

Abstract
Recent studies have identified a subpopulation of highly tumorigenic cells with stem/progenitor cell properties from human breast cancers, and it has been suggested that stem/progenitor cells, which remain after breast cancer therapy, may give rise to recurrent disease. We hypothesized that progenitor cells are resistant to radiation, a component of conventional breast cancer therapy, and that that resistance is mediated at least in part by Wnt signaling, which has been implicated in stem cell survival. To test this hypothesis, we investigated radioresistance by treating primary BALB/c mouse mammary epithelial cells with clinically relevant doses of radiation and found enrichment in normal progenitor cells (stem cell antigen 1-positive and side population progenitors). Radiation selectively enriched for progenitors in mammary epithelial cells isolated from transgenic mice with activated Wnt/β-catenin signaling but not for background-matched controls, and irradiated stem cell antigen 1-positive cells had a selective increase in active β-catenin and survivin expression compared with stem cell antigen 1-negative cells. In clonogenic assays, colony formation in the stem cell antigen 1-positive progenitors was unaffected by clinically relevant doses of radiation. Radiation also induced enrichment of side population progenitors in the human breast cancer cell line MCF-7. These data demonstrate that, compared with differentiated cells, progenitor cells have different cell survival properties that may facilitate the development of targeted antiprogenitor cell therapies.
Keywords: stem cell antigen 1, survivin, MCF-7, lin−CD24+CD29+, side population
It has been speculated that stem cells may represent the cellular origins of cancer because they exist quiescently for long periods of time and could accumulate multiple mutations over the lifespan of an organism, ultimately giving rise to tumors when stimulated to proliferate (1). Recently, it was reported that highly tumorigenic cells with properties consistent with those of stem/progenitor cells can be isolated from human breast cancers (2). These data suggest that cancer stem cells may exist in human breast cancer and that they may have different biologic features than other, more differentiated cells that constitute the majority of the cells in human breast cancers. Conceivably, cancer stem cells may be more resistant to conventional breast cancer therapies, which may ultimately result in recurrence or metastasis even when remarkable initial responses are observed clinically (3).
Although the elucidation of sensitive stem and progenitor cell markers in the mammary gland and in breast tumors has been relatively recent, the normal tissue response of stem and progenitor cells to radiation, an integral component of multidisciplinary breast cancer therapy, has been the subject of several decades of radiobiological data. The development of a simple, reproducible in vivo clonogenic assay in the jejunum, where the effects of radiation on stem cells can be easily measured, led to the conclusions that, in the small intestine, the cells at position 4–5 in the crypt now identified as the stem cells are exquisitely sensitive to radiation (4). However, a second population of potential stem cells, elsewhere called transiently amplifying cells or progenitors, exists that can be called into action in the event of lethal damage to the stem cells. At low doses of radiation the crypt of the small intestine contains four to five clonogenic regenerating cells, but at higher doses of radiation up to 30–40 potential clonogenic regenerators can be called into play (4). The elucidation of stem/progenitor cell markers in the mammary gland now allow this work to be tested at the cellular level in the mammary gland. We hypothesize that mammary gland progenitors may be resistant to radiation and that this resistance is mediated by the β-catenin stem cell survival signaling pathway.
β-Catenin is an essential component of both intercellular junctions and the canonical Wnt signaling pathway, which has been implicated in stem cell survival (5). In recent studies, activation of β-catenin in granulocyte–macrophage progenitors in chronic myelogenous leukemia appeared to enhance their self-renewal activity and leukemic potential (3). In addition, studies in the intestine and mammary gland have linked β-catenin signaling to stem cell survival and tumorigenesis (6–8).
Several methods are currently being used to isolate and study mammary stem/progenitor cells, including long-term bromodeoxyuridine labeling to identify label-retaining cells, Hoechst dye efflux to identify side population (SP) properties, and the potential stem/progenitor cell–cell surface markers, such as stem cell antigen 1 (Sca1) and α6- and β1-integrins (9–11). In the hematopoietic system, cells that efflux Hoechst 33342 dye have been shown to comprise a small fraction of bone marrow, which are capable of recapitulating the bone marrow in irradiated mice, establishing their functional capacity as hematopoietic stem cells (12). These cells are represented as a SP on flow cytometry analysis present in both mouse and human mammary glands (13–16). In hematopoietic stem cells, the efflux of Hoechst dye is due to the presence of a family of drug-effluxing protein pumps, including the breast cancer resistance protein-1 (BCRP1)/ABCG2 transporter, that may be responsible for drug resistance in many types of cancer (17). In the mouse mammary gland, the SP phenotype depends on several members of the ABC transporter family (18). Outgrowth experiments using SP are confounded by the toxicity of the Hoechst dye (14); however, the SP fraction in mouse mammary gland is enriched for long-term bromodeoxyuridine label-retaining cells, as well as Sca1-positive cells, which have the capacity to generate functional mammary outgrowths in transplantation experiments (13). Shackleton et al. (10) recently confirmed outgrowth potential of a Sca1+ population but demonstrated that the majority of outgrowth potential of the Sca1+ population in fact lies in the small, Sca1lo-positive cells. Taken together, these data support the SP and Sca1+ phenotypes as useful surrogates for stem-like/progenitor cells. A thoughtful review of the mammary SP phenotype was published recently (19), and review of the literature regarding both SP and Sca1 suggests that each of these represents markers useful for isolating potential downstream progenitors (9) rather than the more primitive stem cells isolated as described by Shackleton et al. (10) as lin−CD24+CD29+.
