cells resulted in a robust increase in K120 acetylation (Figure 3D). This acetylation was partially inhibited by hMOF depletion (~28% decrease) or by TIP60 depletion (~52% decrease). When hMOF and TIP60 were simultaneously depleted, induction of K120 acetylation was decreased to levels observed in the untreated control cells (compare lanes 1 and 8, Figure 3D).
Figure 6. Depletion of hMOF and TIP60 reduces the ability of p53 to induce BAX and PUMA.
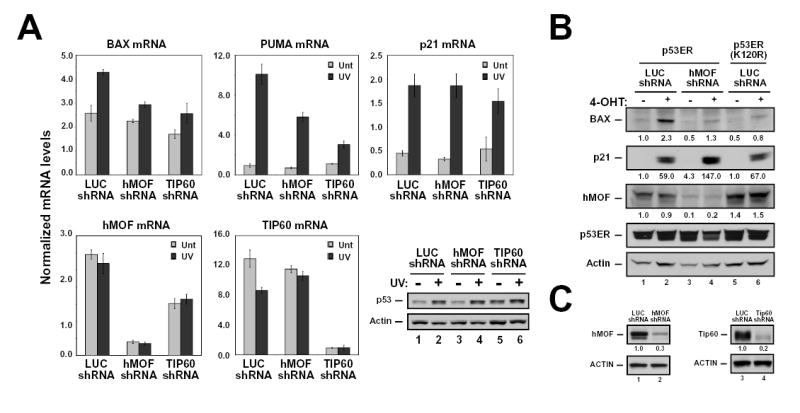
(A) MCF-7 cells were infected with recombinant lentiviruses that express shRNA molecules directed against luciferase (control), hMOF, or TIP60. Seventy-two hours following infection, cells were treated with 25 J/m2 UV and then collected 16 hours thereafter. Harvested cells were divided into two aliquots. One aliquot from each treatment condition was harvested for RNA, which was converted to cDNA as described above. cDNA samples were then analyzed by real-time PCR with primers corresponding to the indicated genes. The other aliquot was lysed and subjected to western blot with the antibodies indicated (lower right panel). (B) H1299-p53ER and –p53ER-(K120R) cells were infected with lentiviruses expressing the shRNA molecules indicated. Seventy-two hours following infection, cells were treated with 100 nM 4-OHT or vehicle for 16 hours. Harvested cells were analyzed by western blot with the antibodies indicated. (C) Lentiviral shRNA vectors efficiently reduce protein levels for both hMOF and TIP60. H1299 cells were transfected with either FLAG-hMOF or FLAG-TIP60. Transfected cells were then split into 2 dishes and infected with recombinant lentiviruses expressing either luciferase (LUC), hMOF, or TIP60 shRNA molecules. Seventy-two hours following infection, cells were harvested, lysed and subjected to western blot with the indicated antibodies. Error bars represent standard deviation of three independent reactions.
Loss of K120 acetylation specifically inhibits p53-mediated apoptosis
Lysine 120 lies within a region of the DNA binding domain of p53 termed the L1 loop (Cho et al., 1994). Although mutations within the L1 loop are infrequent, five mutations where lysine 120 is converted to a glutamate, methionine, or arginine have been reported in human cancer (Deissler et al., 2004; Hashimoto et al., 1999; Hayes et al., 1999; Leitao et al., 2004; Meyers et al., 1993). Each tumor-derived K120 p53 mutant was evaluated for its ability to induce cell cycle arrest and apoptosis. These studies were conducted in the p53ER system in which full-length p53 is expressed as a fusion with a modified version of the estrogen receptor ligand-binding domain. The fusion protein remains inactive until the addition of the synthetic estrogen analog 4-hydroxytamoxifen (4-OHT). An additional advantage of this system is that when stable cell lines expressing p53ER are generated, the p53 expression levels mimic endogenous p53 expression levels (Chipuk et al., 2003). This minimizes artifacts of overexpression that can mask the defects inherent to weak mutant alleles of p53 (Zupnick and Prives, 2006). Cell lines expressing wild-type, K120E, K120M and K120R, versions of p53ER were generated in the p53 null H1299 cell line (Figure 4A and data not shown). Activation of wild-type p53ER in these cells induces a G1 phase cell cycle arrest (Figure 4B). While the K120R mutant was as potent as wild-type p53 at inducing cell cycle arrest, both K120E and K120M mutants were completely defective (Figure 4B and data not shown). This defect is presumably due to an inability of these proteins to bind target gene promoters or induce target gene transcription (Supp. Figure 3).
Figure 4. A conservative mutation at lysine 120 specifically reduces p53 mediated-apoptosis.
