

Abstract
Background: Hypertrophic cardiomyopathy (HCM) is an inherited disease of the sarcomere characterised clinically by myocardial hypertrophy and its consequences. Phenotypic expression is heterogeneous even within families with the same aetiological mutation and may be influenced by additional genetic factors.
Objective: To determine the influence of genetic polymorphisms of the renin-angiotensin-aldosterone system (RAAS) on ECG and two dimensional echocardiographic left ventricular hypertrophy (LVH) in genetically identical patients with HCM.
Patients and methods: Polymorphisms of five RAAS components were determined in 26 gene carriers from a single family with HCM caused by a previously identified myosin binding protein C mutation. Genotypes associated with a higher activation status of the RAAS were labelled “pro-LVH genotypes”.
Results: There was a non-biased distribution of pro-LVH genotypes in the gene carriers. Those without pro-LVH genotypes did not manifest cardiac hypertrophy whereas gene carriers with pro-LVH genotypes did (mean (SD) left ventricular muscle mass 190 (48) v 320 (113), p = 0.002; interventricular septal thickness 11.5 (2.0) v 16.4 (6.7), p = 0.01; pathological ECG 0% (0 of 10) v 63% (10 of 16), respectively). Multivariate analysis controlling for age, sex, and hypertension confirmed an independent association between the presence of pro-LVH polymorphisms and left ventricular mass. When each polymorphism was assessed individually, carriers of each pro-LVH genotype had a significantly greater left ventricular mass than those with no pro-LVH mutation; these associations, with the exception of cardiac chymase A AA polymorphism (p = 0.06), remained significant in multivariate analysis.
Conclusion: Genetic polymorphisms of the RAAS influence penetrance and degree of LVH in 26 gene carriers from one family with HCM caused by a myosin binding protein C mutation.
Keywords: hypertrophic cardiomyopathy, polymorphisms, renin-angiotensin-aldosterone system
Hypertrophic cardiomyopathy (HCM) is a genetically determined myocardial disease, which is characterised by ventricular hypertrophy, arrhythmias, impaired exercise tolerance, and sudden death. Mutations in genes encoding sarcomeric proteins are the molecular basis of this disease.1–3 Aetiological mutations in eight cardiac sarcomeric protein genes have been identified, including the cardiac myosin binding protein C (MyBP-C) gene (associated with late onset disease).2 Pronounced clinical heterogeneity is a feature of HCM.1–3 Even in families affected by an identical mutation, penetrance and phenotypic expression vary considerably.3 The genetic and environmental causes of this phenotypic variability of HCM are unknown. Candidate genes are under investigation.
The renin-angiotensin-aldosterone-system (RAAS) is a plausible candidate for modifying expression of left ventricular hypertrophy (LVH) because of its regulatory role in cardiac function, blood pressure, and electrolyte homeostasis.4 In addition angiotensin II is recognised to influence cardiac myocyte hypertrophy.5 Several polymorphisms are suggested to influence hypertrophy in HCM: (1) an insertion/deletion (I/D) polymorphism in intron 16 of the gene encoding angiotensin converting enzyme (ACE) on chromosome 17q23 6–10; (2) a methionine/threonine exchange in codon 235 of the angiotensinogen gene (AGT) on chromosome 1q42 (M235T, resulting from a T/C exchange in exon 2)11, 12; (3) a polymorphic A/C in position 1166 of the angiotensin II receptor type 1 gene (AGTR1) on chromosome 3q2112, 13; (4) a C/T exchange at position −344 of the aldosterone synthase gene (CYP11B2) on chromosome 8q2214; and (5) an A/G exchange in position −1903 (5` to the transcriptional start) of the cardiac chymase A gene (CMA) on chromosome 14q11.15 The AGT CC and ACE DD polymorphisms are associated with increased gene products16, 17 and the AGTR1 CC polymorphism is associated with a higher angiotensinogen II type 1 receptor responsiveness.18, 19 The aldosterone synthase (CYP11B2) CC polymorphism has been associated with cardiac hypertrophy among healthy subjects14 and the CMA AA polymorphism has been associated with HCM.15 However, the molecular effects of these two polymorphisms are unknown.