Although it has been speculated that stem or progenitor cells in the mammary gland are more resistant to conventional cancer therapies, this relationship has not been explicitly demonstrated. Here we demonstrate that progenitors in murine mammary epithelial cell (MEC) culture are enriched by clinically relevant doses of ionizing radiation and that this enrichment is enhanced by β-catenin stabilization. In human breast cancer MCF-7 cells, radiation induced enrichment of both SP progenitors and a lin−CD24+CD29+ subpopulation. These findings from normal and genetically manipulated mouse mammary glands may have important implications for the evaluation of new and current cancer therapies and the development of future new anti-stem-cell-targeted therapies.
Results
Primary MEC Progenitor Cells Are Radioresistant.
To test our hypothesis that progenitor cells in the mammary gland are resistant to radiation compared with the nonprogenitors, cultured primary MECs from BALB/c mice were irradiated and the percentage of SP progenitor cells in the total population after treatment was analyzed by flow cytometry. MECs were isolated, cultured for 72 h, and irradiated 24 h before analysis. Irradiation of BALB/c MECs lead to a 4-fold increase in the percent SP (%SP) cells at 2 Gy to a nearly 6-fold increase at 4 Gy. The increase in %SP decreased at 6 Gy but was still >3-fold higher than baseline (Fig. 1A). Irradiation of sorted NSP cells does not lead to SP cells, suggesting that this increase in the SP fraction is not a result of radiation induction of breast cancer resistance protein (BCRP) in MECs (data not shown). To further explore the potential clinical relevance of these findings we examined the human breast cancer cell line MCF-7, which has been reported previously to contain a subpopulation with stem/progenitor characteristics (20, 21). The %SP in MCF-7 cells was significantly increased by radiation (0.08 at 0 Gy vs. 0.19 at 4 Gy; P = 0.05) (Fig. 1B).
Fig. 1.

Clinically relevant doses of radiation increased the percentage of progenitor cells (%SP and Sca1+) in primary MEC culture and human MCF-7 cells. (A) MECs were isolated from BALB/c mice, cultured for 3 days, irradiated, and analyzed for %SP by Hoechst 33342 staining and flow cytometry. Radiation selectively increased the progenitor fraction (%SP) (P = 0.015 for 2 Gy, 0.008 for 4 Gy, and 0.05 for 6 Gy by the two-tailed t test). (B) MCF-7 cells were analyzed for %SP by Hoechst 33342 staining and flow cytometry. Radiation selectively increased the progenitor fraction (%SP) (P = 0.05 for 0 Gy vs. 4 Gy by the two-tailed t test). (C) Cells were analyzed for Sca1 in the SP 24 h after irradiation. Radiation selectively increased the Sca1+ (progenitor) fraction within the SP by killing the more sensitive Sca1− (nonprogenitor) cells (P < 0.05 for Sca1+ to Sca1− at 0 Gy vs. 2–8 Gy). The differences in effects of doses of 2 Gy vs. higher doses were not significant. (D) Anesthetized BALB/c mice were immobilized supine, and mammary glands (entire ventral surface) were irradiated. MECs were isolated 48 h after irradiation and analyzed immediately for Sca1 by flow cytometry. Radiation selectively increased the Sca1+ (progenitor) fraction and decreased the Sca1− (nonprogenitor) cells. ∗, P < 0.0001.
Welm et al. (13) showed that the SP population is enriched for Sca1+ cells and that Sca1+ progenitors give rise to outgrowths when transplanted. In our study, the percentage of Sca1+ cells within the SP increased with radiation whereas the percentage of Sca1− cells was selectively decreased with radiation (Fig. 1C). Stingl et al. (11) recently reported very high levels of Sca1+ cells after culture of primary MECs similar to the methods used in this study and as opposed to analysis of freshly isolated MECs. To evaluate the impact of culturing, we radiated the mammary glands of anesthetized BALB/c mice in vivo, dissected the glands, and performed Sca1 analysis on freshly dissociated MECs (Fig. 1D). In vivo radiation (4 Gy) significantly decreased the percentage of Sca1− cells (88% at 0 Gy vs. 70% at 4 Gy; P < 0.0001) and increased the percentage of Sca1+ cells (12% vs. 30%; P < 0.0001). The percentage of Sca1+ cells after fresh digestion ranged from 10% to 25% and consistently increased ≈3-fold after 4 Gy (n = 3).
The CD24+CD29+ population recently characterized for its ability to give rise to mammary outgrowths from a single cell (10) was examined in parallel by using freshly isolated MECs. Using freshly isolated MECs, we observed a level of CD24+CD29+ cells similar to that published by Shackleton et al. (10) (< 10%). In vivo radiation (4 Gy) did not enrich for this stem cell population and in fact decreased this population by approximately one-third (lin−CD24+CD29+ 12.5% at 0 Gy vs. 8% at 4 Gy; P = 0.01) (Fig. 2A). Radiation decreased the brightest double positive cells in this population by approximately two-thirds (P = 0.002) (Fig. 2A). For comparison, the radiation resistance of this population was also examined in MCF-7 cells. Radiation dramatically increased the lin−CD24+CD29+ population in MCF-7 cells (34% at 0 Gy vs. 53% at 2 Gy and 71% at 4 Gy; P = 0.003 for 0 Gy vs. 2 Gy and P = 0.0002 for 0 Gy vs. 4 Gy) (Fig. 2B). Both the CD24+CD29lo and double negative populations were significantly diminished after irradiation.