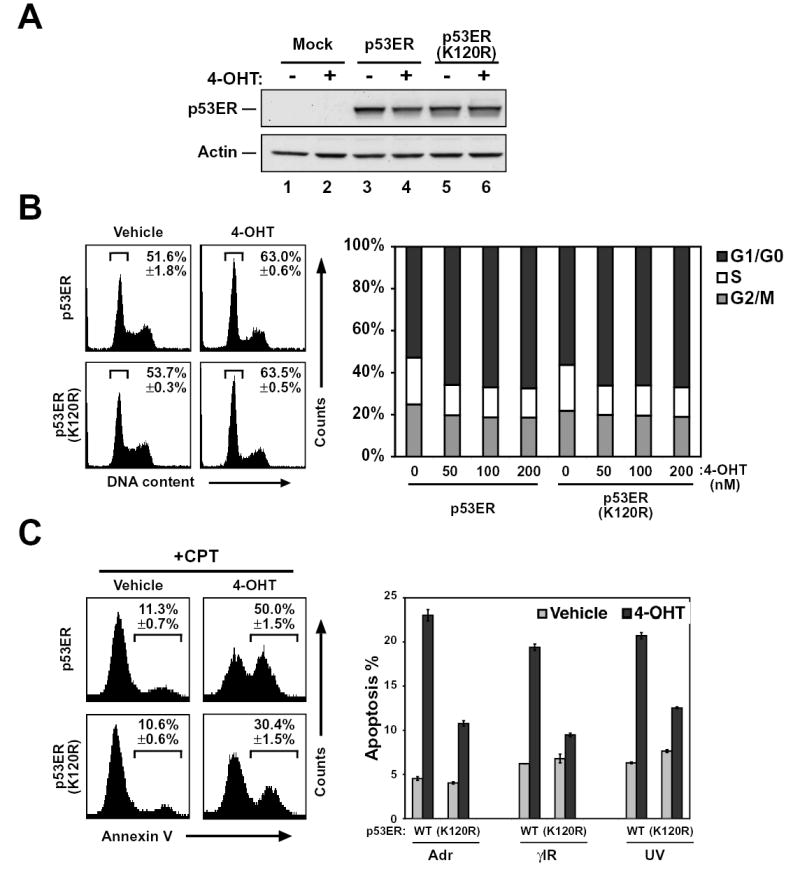
(A) H1299 cells were infected with recombinant retroviruses expressing either p53ER or p53ER-(K120R). Mock infected cells served as a control. Infected cells were lysed and subjected to western blot to assess the expression of each construct. (B) H1299-p53ER and H1299-p53ER-(K120R) cells were treated with increasing concentrations of 4-hydroxytamoxifen (4-OHT) for 24 hours. Following treatment cells were fixed, stained with propidium iodide (PI), and analyzed by FACS to assess cellular DNA content. Left panel displays results obtained from FACS analysis of cells treated with and without 50 nM 4-OHT. Right panel is a graphical representation of FACS data obtained from all treatment conditions. (C) H1299-p53ER and H1299-p53ER-(K120R) cells were treated with 50 nM 4-OHT for 24 hours. Cells were then treated with 5 μM CPT, 0.5 μM Adr, 12 Gy γIR, or 50 J/m2 UV. Twenty-four hours after DNA damage, cells were stained with annexin V-FITC and PI and then analyzed by FACS. Left panel displays plots obtained from FACS analysis of cells treated with CPT. Right panel is a graphical representation of annexin V positive (PI negative) cells collected after the treatment indicated. Error bars represent standard deviation of three independent cell counts.
When combined with DNA damage stimuli, activation of p53ER by 4-OHT results in apoptosis induction (Figure 4C), as assessed by Annexin V staining. As expected, both the K120E and K120M mutants were defective for apoptosis (data not shown). Remarkably, the K120R mutant, which was fully competent for cell cycle arrest, was unable to induce a full apoptotic response (Figure 4C, left panel). Further analysis revealed that the apoptotic response to a variety of other DNA damaging agents was also debilitated by the K120R mutation (Figure 4C, right panel). Thus, while p53 mutants carrying the non-conservative K120M or K120E mutations are defective for both cell cycle arrest and apoptosis, the conservative K120R mutation is selectively defective for apoptosis induction while remaining competent for cell cycle arrest.
Loss of K120 acetylation diminishes p53-initiated transcription of BAX and PUMA
As mentioned above, p53-mediated apoptosis depends on the ability of p53 to up-regulate the expression proteins that directly promote apoptosis. In human cells, p53 requires both BAX and PUMA to efficiently induce apoptosis (Chipuk et al., 2003; Jeffers et al., 2003; Villunger et al., 2003; Yu et al., 2003; Zhang et al., 2000). Consequently, studies were conducted to determine whether selective defects in p53 target gene induction were responsible for the defect in apoptosis evident with the tumor-derived, acetylation-defective mutant K120R. For this purpose, the induction of target gene protein products by wild-type p53 and the K120R mutant was compared. As expected, activation of wild-type p53 resulted in the induction of all target gene products examined (Figure 5A). Consistent with its ability to promote cell cycle arrest, K120R is competent for induction of the cell cycle arrest target p21 and the regulatory enzyme hMDM2 (Figure 5A). However, K120R was severely impaired for BAX induction (~60% decrease) and mildly impaired for PUMA induction (~20% decrease) (Figure 5A). Quantitative RT-PCR confirmed that this selective defect occurred at the level of transcription (Figure 5B and Supp. Figure 4A). These findings are consistent with the biological functions of the K120R mutant (i.e. cell cycle arrest competent but apoptosis defective).
Figure 5. p53-(K120R) is specifically defective for BAX and PUMA induction.