Results of investigations of the clinical effects of these polymorphisms have been conflicting and the role of the RAAS system in modifying the phenotype in HCM remains controversial. However, such studies may be compromised by three potential confounding factors. Firstly, the major determinant of LVH in HCM is the primary aetiological gene, which in genetically heterogenous HCM populations may make the influence of RAAS polymorphisms undetectable. Secondly, the RAAS system has several components and investigation of a single polymorphism may result in a β error unless the population investigated is sufficiently large, since genetic heterogeneity in other RAAS components may mask any effects of the polymorphism being investigated. Thirdly, normal regulatory mechanisms within the RAAS may correct for changes in concentrations of one component by a counterbalancing change in the concentrations of another component.20, 21 This mechanism may function less efficiently if more than one polymorphism associated with LVH is present.
The aim of this study was to investigate the relation between RAAS gene polymorphisms and cardiac hypertrophy as assessed by echocardiography and ECG, by measuring five RAAS polymorphisms in one family affected by a single mutation in the MyBP-C gene.22
METHODS
Subjects
In the course of a clinical and genetic investigation of a family of 48 adults, 26 were shown to be gene carriers, while 22 were unaffected. Mutation analysis identified a single G nucleotide insertion in exon 24 of the MyBP-C gene. Additional data have been presented in a study investigating the functional consequences of this mutation.22 In brief, nine of 26 gene carriers fulfilled World Health Organization diagnostic criteria for HCM.23–25 Five carriers had a borderline phenotype with electrocardiographic and echocardiographic abnormalities fulfilling recently proposed diagnostic criteria for HCM within families,26 while 12 gene carriers did not fulfil the criteria for HCM.
Electrocardiography
Twelve lead ECGs were assessed for the presence of abnormalities including LVH and repolarisation changes; T wave inversion in leads I and aVL (≥ 3 mm, with QRS-T wave axis difference ≥ 30°), V3–V6 (≥ 3 mm), or II and III and aVF (≥ 5 mm); abnormal Q (> 40 ms or > 25% R wave) in at least two leads from II, III, aVF (in the absence of left anterior hemiblock), V1–V4, or I, aVl and V5–V6; minor repolarisation changes in left ventricular (LV) leads; and a deep S in V2 (> 25 mm).26, 27
Echocardiography
Two dimensional echocardiograms were recorded with a Hewlett Packard Sonos 2000 (Hewlett Packard, Palo Alto, California) using 2.25 and 2.5 MHz transducers. Transthoracic images were obtained in standard cross sectional planes. M mode echocardiograms were derived from two dimensional images under direct anatomic visualisation. The extent and localisation of LVH were assessed as previously described.23–25 LV mass was calculated by the cube function formula:
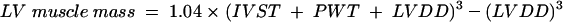 |
where LVDD is LV diastolic dimension, IVST is interventricular septal thickness, and PWT is LV posterior wall thickness.
Phenotyping
ECGs and echocardiograms were analysed by two cardiologists who were blinded to the genetic analysis.
Analysis of polymorphic genotypes
The genomic DNA of 26 gene carriers was extracted from whole blood using the QIAmp Blood Kit 250 (Quiagen Valencia, California, USA). Polymerase chain reaction of genomic DNA segments was achieved using a standard thermocycler (Hybaid PCR Express, Hybaid, Ashford, Kent). Details of the amplification protocols, oligonucleotide primer sequences, and diagnostic restriction enzymes were described previously.14–16, 28, 29 DNA sequence polymorphisms were tested in the five RAAS genes outlined above. An additional set of primers was used to evaluate the I/D polymorphism of the ACE gene, with an “insertion” sequence specific oligonucleotide to prevent misclassification of I/D as D/D.29 Alleles previously associated with LVH or with increased RAAS activity (that is, ACE DD, AGT CC, AGTR1 CC, CYP11B2 CC, CMA AA), as outlined above, were tentatively labelled “pro-LVH”. DNA extraction and genotyping were performed blinded in two different laboratories (Aachen and Bad Nauheim, Germany). Each polymorphic locus was tested in 48 adult members (26 gene carriers and 22 non-gene carriers) of the family. In addition, 100 unrelated patients (with HCM or a family history of HCM excluded) living in the same area as the study family served as a control for the assessment of allele frequencies and Hardy-Weinberg distributions of polymorphic genotypes.