Fig. 2.

In vivo radiation increased the percentage of CD24+CD29+ positive cells from MCF-7 cells but not uncultured MECs. (A) Freshly digested MECs were analyzed for lin−CD24+CD29+ 48 h after in vivo irradiation. The CD24+CD29+ population is sensitive to radiation. (B) MCF-7 cells were irradiated and analyzed for lin−CD24+CD29+ by flow cytometry. Radiation selectively decreased the lin−CD24+CD29lo fraction cells (P = 0.003 for 0 Gy vs. 2 Gy, and P = 0.0002 for 0 Gy vs. 4 Gy).
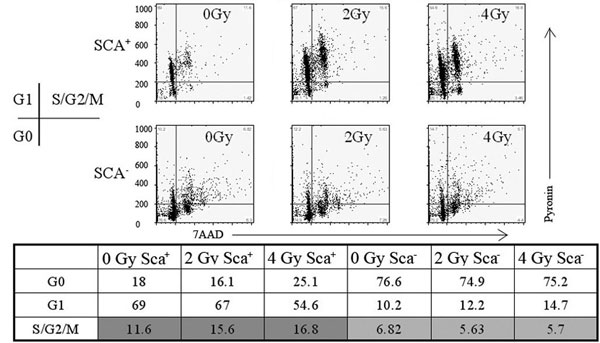
Because radiation cell killing is generally attributed to mitotic cell death, clonogenic assays to assess the replicative competence of radiated subpopulations were performed [see supporting information (SI) Methods]. Sca1− cells generally failed to form colonies when sorted into Matrigel and were allowed to grow for 2 weeks in standard radiation clonogenic assays, whereas Sca1+ cells readily formed colonies. Irradiating cells (2 Gy) before clonogenic assays yielded no reduction in the number of clonogens (SI Fig. 5). To substantiate that the Sca1+ cells are cycling, we performed cell cycle analysis after radiation treatment (SI Methods). Primary BALB/c MECs were sorted into Sca1+ and Sca1− populations and stained with 7-amino-actinomycin D and pyronin Y to distinguish between G0 and G1 (SI Fig. 6). At baseline both populations contain a significant cycling population (Sca1+, 11.6%; Sca1−, 6.82%) but inversely related G0 and G1 populations (Sca1+, G0 = 18% and G1 = 69%; Sca1−, G0 = 76.6% and G1 = 12.2%). Although Sca1− cells exhibited no redistribution in response to radiation treatment, both the G0 and S/G2/M populations among Sca1+ cells increased after radiation treatment.
Because double-strand DNA breaks lead to lethal radiation damage more often than single-strand breaks (22), we examined radiation-induced DNA double-strand breaks using immunofluorescent staining with a γ-phospho-H2AX antibody. This antibody binds to DNA flanking the double-strand breaks, creating discrete foci. Phosphorylation of the histone variant H2AX at the site of DNA damage occurs rapidly after ionizing radiation, and the formation of DNA damage foci is ATM-dependent (23). Two hours after radiation treatment, significantly more Sca1− cells contained foci, and there were more foci per damaged cell in Sca1− cells (Fig. 3).
Fig. 3.

Radiation induced more DNA damage foci in Sca1− cells 2 h after irradiation. Sca1+ and Sca1− cells from BALB/c MECs were sorted onto glass slides after irradiation with 2 Gy and immunostained with anti-phospho-H2AX. (Scale bar: 10 μm.) There were significantly more DNA-damaged foci in the Sca1− population than in the Sca1+ population (3.7-fold difference, ∗∗, P < 0.05).
Wnt/β-Catenin Signaling Mediates Progenitor Cell Resistance.
To determine the role of the putative mammary stem cell survival factor Wnt/β-catenin in mediating radioresistance of SP cells, MECs were isolated from mouse mammary tumor virus (MMTV)-driven Wnt-1 transgenic mice at 10–12 weeks of age and expanded in tissue culture for 72 h. Cells were then irradiated and stained with Hoechst 33342 for SP analysis 24 h later. MECs isolated from Wnt-1-induced hyperplasias exhibited a trend toward higher radiation-induced increase in %SP than MECs isolated from mice of a matched background (P = 0.08) (Fig. 4A). Consistent with data reported by Liu et al. (24), the %SP in MECs from Wnt-induced hyperplasias was significantly higher (>2-fold) than those in background-matched controls (0.75 vs. 0.32, respectively; P < 0.05). The radiation-induced increase in the %SP in the wild-type FVB mice was less remarkable than that observed in MECs from BALB/c mice, consistent with previous studies demonstrating marked differences in radiation response between mouse strains including increased radiation-induced genomic instability and increased susceptibility to radiation-induced mammary epithelial tumors in BALB/c mice because of a functional polymorphism (25, 26).
Fig. 4.