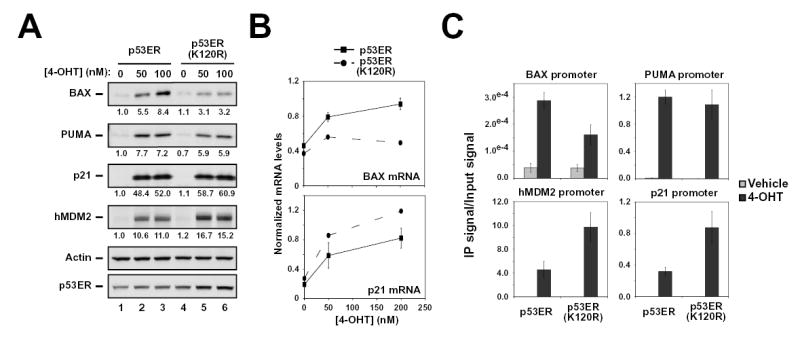
(A) H1299-p53ER and –p53ER-(K120R) cells were treated with 50 or 100 nM 4-OHT or vehicle for 24 hours. Cells were collected, lysed and subjected to western blot with the antibodies indicated. (B) H1299-p53ER and –p53ER-(K120R) cells were treated with 50 or 200nM 4-OHT for 24 hours and subsequently harvested for RNA at 0, 6, 12, 18 and 24 hours. cDNA generated from recovered RNA was amplified with primers to the target genes indicated and quantified by real-time PCR. (C) H1299-p53ER and –p53ER-(K120R) cells were treated with 100nM 4-OHT or vehicle for 6 hours. Cells from each condition were cross-linked and subjected to ChIP analysis with a p53 antibody. Precipitated DNA was recovered and analyzed by real-time PCR with primers that amplify the p53-binding site within the BAX, PUMA, hMDM2, and p21 promoters. Error bars represent standard deviation of three independent reactions.
Using Chromatin Immunoprecipitation (ChIP), wild-type p53 and the K120R mutant were compared for binding to endogenous target gene loci in H1299 cells. This analysis revealed a modest defect in the ability of the K120R mutant to bind the BAX promoter (Figure 5C). In contrast, the mutant was not defective for binding to the PUMA, p21 or hMDM2 promoters. Therefore, the transcriptional defect that the K120R mutant displays for BAX and PUMA is not explained by a structural defect that globally compromises DNA binding by p53. The contribution of the modest defect exhibited by the K120R mutant in binding to BAX, to the more severe transcriptional defect observed in Figure 5B is addressed below.
Depletion of MYST family members reduces BAX and PUMA transcription
As the K120R mutation may result in structural defects in addition to a loss of acetylation potential, it was difficult to discern from mutational analysis which effect was responsible for the decrease in BAX and PUMA expression. Therefore the ability of wild-type p53ER to induce BAX expression after shRNA-mediated depletion of either hMOF or TIP60 was examined. Depletion of either hMOF or TIP60 partially blocked the induction of BAX and PUMA mRNA by endogenous p53 in MCF7 cells treated with UV light (Figure 6A). In contrast, neither enzyme was absolutely required for the induction of p21 by p53 in these cells. (A slight defect in p21 transcription was observed upon TIP60 depletion, consistent with suggestions that TIP60 is required for the acetylation of histones at the p21 promoter (Doyon et al., 2004).) Western blot analysis revealed that the inhibition of BAX and PUMA induction was not due to an effect of hMOF or TIP60 on the level of p53 protein induced by UV light in these cells.
In order to directly compared to the level of inhibition observed upon knockdown of hMOF with that observed with the K120R mutant, the p53ER system was used. This analysis revealed that decreasing hMOF levels inhibited BAX transactivation by wild-type p53 to a level similar to the inhibition observed upon introduction of the tumor-derived K120R mutation (Figure 6B). Neither the depletion of hMOF nor the K120R mutation had an effect on p21 induction. shRNA constructs targeting hMOF and TIP60 were efficient at depleting both mRNA and protein (Figure 6A–C).
K120-acetylated p53 specifically accumulates at the BAX and PUMA promoters
Conversion of K120 to an arginine leads to a loss of acetylation potential, a defect in BAX and PUMA induction, and a defect in apoptosis. Given the structural evidence that K120 may play a role in target gene selectivity (Kitayner et al., 2006), one of the mechanistic models linking these findings predicts that the K120 acetylated form of p53 would be enriched at the BAX and PUMA promoters versus non-apoptotic promoters, such as p21 and MDM2. To address this possibility, both untreated and camptothecin treated LNCaP cells were subjected to ChIP with either the acetyl-K120-p53 specific antibody or an antibody directed against total p53. Consistent with the transcriptional defect observed with the K120R mutant, the acetyl-K120 form of endogenous p53 accumulated to high levels at the BAX and PUMA promoters (Figure 7A). In marked contrast, the acetyl-K120 form of p53 failed to accumulate to significant levels at the non-apoptotic p21 promoter and hMDM2 promoters. The selective accumulation of the acetyl-K120 form of endogenous p53 at the BAX and PUMA promoters was also observed in the breast cancer line MCF-7 (Figure 7B and data not shown). As in LNCaP cells, this analysis revealed no evidence for the accumulation of acetyl-K120 p53 at the non-apoptotic p21 and MDM2 promoters (Figure 7B). These data combined with the transcriptional defect observed with the K120R mutant strongly suggest that the acetyl-K120 form of p53 selectively functions at pro-apoptotic p53 target genes such as BAX and PUMA.
Figure 7. p53 acetylated at K120 selectively accumulates at pro-apoptotic target gene promoters.