Clinically relevant doses of radiation led to an increased percentage of SP cells in primary mouse MECs isolated from mice with a gain-of-function, conditionally stabilized β-catenin allele and from Wnt-1 transgenic mice compared with in control cells. (A) MECs from Wnt-1 transgenic mice at 16 weeks of age and wild-type mice of the same background were stained with Hoechst 33342, and the %SP was analyzed by using flow cytometry as described. ∗, P = 0.08 for 0 Gy Wnt vs. 2 Gy Wnt, P = 0.001 for 0 Gy wild type vs. Wnt, and P = 0.04 for 2 Gy wild type vs. Wnt by two-tailed t test. MECs from mice treated with AdCre recombinase to generate stabilized β-catenin or an AdLacZ control vector were stained with Hoechst 33342, and the %SP was analyzed by using flow cytometry as described (P < 0.05 for 0 Gy vs. 2 Gy, and P < 0.05 for 0 Gy vs. 4 Gy). Radiation selectively activated β-catenin and survivin in Sca1+ cells. (B) Quantitative assessment of activated β-catenin signaling was assessed by flow cytometry after staining for Sca1 and unphosphorylated β-catenin. Real-time PCR for survivin expression was performed 24 h after irradiation in Sca1+ and Sca1− cells.
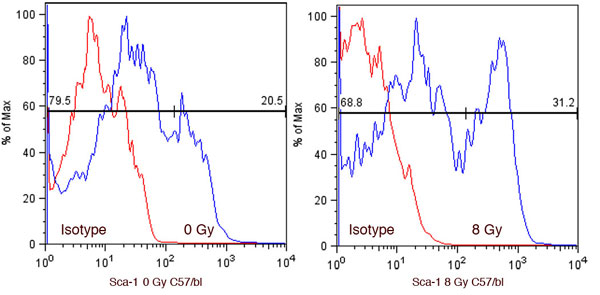
The effect of increased Wnt signaling on SP cell radioresistance was also determined by using MECs from C57BL/6 transgenic mice that contained a floxed allele of β-catenin exon III (27) isolated and transduced with an adenovirus encoding Cre recombinase (AdCre) or a comparable titer of a control adenovirus encoding β-galactosidase (AdLacZ) on day 3 of culture. Cells were irradiated on day 4 and stained with Hoechst 33342 for SP analysis 24 h later. PCR was used to demonstrate efficient recombination (>75%) in the primary MEC cultures transduced with AdCre (data not shown). Primary MECs containing stabilized β-catenin exhibited a higher radiation-induced increase in %SP than AdLacZ-treated controls (P < 0.05 for 0 Gy vs. 2 Gy, and P < 0.05 for 0 Gy vs. 4 Gy) (Fig. 4A). The lack of radiation-induced change in %SP in the C57BL/6 MECs compared with the BALB/c MECs at low doses may be due to strain differences in mice. Modest but nonstatistical increases in SP are appreciable in untreated controls at 4 Gy (data not shown), whereas a statistically significant increase after radiation is seen for Sca1+ cells in C57BL/6 mice but not at doses <8 Gy (SI Fig. 7).
We examined the role of Wnt/β-catenin signaling in response to radiation in wild-type MECs from BALB/c mice with flow cytometry on fixed cells. Staining with the anti-nonphospho-β-catenin-phycoerythrin (PE) antibody that binds to activated β-catenin showed that β-catenin is selectively activated in Sca1+ cells in response to radiation, whereas β-cat staining in Sca1− cells is unchanged in response to radiation (Fig. 4B). Survivin, a bifunctional member of the inhibitor of apoptosis gene family, has been shown to be up-regulated by TCF/β-catenin in intestinal progenitor cells upon UV-B irradiation (28). In addition, survivin has been shown to play an essential role in mitosis, in both the segregation of sister chromatids and the assembly stabilization of microtubules in late mitosis (29). This finding suggests that overexpression of β-catenin may enhance cell survival on radiation treatment at least in part by regulating survivin. Using real-time PCR, we demonstrated that survivin mRNA expression was selectively enhanced in Sca1+ cells in response to radiation (P = 0.01) (Fig. 4B). Because lin−CD24+CD29+ MCF-7 cells are enriched after radiation, we examined the expression of activated β-catenin and γ-phospho-H2AX in mammospheres derived from lin−CD24+CD29+ and lin−CD24−CD29− MCF-7 cells. Distinct patterns of activated β-catenin staining are observed in cells from double positive mammospheres, and discrete γ-phospho-H2AX foci resolve more rapidly in double positive mammospheres (SI Fig. 8).
Discussion
This study demonstrates that progenitor cells in the mammary gland are more resistant to clinically relevant doses of radiation than are nonprogenitors, which constitute the bulk of the mammary gland, and that overexpression of the Wnt/β-catenin pathway can enhance the radioresistance of progenitor cells. In wild-type mice, this effect varies by mouse strain and is most pronounced in the radiosensitive BALB/c strain, where enrichment of the progenitor population is statistically significant at 2 Gy. These experiments also demonstrate that radiation resistance in primary MECs can be altered through manipulation of the Wnt/β-catenin stem cell survival pathway. Understanding the mechanisms of resistance in normal MECs is an important step toward designing novel therapies for tumor progenitor cells. Indeed, Bao et al. (30) have shown recently that human glioma stem/progenitors demonstrate chk1-mediated resistance to radiation treatment.