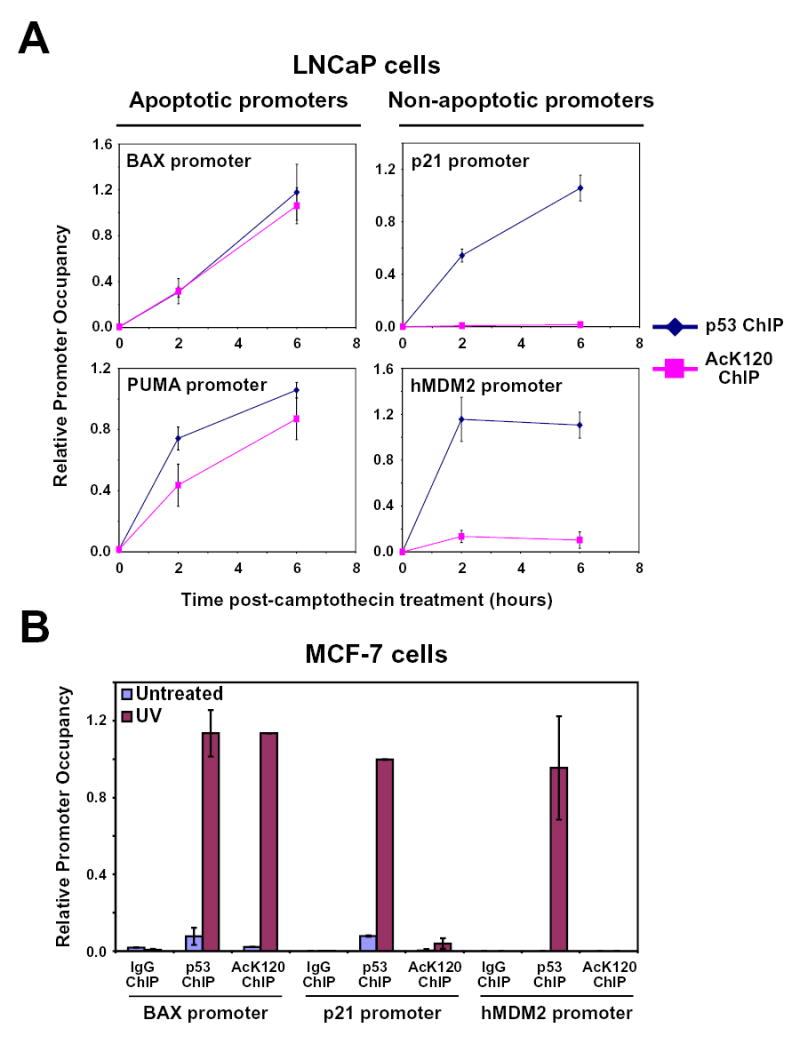
(A) LNCaP cells treated with 5 μM CPT for 0, 2, and 6 hours were cross-linked and harvested for ChIP. Lysates from each condition were divided into equal aliquots and incubated with either p53 or AcK120-p53 antibodies. Precipitated DNA was recovered and analyzed by real-time PCR with primers that amplify the area encompassing the p53 binding sites within the promoters indicated. (B) Six hours following 20 J/m2 UV exposure, MCF-7 cells were harvested and analyzed as described in (A). Error bars represent standard deviation of three independent reactions.
DISCUSSION
The studies reported here demonstrate that endogenous p53 is acetylated at K120 within the DNA binding domain. Rare mutations that convert K120 either to a conservative arginine or to a non-conservative glutamate or methionine residue have been reported in human cancer (Deissler et al., 2004; Hashimoto et al., 1999; Hayes et al., 1999; Leitao et al., 2004; Meyers et al., 1993). We show here that acetylation of K120 is catalyzed by the closely related MYST family enzymes hMOF and TIP60 and that K120 acetylation is rapidly induced in cells exposed to genotoxic stress. Interestingly, the tumor-derived K120R mutation both blocks K120 acetylation and inhibits apoptosis induction by p53. However, this mutant retains the ability to promote cell cycle arrest. Consistent with this, loss of K120 acetylation, by direct mutation or depletion of hMOF and TIP60 specifically inhibits the transcription of key pro-apoptotic p53 targets, while non-apoptotic targets are still robustly induced. As a potential mechanism explaining why K120 acetylation is critical for apoptosis but not cell cycle arrest, we demonstrate that the acetyl-K120 form of endogenous p53 selectively accumulates at the promoters of certain pro-apoptotic target genes.
The inability of the K120R mutation to promote apoptosis and induce BAX and PUMA transcription could result from a loss of acetylation potential or from structural changes in the protein. Several lines of evidence support the model in which the K120R defect results primarily from a loss of acetylation potential. First, the K120R mutant retains the ability to both bind and transactivate the target genes p21 and hMDM2 (Figure 5). Second, depletion of the enzymes responsible for K120 acetylation in response to DNA damage yields a phenotype similar to mutation of K120 to arginine (Figure 6). Third, the acetyl-K120 form of p53 selectively accumulates at pro-apoptotic promoters (Figure 7), consistent with it having an active functional role at these genes. Collectively, these findings support the model that the K120R mutation is unable to promote apoptosis due to a lack of K120 acetylation and not due to structural defects.