Hematopoietic SP cells have been shown to possess “stemness” through their ability to recapitulate bone marrow (12). SP cells in the mammary gland represent a heterogeneous population (19) but are enriched for long-term bromodeoxyuridine label-retaining cells as well as cells positive for Sca1, a putative stem cell marker, which are capable of producing mammary outgrowths after transplantation into cleared murine mammary fat pads (13). Sca1 cells from the BALB/c-derived Comma-Dβ cell line have been characterized by Deugnier et al. (31) and were similarly found to generate in vivo outgrowths whereas Sca1lo cells exhibited decreased outgrowth potential. They reported that Sca1+ cells stain for the putative markers phenotypes reported to represent primary mouse stem cells (CD24 and α6-integrin) (11) and breast tumor progenitors (CD44) (2).
Selection and identification of putative progenitor markers has been complicated, and few comparisons have been made between different laboratories studying these phenotypes. In contrast to findings reported here and previously from our laboratory regarding the outgrowth potential of Sca1+ cells (13), Shackleton et al. (10) report that mammary gland repopulating units are best characterized by CD24+Sca1loCD29+ and that Sca1hi cells did not display outgrowth potential in vivo. The population included in the Sca1lo gate described in the Stingl et al. article (11) would typically be included in the Sca1+ population isolated either by magnetic bead sorting or from Sca-EGFP knockin mice by FACS sorting for EGFP used previously and here that have generated outgrowths in vivo in our hands. Indeed, Shackleton et al. (10) report a 3-fold enrichment in outgrowth potential among Sca1lo vs. Sca1hi populations. This suggests that the critical Sca1 population is Sca1lo rather than Sca1hi. Using magnetic bead sorting to distinguish Sca1+ and Sca1− isolated from the COMMA-D cell line Deugnier et al. (31) also reported recently that Sca1+ cells also display increased outgrowth potential. Similar discrepancies exist regarding the reported outgrowth potential of CD24hi cells (10, 11) vs. CD24lo cells (32), and it is likely that differences in antibodies and staining protocol account for these apparent discrepancies.
Stingl et al. (11) also report that culturing primary MECs leads to 100% Sca1 positivity, which has not been observed in our studies. Stingl et al. (11) use a longer digestion process (8 h vs. 1 h) with different enzymatic solutions, including cholera toxin, which may select for a somewhat different population of cells and impact the outgrowth numbers. To rule out the possibility that culturing the cells confounds the results, we analyzed freshly isolated primary MECs after in vivo irradiation and demonstrated that 4 Gy increases the Sca1+ population. These data suggest that, although differences in isolation protocols may have significant impact on absolute marker percentages, this may have less impact on relative difference between samples.
Building on our experience with putative progenitors using established protocols, we used both the SP phenotype and Sca1 as surrogate progenitor cell markers and the standard daily dose of radiation that would be delivered during a course of radiation therapy for breast cancer, i.e., 2 Gy, and found that the percentages of both SP and Sca1+ cells increased in primary BALB/c MECs. At higher doses of radiation, the percentage of progenitor cells declined, suggesting that 6 Gy is sufficient to kill both progenitor and nonprogenitor cells. Our findings demonstrate that, almost exclusively, Sca1+ cells give rise to colonies in Matrigel, and clinically relevant doses of radiation failed to reduce the number of colonies formed by Sca1+ cells. Of interest is the observation that, in preliminary experiments using MECs from either mammary tumors from p53-null mice or mammary hyperplasias from Wnt-1 transgenic mice, Sca1− cells were capable of limited colony formation that is not diminished by 2–4 Gy of radiation. (Mei Zhang and J.M.R., unpublished data).
These data are consistent with observations regarding stem cell irradiation in the small intestine summarized eloquently by Christopher Potten (4). Previous findings from our laboratory and others suggested that, although the SP and Sca1+ population may contain lineage ancestor stem cells, most SP and Sca1+ populations represent progenitors (9, 14, 31): i.e., potential stem cells or transiently amplified cells that may be more resistant to radiation than the lineage ancestor stem cells. Similar to the findings in the intestine, the single-cell stem cell phenotype in the normal mouse mammary gland, CD24+CD29+, is sensitive to clinically relevant doses of radiation. Interestingly, a similar subpopulation in human breast tumor MCF-7 cells is enriched after irradiation, highlighting a potential difference between normal and tumor stem cells. However, further studies are required to determine whether this subpopulation of MCF-7 cells will actually exhibit increased tumorigenicity in xenografts. The functional importance of this subpopulation in MCF-7 cells has yet to be demonstrated.
β-Catenin has been implicated as a stem cell survival factor in several systems including neural crest cells, gastrointestinal crypts, epidermal follicles, and hematopoietic stem cells (33–37). Inhibition of β-catenin signaling in mammary alveolar progenitors leads to the inhibition of mammary development and pregnancy-induced proliferation, implicating β-catenin as a stem cell survival factor in the mammary gland (38). In addition, it has been shown that the SP-enriched fraction is increased in the mammary gland of MMTV-Wnt-1 and MMTV-ΔNβ-catenin transgenic mice and that ectopic Wnt ligands increase the SP fraction in MECs after 3 days in culture (24). A recent study reported the initial development of a small-molecule Wnt/β-catenin pathway inhibitor that down-regulates β-catenin/TCF-mediated gene expression through its interaction with the cAMP-responsive element binding protein (39). This molecule, ICG-001, has been shown to inhibit β-catenin/TCF-mediated transcription of survivin, which has been shown to be up-regulated in many cancers. Survivin is regulated by the gene product of the adenomatous polyposis coli gene in intestinal crypts, where it may limit the stem cell population on the basis of its localization and regulation (40). We showed that survivin is selectively up-regulated by radiation in Sca1+ cells. Because apoptosis is a minor component of radiation-induced cell death in solid tumors at low doses (22), this finding potentially suggests a nonapoptosis-related role for survivin in these cells. Indeed, such a role has recently been described in colon cancer cell lines where survivin was shown to assist cancer cells in escaping replicative senescence by enhancing telomerase activity (41).