The overlapping ability of hMOF and TIP60 to acetylate K120 is similar to the overlapping lysine substrates on p53 shared by p300 and CBP as well as PCAF and GCN5. It remains unclear why cells express two enzymes that share the same substrate. One potential explanation is that the two enzymes are redundant so that one can protect against the loss of the other. However, both hMOF and TIP60 appear to contribute independently to K120 acetylation as depletion of either enzyme alone is sufficient to partially reduce K120 acetylation. An alternative explanation may be that distinctions in the type of genotoxic stress or tissue being examined dictate which enzyme catalyzes K120 acetylation. In support of this notion, depletion of TIP60 in the osteosarcoma cell line U2OS and the breast cancer cell line MCF-7 results in a greater reduction of K120 acetylation and pro-apoptotic gene transcription than does hMOF depletion (Figures 3D and 6A). However, in the lung cancer cell line H1299, hMOF displays greater catalytic activity over TIP60 in acetylating K120 (Figure 1). Furthermore, depletion of hMOF in H1299 cells reduces the ability of p53ER to activate BAX expression, where as TIP60 shRNA has a minimal affect (Figure 6B and data not shown). It should be noted that interpretations drawn from experiments involving TIP60 and hMOF shRNA regarding p53 signaling should consider that both TIP60 and hMOF also participate in the DNA damage signaling pathway at points that lie well upstream of p53 activation and K120 acetylation (Gupta et al., 2005; Ikura et al., 2000; Kusch et al., 2004; Legube et al., 2002; Sun et al., 2005). Therefore, further analysis is required to elucidate under which scenarios K120 is acetylated by these enzymes.
Although we have shown that acetylation of K120 is required for the activation of the p53 pro-apoptotic target genes BAX and PUMA, K120 acetylation does not appear to affect all p53 pro-apoptotic target genes. For example, the NOXA gene is still induced by the K120R mutant of p53 in the H1299 cell line and the acetyl-K120 form of endogenous p53 fails to accumulate to significant levels at the NOXA promoter after DNA damage treatment of LNCaP cells (Supp. Figure 4). While the role of NOXA in p53-mediated apoptosis is less universal and more cell-type specific than that of BAX and PUMA (Villunger et al., 2003), this finding does suggest that not all apoptotic targets have equal requirements for the K120-acetyl form of p53. This differential requirement may reflect differences in the promoter structure and/or cofactors needed for transactivation of the different subsets of target genes.
As discussed above, the finding that the K120R mutation occurs in human cancer suggests that acetylation by MYST family proteins may be critical to the tumor suppressor function of p53. Consistent with this, hMOF levels are frequently decreased in human tumor samples (S. Rea and A. Akhtar, personal communication). A role for TIP60 in tumorigenesis has also been suggested by recent studies (Kim et al., 2005; Squatrito et al., 2006). It is therefore tempting to speculate that loss of K120 acetylation is partially responsible for the roles played by hMOF or TIP60 in cancer. Furthermore, a role for TIP60 in the UV-induced transcription of several p53 targets was recently reported (Tyteca et al., 2006).
The next series of challenges that are presented by the identification of this pathway relate to understanding precisely why K120 acetylation is required for p53-mediated transcription of apoptotic target genes, BAX and PUMA. It is possible that the presence of an acetyl group on K120 alters the interaction this residue makes with DNA. This is supported by structural studies suggesting that K120 indeed participates in dictating the preference of p53 for distinct classes of binding sites (Kitayner et al., 2006). However, the finding that the non-acetylated K120R mutant retains significant binding to the PUMA promoter and exhibits only a partial defect at the BAX promoter suggests that a direct effect on DNA binding may not explain the critical role of this residue (Figure 5). More likely is a model in which increased transcription of pro-apoptotic targets results from the ability of the acetyl-K120 form of p53 to recruit essential transcriptional cofactors that modify nucleosomal histones, stabilize the p53-DNA interaction or otherwise augment mRNA synthesis. For example, the ASPP family of proteins (ASPP1 and ASPP2) preferentially localize to p53 pro-apoptotic promoters, such as BAX (Samuels-Lev et al., 2001), similar to the acetyl-K120 form of p53. Furthermore, the ASPP proteins specifically enhance the binding of p53 to the BAX promoter and consequently the apoptotic function of p53 (Samuels-Lev et al., 2001). Therefore the modest defect in p53 K120R binding to the BAX promoter may result from its inability to bind cofactors that enhance p53-DNA interactions, such as the ASPP proteins. Finally, although the ASPP proteins do not directly interact with the L1 loop, it has been proposed that the L1 loop influences the binding of ASPP2 to p53 (Friedler et al., 2005). Thus, acetylation of K120 may result in enhanced recruitment of the ASPP proteins or other essential cofactors to pro-apoptotic promoters.
Previous studies have identified other post-translational modifications of p53 that are specifically associated with the induction of apoptosis, but not cell cycle arrest. These modifications include the phosphorylation of serines 20 and 46 and the acetylation of lysine 373 (Bulavin et al., 1999; Jack et al., 2002; Knights et al., 2006; Oda et al., 2000). While the role of these modifications in apoptosis induction remains unclear (Thompson et al., 2004), there may be some interplay between them and K120 acetylation. This type of crosstalk between distinct post-translational modifications occurs on histones (Sun and Allis, 2002) and has been suggested to occur on p53 as well (Sakaguchi et al., 1998). Current efforts are aimed at determining whether the acetylation of K120 requires or influences other p53 post-translational modifications.
The acetylation of lysine 120 within the DNA binding domain of p53 is distinct from most other sites of p53 modification in that it is disrupted by mutation in human cancer. Also unusual is that this naturally occurring p53 mutation selectively affects the ability of p53 to induce apoptosis without blocking cell cycle arrest. Similarly, the MYST family of enzymes that catalyze K120 acetylation are implicated in the DNA damage response pathway and appear to be deregulated in cancer as well. Considered with the finding that the acetyl-K120 form of p53 selectively accumulates at key pro-apoptotic promoters, these studies define a pathway of p53 regulation that likely participates in determining whether the cellular response to DNA damage elicits cell cycle arrest or apoptosis. Further analysis will be required to gain a complete understanding of the contribution made by the genetic lesions in this pathway to human cancer.