In conclusion, this study demonstrates radioresistance of progenitor cells in the mouse mammary gland. On the basis of our findings and of the studies presented here, we suggest that the Wnt/β-catenin signaling pathway may be an attractive target for directed anti-stem cell therapeutics. Although β-catenin is not commonly mutated in human breast cancers, several studies have implicated components of the Wnt signaling pathway in human breast cancer pathogenesis and prognosis (42–45), and Ayyanan et al. (46) have recently reported a direct link between Wnt-1 signaling and the DNA damage response primary human epithelial cells. These studies underscore the potential for treatment strategies that target pathways such as Wnt/β-catenin that are responsible for self-renewal (47).
Materials and Methods
Cell Culture.
All animals used were used in accordance with an Institutional Review Board-approved protocol and killed before gland collection. MECs were isolated from 6- to 8-week-old wild-type BALB/c mice and from C57BL/6 mice (Harlan, Indianapolis, IN) containing a floxed exon III β-catenin allele (CatnbloxP(ex3); Makoto Taketo, Kyoto University, Kyoto, Japan). The MECs generated stabilized β-catenin on excision in culture with an adenovirus-driven Cre recombinase, AdCre1 (48), at a multiplicity of infection of 50, as determined by an adenovirus-expressing Escherichia coli β-galactosidase (Adβ-gal) (AdCre1 and Adβ-gal; M. Abdelative and M. Schneider, Baylor College of Medicine). In addition, MECs were isolated from transgenic Wnt-1 mice with mammary hyperplasias (Yi Li, Baylor College of Medicine) and from wild-type MECs from mice of the same genetic background as the Wnt-1 transgenic mice. All 10 glands were isolated. For primary MEC culture (Figs. 1 A and C, 2, and 3), the epithelial cell fraction was isolated as described previously (49). Primary MECs were plated at a density of 2.5 × 105 cells per cm2 in six-well plates that had been coated with 100 μl/cm2 serum/fetuin (20% FCS; Summit Biotechnology, Fort Collins, CO) and 1 mg/ml fetuin (Sigma, St. Louis, MO). Cells were left for 2 days in F12 plating medium (5 μg/ml insulin, 2 μg/ml hydrocortisone, 5 ng/ml epidermal growth factor, 50 μg/ml gentamycin, 100 units penicillin/streptomycin, and 10% FCS) (GIBCO/BRL, Grand Island, NY). After 48 h in the plating medium, MECs were maintained in stem cell-promoting neurobasal medium (GIBCO/BRL) containing 4 μg/ml B-27 supplement (GIBCO/BRL), 20 ng/ml basic fibroblast growth factor (Invitrogen, Carlsbad, CA), 20 ng/ml insulin-like growth factor I (Invitrogen), and 20 ng/ml epidermal growth factor (Invitrogen). Cells were irradiated on day 4. Primary MECs were irradiated in 60-mm culture dishes by using a 137cesium cell irradiator at doses of 2, 4, 6, or 8 Gy. Control cells were sham-irradiated. On day 5, cells were trypsinized (10×) (JRH Biosciences, Lenexa, KS) for 3 min; washed in Hanks' balanced salt solution (HBSS) (GIBCO/BRL), 2% FBS, and 10 mM Hepes (HBSS+); and stained with Hoechst 33342 (Sigma) at a final concentration of 5–15 μg/ml for 60 min (12) before being analyzed by flow cytometry.
Fresh digestion was performed by using the following protocol: in vivo irradiation (sham or 4 Gy) (Fig. 1 D and E) was performed 48 h before isolation of MECs. After administration of 0.2 ml of 1.2% Avertin, mice were immobilized in the supine position to allow uniform dosimetry to the glands by using a small animal 137cesium irradiator. Glands were isolated from 7- to 10-week-old BALB/c female mice and minced into small pieces. The minced tissue was digested with 2 mg/ml collagenase (Roche, Indianapolis, IN) and 300 units/ml hyuronidase (Sigma) in HBSS+ buffer by using 1 g of tissue per 10 ml of digestion solution at 37°C with shaking for 2 h. The tissue slurry was further digested in 0.25% trypsin-EGTA for 1 min and 5 mg/ml dispase (Roche Diagnostics) for 5 min. The suspension was washed and pelleted twice at 800 rpm (110 × g). The cell pellet was collected for antibody staining with 2 μl each of the Biotin-Conjugated Mouse Lineage Panel (BD Pharmingen, San Diego, CA) containing mouse CD3e, CD11b, CD45R, Ly-6G, and Ly-6C, and TER-199 was used to stain 106 cells in 10 μl on ice for 15 min. Streptavidin-conjugated PE-Cy5.5 (Caltag Laboratories) was used at a 1:200 dilution on ice for 15 min. CD29 (BD Pharmingen) was used at 1:200 at room temperature for 60 min. A goat anti-rat Alexa Fluor 488 secondary antibody (Molecular Probes, Carlsbad, CA) was used at a 1:200 dilution at room temperature for 30 min. CD24-PE (BD Pharmingen) was used on ice for 15 min at 1:200. MCF-7 cells were radiated on plastic 48 h before analysis and examined for SP or marker expression by flow cytometry.