EXPERIMENTAL PROCEDURES
Plasmids- pcDNA3-FLAG-hMOF was generated by PCR amplification of the hMOF cDNA with subsequent ligation into the pcDNA3.1-TOPO-TA cloning vector (Invitrogen). pcDNA3-FLAG-TIP60 was obtained from Saadi Khochbin (Institut Albert Bonniot-France). pcDNA3-FLAG-p53 was obtained from K. Adler-Storthz (The University of Texas-Houston). pRC-p53 was obtained from M. Murphy (Fox Chase Cancer Center-Philadelphia). p53 mutants were generated by site-directed mutagenesis (Stratagene). Primer sequences are available upon request. The retroviral pBABE-puro-p53/ER construct was obtained M. Schuler (Johannes Gutenberg University, Germany). Retroviral packaging plasmid (SVΨ-A-MLV) was described previously (Patel and McMahon, 2006). Lentiviral shRNA plasmids corresponded to clone 423 (hMOF) and clone 1633 (TIP60) and control luciferase shRNA were obtained from the TRC collection (Sigma). Lentiviral packaging plasmids (pCMV-ΔR8.2 and pCMV-VSV-G) were described previously (Budanov et al., 2004).
Cell culture
H1299, U2OS, 293T and MCF-7 cells (ATCC) were maintained in DMEM supplemented with 10% FBS (Hyclone). LNCaP cells (ATCC) were maintained in RPMI with 10% FBS . Transfections were performed using Lipofectamine 2000 (Invitrogen). For retroviral packaging, pBABE-puro-p53/ER or pBABE-puro-p53/ER-(K120R) were co-transfected into 293T cells with a retroviral packaging plasmid (SVΨ-A-MLV). Pooled stable cell lines expressing these constructs were generated by puromycin selection of infected H1299 cells. For lentiviral packaging, the shRNA vectors were cotransfected into 293T cells with the lentiviral packaging plasmids (pCMV-ΔR8.2 and pCMV-VSV-G). Viral supernatants were collected, filtered and added directly to target cells in the presence of 8μg/ml polybrene.
Antibodies, Western Blotting, and Immunoprecipitation
Whole cell extracts were generated using an NP-40 based lysis buffer supplemented with 30mM Sodium butyrate (Ard et al., 2002). The following antibodies were used to detect protein expression by western blot, p53 (FL-393G), p53 (DO-1), Actin (C-2), p21 (C-19) (Santa Cruz), BAX (Ab-3), PUMA (Ab-1) (Calbiochem), and hMDM2 (SMP14, BD PharMingen). Anti-hMOF antibody was provided by E. Smith and J. Lucchessi (Emory University). Total p53 acetylation was assessed using a pan acetyl-lysine antibody (Upstate Biotech.). AcK120-p53 antibodies were generated in rabbits. For immunoprecipitations, antibody-bound protein complexes were then precipitated with protein A/G sepharose (Santa Cruz). Precipitated proteins were subjected to western blotting with anti-p53 (FL-393G).
Mass spectrometry
hMOF- or TIP60- acetylated p53 was subjected to SDS-PAGE and excised. After analysis with EnzymeOpt, a custom program to maximize recovery and detection of proteotypic peptides, bands were split in two and digested with chymotrypsin or trypsin. Peptide sequence analysis on each was performed by microcapillary reverse-phase HPLC nanoelectrospray tandem mass spectrometry (μLC-MS/MS) on a Finnigan LCQ DECA XP Plus quadrupole ion trap mass spectrometer (Thermo). The ion trap surveyed m/z 395 to 1,600, acquiring data-dependent MS/MS spectra, (CE:30%, isolation: 2.5amu), for peptide sequence information on the four most abundant ionssurveyed. Sequencing of peptides was performed with SEQUEST (Eng et al., 1994) against the NCBInr protein database with appropriate restriction to chymotrypsin or trypsin specificity; static modification to carboxyamidomethylcys and differential modification to methionine sulfoxide. In silico combined data sets for both digest results, culled of minor contaminating keratin or autolytic peptide spectra, were re-searched against the FLAG-tag p53 sequence with no enzyme specificity. Differential modifications of acetylated lysine, proprionamidocysteine and oxidized tryptophan were included. The discovery of peptides carrying acetylation and subsequent manual analysis of their MS/MS spectra were facilitated with the in-house programs MuQuest and FuzzyIons (Chittum et al., 1998).
In vitro acetylation assay
In vitro acetylation assays were performed as described previously (Lo et al., 2000). In brief, recombinant full length hMOF (Dou et al., 2005), and p53 and p53K120R core domains (Zhao et al., 2001) were purified from bacteria and H4p19 (S1GRGKGGKGLGKGGAKRHR19) was prepared as previously described (Lo et al., 2000). hMOF was incubated with radiolabeled acetyl-CoA and substrate (H4p16, p53 or p53K120R) within the predetermined linear range of enzyme and substrate concentration and acetylation was measured by filter blotting and scintillation counting. Error bars represent the standard deviation of three individual reactions.
Cell cycle analysis
Following treatment cells were collected and fixed with 70% ethanol for at least 4 hours at −20°C. After fixation, cells were washed 2-3 x with PBS and then stained with 10ug/ml Propidium Iodide (BD PharMingen) and 100ug/ml RNAse A (Fisher).