Immunofluorescence.
Primary BALB/c MECs were cultured, treated, and stained with PE-conjugate Sca1 antibody (BD Pharmingen) diluted at 1:200. Sca1+ and Sca1− were sorted directly onto glass slides (ProbeOn Plus; Fisher Biotech, Hampton, NH) at 500 cells each. The sorted cells were fixed in 4% paraformaldehyde for 15 min and stained with phospho-H2AX antibody (1:200) and a secondary Texas red-conjugated anti-rabbit antibody (Molecular Probes). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole and dihydrochloride (Vector Laboratories, Burlingame, CA), and coverslips were mounted with a SlowFade light antifade kit (Molecular Probes). Images were captured at ×100 by using a Zeiss CCD camera.
Flow Cytometry.
Samples were prepared for flow cytometry by using antibodies or Hoescht 33342, resuspended in HBSS+, and filtered through a 0.45-μm cell filter into polypropylene tubes (Fisher Biotech) containing 0.5 μg/ml propidium iodide (Sigma) to exclude dead cells. Analysis and sorting were performed on a triple-laser MoFlo (Cytomation, Fort Collins, CO). The Hoechst dye was excited at 350 nm, and its fluorescence was measured at 450 nm/20 band-pass filter blue and 675 nm long-pass edge optical filter red, as described previously (12). Activated β-catenin was measured by using the anti-nonphospho-β-catenin antibody, clone 8E4 (Upstate Cell Signaling Solutions, Charlottesville, VA). Data analysis was performed with FlowJo software, version 4 (Tree Star, Ashland, OR).
Survivin PCR Methods.
The survivin primer sequences were 5′-AAGAACTACCGCATCGCCACC for survivin and 5′-AGCCAGCTCCGCCATT for survivin reverse. Cells were harvested 24 h after irradiation. SYBR green quantitative PCR was performed by using the ABI 7500 real-time PCR system (Applied Biosystems).
Statistical Analysis.
Statistical comparisons were performed by using Student's t test in Excel (Microsoft, Redmond, WA). All P values are two-sided.
Supplementary Material
Acknowledgments
We thank the Baylor College of Medicine Flow Core Laboratory and the Texas Children's Hospital Flow Laboratory for flow cytometry assistance. We acknowledge Frances Kittrell and Jessica Li for assistance. We also thank Drs. Daniel Medina and Peggy Goodell for their advice and helpful criticisms. This work is supported in part by National Cancer Institute Grant CA-16303 (to J.M.R.) and Department of Defense Grant Predoctoral Fellowship DAMD 17-03-1-0699 (to M.S.C.).
Abbreviations
- MEC
mammary epithelial cell
- SP
side population
- %SP
percent SP
- Sca1
stem cell antigen 1
- PE
phycoerythrin.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0606599104/DC1.
References
- 1.Sell S. Crit Rev Oncol Hematol. 2004;51:1–28. doi: 10.1016/j.critrevonc.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, et al. N Engl J Med. 2004;351:657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 4.Potten CS. Radiat Res. 2004;161:123–136. doi: 10.1667/rr3104. [DOI] [PubMed] [Google Scholar]
- 5.Polakis P. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 6.Theodosiou NA, Tabin CJ. Dev Biol. 2003;259:258–271. doi: 10.1016/s0012-1606(03)00185-4. [DOI] [PubMed] [Google Scholar]
- 7.Taipale J, Beachy PA. Nature. 2001;411:349–354. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Welm B, Podsypanina K, Huang S, Chamorro M, Zhang X, Rowlands T, Egeblad M, Cowin P, Werb Z, et al. Proc Natl Acad Sci USA. 2003;100:15853–15858. doi: 10.1073/pnas.2136825100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woodward WA, Chen MS, Behbod F, Rosen JM. J Cell Sci. 2005;118:3585–3594. doi: 10.1242/jcs.02532. [DOI] [PubMed] [Google Scholar]
- 10.Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, Wu L, Lindeman GJ, Visvader JE. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- 11.Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, Li HI, Eaves CJ. Nature. 2006;439:993–997. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- 12.Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. J Exp Med. 1996;183:1797–1806. doi: 10.1084/jem.183.4.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Welm BE, Tepera SB, Venezia T, Graubert TA, Rosen JM, Goodell MA. Dev Biol. 2002;245:42–56. doi: 10.1006/dbio.2002.0625. [DOI] [PubMed] [Google Scholar]
- 14.Alvi AJ, Clayton H, Joshi C, Enver T, Ashworth A, Vivanco MM, Dale TC, Smalley MJ. Breast Cancer Res. 2003;5:R1–R8. doi: 10.1186/bcr547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clayton H, Titley I, Vivanco M. Exp Cell Res. 2004;297:444–460. doi: 10.1016/j.yexcr.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 16.Clarke RB, Spence K, Anderson E, Howell A, Okano H, Potten CS. Dev Biol. 2005;277:443–456. doi: 10.1016/j.ydbio.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 17.Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA, Brenner MK. Proc Natl Acad Sci USA. 2004;101:14228–14233. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jonker JW, Freeman J, Bolscher E, Musters S, Alvi AJ, Titley I, Schinkel AH, Dale TC. Stem Cells. 2005;23:1059–1065. doi: 10.1634/stemcells.2005-0150. [DOI] [PubMed] [Google Scholar]
- 19.Smalley MJ, Clarke RB. J Mamm Gland Biol Neoplasia. 2005;10:37–47. doi: 10.1007/s10911-005-2539-0. [DOI] [PubMed] [Google Scholar]
- 20.Kondo T, Setoguchi T, Taga T. Proc Natl Acad Sci USA. 2004;101:781–786. doi: 10.1073/pnas.0307618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Cancer Res. 2005;65:6207–6219. doi: 10.1158/0008-5472.CAN-05-0592. [DOI] [PubMed] [Google Scholar]
- 22.Hall EJ. Radiobiology for the Radiologist. Philadelphia: Lippincott, Williams & Wilkins; 2000. [Google Scholar]
- 23.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. J Biol Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 24.Liu BY, McDermott SP, Khwaja SS, Alexander CM. Proc Natl Acad Sci USA. 2004;101:4158–4163. doi: 10.1073/pnas.0400699101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okayasu R, Suetomi K, Yu Y, Silver A, Bedford JS, Cox R, Ullrich RL. Cancer Res. 2000;60:4342–4345. [PubMed] [Google Scholar]
- 26.Storer JB, Mitchell TJ, Fry RJ. Radiat Res. 1988;114:331–353. [PubMed] [Google Scholar]
- 27.Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM. EMBO J. 1999;18:5931–5942. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim PJ, Plescia J, Clevers H, Fearon ER, Altieri DC. Lancet. 2003;362:205–209. doi: 10.1016/S0140-6736(03)13910-4. [DOI] [PubMed] [Google Scholar]
- 29.Yang D, Welm A, Bishop JM. Proc Natl Acad Sci USA. 2004;101:15100–15105. doi: 10.1073/pnas.0406665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 31.Deugnier MA, Faraldo MM, Teuliere J, Thiery JP, Medina D, Glukhova MA. Dev Biol. 2006;293:414–425. doi: 10.1016/j.ydbio.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 32.Sleeman KE, Kendrick H, Ashworth A, Isacke CM, Smalley MJ. Breast Cancer Res. 2006;8:R7. doi: 10.1186/bcr1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pardal R, Clarke MF, Morrison SJ. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 34.Lee HY, Kleber M, Hari L, Brault V, Suter U, Taketo MM, Kemler R, Sommer L. Science. 2004;303:1020–1023. doi: 10.1126/science.1091611. [DOI] [PubMed] [Google Scholar]
- 35.Huelsken J, Birchmeier W. Curr Opin Genet Dev. 2001;11:547–553. doi: 10.1016/s0959-437x(00)00231-8. [DOI] [PubMed] [Google Scholar]
- 36.Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL. Nature. 2003;423:409–414. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- 37.van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, et al. Cell. 2002;111:241–250. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 38.Tepera SB, McCrea PD, Rosen JM. J Cell Sci. 2003;116:1137–1149. doi: 10.1242/jcs.00334. [DOI] [PubMed] [Google Scholar]
- 39.Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, Moon RT, Teo JL, Kim HY, Moon SH, et al. Proc Natl Acad Sci USA. 2004;101:12682–12687. doi: 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim KM, Calabrese P, Tavare S, Shibata D. Am J Pathol. 2004;164:1369–1377. doi: 10.1016/S0002-9440(10)63223-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Endoh T, Tsuji N, Asanuma K, Yagihashi A, Watanabe N. Exp Cell Res. 2005;305:300–311. doi: 10.1016/j.yexcr.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 42.Wong SC, Lo SF, Lee KC, Yam JW, Chan JK, Wendy Hsiao WL. J Pathol. 2002;196:145–153. doi: 10.1002/path.1035. [DOI] [PubMed] [Google Scholar]
- 43.Klopocki E, Kristiansen G, Wild PJ, Klaman I, Castanos-Velez E, Singer G, Stohr R, Simon R, Sauter G, Leibiger H, et al. Int J Oncol. 2004;25:641–649. [PubMed] [Google Scholar]
- 44.Lin SY, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y, Pestell RG, Hung MC. Proc Natl Acad Sci USA. 2000;97:4262–4266. doi: 10.1073/pnas.060025397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, Sundberg CD, Bishop JM, Felsher DW. Science. 2002;297:102–104. doi: 10.1126/science.1071489. [DOI] [PubMed] [Google Scholar]
- 46.Ayyanan A, Civenni G, Ciarloni L, Morel C, Mueller N, Lefort K, Mandinova A, Raffoul W, Fiche M, Dotto GP, Brisken C. Proc Natl Acad Sci USA. 2006;103:3799–3804. doi: 10.1073/pnas.0600065103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Behbod F, Rosen JM. Carcinogenesis. 2005;26:703–711. doi: 10.1093/carcin/bgh293. [DOI] [PubMed] [Google Scholar]
- 48.Anton M, Graham FL. J Virol. 1995;69:4600–4606. doi: 10.1128/jvi.69.8.4600-4606.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pullan S, Wilson J, Metcalfe A, Edwards GM, Goberdhan N, Tilly J, Hickman JA, Dive C, Streuli CH. J Cell Sci. 1996;109:631–642. doi: 10.1242/jcs.109.3.631. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}