Apoptosis Assays
Following treatments, cells were collected by trypsinization, washed with phosphate-buffer saline and stained using an Annexin V-FITC apoptosis detection kit (BD PharMingen) as described previously (Patel and McMahon, 2006).
RNA analysis and Chromatin Immunoprecipitation
Treated cells were collected and RNA was recovered using TriZol (Invitrogen). RNA was then converted to cDNA using SSII Reverse Transcriptase (Invitrogen) and random Decamers (Ambion). cDNA samples were analyzed by RealTime PCR (ABI prism 7000) with primers that amplify the corresponding mRNAs. Real-time signals were normalized to ACTIN, GAPDH or 18S ribosomal RNA levels, as indicated. ChIP was performed as described previously (Zhang et al., 2005) with antibodies against p53 (FL-393R, Santa Cruz) or the acetyl-K120 antisera described above. Primer sequences available upon request.
Supplementary Material
Acknowledgments
The authors thank Drs. E. Smith, K. Adler-Storthz, M. Schuler and L. Zhang for kindly providing reagents. We also thank Drs. D. George, A. Akhtar, T. Halazonetis, A. Diehl, Xianxin Hua and A. Norvell for helpful discussions. This work was supported by grants from the NIH (CA090465 and CA098172) to SBM. In addition, this work was supported by the Commonwealth Universal Research Enhancement Program, Pennsylvania Dept. of Health.
References
- Ard PG, Chatterjee C, Kunjibettu S, Adside LR, Gralinski LE, McMahon SB. Transcriptional regulation of the mdm2 oncogene by p53 requires TRRAP acetyltransferase complexes. Molecular and Cellular Biology. 2002;22:5650–5661. doi: 10.1128/MCB.22.16.5650-5661.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B, et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428:431–437. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science. 2004;304:596–600. doi: 10.1126/science.1095569. [DOI] [PubMed] [Google Scholar]
- Bulavin DV, Saito S, Hollander MC, Sakaguchi K, Anderson CW, Appella E, Fornace AJ., Jr Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. Embo J. 1999;18:6845–6854. doi: 10.1093/emboj/18.23.6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Maurer U, Green DR, Schuler M. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell. 2003;4:371–381. doi: 10.1016/s1535-6108(03)00272-1. [DOI] [PubMed] [Google Scholar]
- Chittum HS, Lane WS, Carlson BA, Roller PP, Lung FD, Lee BJ, Hatfield DL. Rabbit beta-globin is extended beyond its UGA stop codon by multiple suppressions and translational reading gaps. Biochemistry. 1998;37:10866–10870. doi: 10.1021/bi981042r. [DOI] [PubMed] [Google Scholar]
- Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- Deissler H, Kafka A, Schuster E, Sauer G, Kreienberg R, Zeillinger R. Spectrum of p53 mutations in biopsies from breast cancer patients selected for preoperative chemotherapy analysed by the functional yeast assay to predict therapeutic response. Oncol Rep. 2004;11:1281–1286. [PubMed] [Google Scholar]
- Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J, Allis CD, Chait BT, Hess JL, Roeder RG. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121:873–885. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Doyon Y, Selleck W, Lane WS, Tan S, Cote J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol Cell Biol. 2004;24:1884–1896. doi: 10.1128/MCB.24.5.1884-1896.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JRI. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Friedler A, Veprintsev DB, Rutherford T, von Glos KI, Fersht AR. Binding of Rad51 and other peptide sequences to a promiscuous, highly electrostatic binding site in p53. J Biol Chem. 2005;280:8051–8059. doi: 10.1074/jbc.M411176200. [DOI] [PubMed] [Google Scholar]
- Green DR. Apoptotic pathways: paper wraps stone blunts scissors. Cell. 2000;102:1–4. doi: 10.1016/s0092-8674(00)00003-9. [DOI] [PubMed] [Google Scholar]
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- Gupta A, Sharma GG, Young CS, Agarwal M, Smith ER, Paull TT, Lucchesi JC, Khanna KK, Ludwig T, Pandita TK. Involvement of human MOF in ATM function. Mol Cell Biol. 2005;25:5292–5305. doi: 10.1128/MCB.25.12.5292-5305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Tokuchi Y, Hayashi M, Kobayashi Y, Nishida K, Hayashi S, Ishikawa Y, Tsuchiya S, Nakagawa K, Hayashi J, Tsuchiya E. p53 null mutations undetected by immunohistochemical staining predict a poor outcome with early-stage non-small cell lung carcinomas. Cancer Res. 1999;59:5572–5577. [PubMed] [Google Scholar]
- Hayes VM, Dirven CM, Dam A, Verlind E, Molenaar WM, Mooij JJ, Hofstra RM, Buys CH. High frequency of TP53 mutations in juvenile pilocytic astrocytomas indicates role of TP53 in the development of these tumors. Brain Pathol. 1999;9:463–467. doi: 10.1111/j.1750-3639.1999.tb00535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000;102:463–473. doi: 10.1016/s0092-8674(00)00051-9. [DOI] [PubMed] [Google Scholar]
- Jack MT, Woo RA, Hirao A, Cheung A, Mak TW, Lee PW. Chk2 is dispensable for p53-mediated G1 arrest but is required for a latent p53-mediated apoptotic response. Proc Natl Acad Sci U S A. 2002;99:9825–9829. doi: 10.1073/pnas.152053599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- Kim JH, Kim B, Cai L, Choi HJ, Ohgi KA, Tran C, Chen C, Chung CH, Huber O, Rose DW, et al. Transcriptional regulation of a metastasis suppressor gene by Tip60 and beta-catenin complexes. Nature. 2005;434:921–926. doi: 10.1038/nature03452. [DOI] [PubMed] [Google Scholar]
- Kitayner M, Rozenberg H, Kessler N, Rabinovich D, Shaulov L, Haran TE, Shakked Z. Structural basis of DNA recognition by p53 tetramers. Mol Cell. 2006;22:741–753. doi: 10.1016/j.molcel.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Knights CD, Catania J, Di Giovanni S, Muratoglu S, Perez R, Swartzbeck A, Quong AA, Zhang X, Beerman T, Pestell RG, Avantaggiati ML. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J Cell Biol. 2006;173:533–544. doi: 10.1083/jcb.200512059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates JR, 3rd, Abmayr SM, Washburn MP, Workman JL. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science. 2004;306:2084–2087. doi: 10.1126/science.1103455. [DOI] [PubMed] [Google Scholar]
- Legube G, Linares LK, Lemercier C, Scheffner M, Khochbin S, Trouche D. Tip60 is targeted to proteasome-mediated degradation by Mdm2 and accumulates after UV irradiation. Embo J. 2002;21:1704–1712. doi: 10.1093/emboj/21.7.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legube G, Linares LK, Tyteca S, Caron C, Scheffner M, Chevillard-Briet M, Trouche D. Role of the histone acetyl transferase Tip60 in the p53 pathway. J Biol Chem. 2004;279:44825–44833. doi: 10.1074/jbc.M407478200. [DOI] [PubMed] [Google Scholar]
- Leitao MM, Soslow RA, Baergen RN, Olvera N, Arroyo C, Boyd J. Mutation and expression of the TP53 gene in early stage epithelial ovarian carcinoma. Gynecol Oncol. 2004;93:301–306. doi: 10.1016/j.ygyno.2004.01.043. [DOI] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Lo WS, Trievel RC, Rojas JR, Duggan L, Hsu JY, Allis CD, Marmorstein R, Berger SL. Phosphorylation of serine 10 in histone H3 is functionally linked in vitro and in vivo to Gcn5-mediated acetylation at lysine 14. Mol Cell. 2000;5:917–926. doi: 10.1016/s1097-2765(00)80257-9. [DOI] [PubMed] [Google Scholar]
- Meyers FJ, Chi SG, Fishman JR, deVere White RW, Gumerlock PH. p53 mutations in benign prostatic hyperplasia. J Natl Cancer Inst. 1993;85:1856–1858. doi: 10.1093/jnci/85.22.1856. [DOI] [PubMed] [Google Scholar]
- Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, Taya Y. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 2000;102:849–862. doi: 10.1016/s0092-8674(00)00073-8. [DOI] [PubMed] [Google Scholar]
- Patel JH, McMahon SB. Targeting of MIZ-1 is essential for MYC mediated apoptosis. J Biol Chem. 2006;281:3283–3289. doi: 10.1074/jbc.M513038200. [DOI] [PubMed] [Google Scholar]
- Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998;12:2831–2841. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels-Lev Y, O'Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, Campargue I, Naumovski L, Crook T, Lu X. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell. 2001;8:781–794. doi: 10.1016/s1097-2765(01)00367-7. [DOI] [PubMed] [Google Scholar]
- Squatrito M, Gorrini C, Amati B. Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol. 2006;16:433–442. doi: 10.1016/j.tcb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A. 2005;102:13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ZW, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002;418:104–108. doi: 10.1038/nature00883. [DOI] [PubMed] [Google Scholar]
- Taipale M, Rea S, Richter K, Vilar A, Lichter P, Imhof A, Akhtar A. hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol Cell Biol. 2005;25:6798–6810. doi: 10.1128/MCB.25.15.6798-6810.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson T, Tovar C, Yang H, Carvajal D, Vu BT, Xu Q, Wahl GM, Heimbrook DC, Vassilev LT. Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J Biol Chem. 2004;279:53015–53022. doi: 10.1074/jbc.M410233200. [DOI] [PubMed] [Google Scholar]
- Tyteca S, Vandromme M, Legube G, Chevillard-Briet M, Trouche D. Tip60 and p400 are both required for UV-induced apoptosis but play antagonistic roles in cell cycle progression. Embo J. 2006;25:1680–1689. doi: 10.1038/sj.emboj.7601066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci U S A. 2003;100:1931–1936. doi: 10.1073/pnas.2627984100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science. 2000;290:989–992. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]
- Zhang XY, DeSalle LM, Patel JH, Capobianco AJ, Yu D, Thomas-Tikhonenko A, McMahon SB. Metastasis-associated protein 1 (MTA1) is an essential downstream effector of the c-MYC oncoprotein. Proc Natl Acad Sci U S A. 2005;102:13968–13973. doi: 10.1073/pnas.0502330102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Chai X, Johnston K, Clements A, Marmorstein R. Crystal structure of the mouse p53 core DNA-binding domain at 2.7 A resolution. J Biol Chem. 2001;276:12120–12127. doi: 10.1074/jbc.M011644200. [DOI] [PubMed] [Google Scholar]
- Zupnick AE, Prives C. Mutational analysis of the p53 core domain L1 loop. J Biol Chem. 2006 doi: 10.1074/jbc.M603387200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.