Statistical analysis
Statistical analysis was performed using GraphPad InStat version 2.05 (GraphPad Software Inc, San Diego, California, USA) and SPSS for windows version 7.0 (SPSS Inc, Chicago, Illinois, USA). Continuous variables were formally tested for skewness with a one sample Kolmogorov-Smirnov test and, if normally distributed, are expressed as mean (SD). Carriers of pro-LVH genotypes were compared with non-carriers using independent samples t test for continuous variables and χ2 and Fisher's exact test as appropriate for categorical variables. Multivariate analysis was performed by general linear modelling and logistic regression analysis for continuous and categorical variables, respectively.
RESULTS
Allele and genotype frequencies
The frequencies of polymorphic alleles in the gene carriers and in the carriers and non-carriers (combined) in the study family were similar to the white control population from the same area (data not shown). The fraction of pro-LVH genotypes among gene carriers (DCP1 DD 23%, CMA AA 23%, AGT CC 23%, CYP11B2 CC 15%, AGTR1 CC 3.8%; table 1) was similar to and not more frequent than that in the control population. These data indicate normal allele frequencies in the family and a non-biased distribution of pro-LVH genotypes among gene carriers.
Table 1.
Distribution of pro-left ventricular hypertrophy (LVH) genotypes in patients with hypertrophic cardiomyopathy (HCM) and controls
| ACE | AGT | AGTR1 | CMA | CYP11B2 | |
| HCM (n=26) | |||||
| II=6 | CC=6 | CC=1 | AA=6 | CC=4 | |
| ID=14 | TC=17 | AC=10 | GG=13 | TC=14 | |
| DD=6 | TT=3 | AA=15 | AG=7 | TT=8 | |
| Allele frequency | I=0.5 | C=0.54 | C=0.22 | A=0.46 | C=0.41 |
| Prevalence of pro-LVH genotype | D=0.5 | T=0.46 | A=0.78 | G=0.54 | T=0.59 |
| DD=23% | CC=23% | CC=4% | AA=23% | CC=15% | |
| Control (n=100) | |||||
| II=28 | CC=17 | CC=9 | AA=23 | CC=23 | |
| ID=43 | TC=58 | AC=34 | GG=58 | TC=40 | |
| DD=29 | TT=25 | AA=57 | AG=19 | TT=37 | |
| Allele frequency | I=0.5 | C=0.46 | C=0.26 | A=0.52 | C=0.43 |
| D=0.5 | T=0.54 | A=0.74 | G=0.48 | T=0.57 | |
| Prevalence of pro-LVH genotype | DD=29% | CC=17% | CC=9% | AA=23% | CC=23% |
ACE, angiotensin converting enzyme gene; AGT, angiotensinogen gene; AGTR1, angiotensin II receptor type 1 gene; CMA, chymase A gene; CYP11B2, aldosterone synthase gene.
Influence of polymorphisms on expression of cardiac hypertrophy
Family members were classified according to the presence or absence of pro-LVH polymorphisms (table 2). Those with one or more pro-LVH polymorphism (n = 16) had significantly greater LV mass and LV wall thickness and a significantly increased frequency of abnormal ECG than family members with no pro-LVH polymorphisms. There was a direct relation between the number of pro-LVH polymorphisms present and the degree of LVH (table 3).
Table 2.
Influence of pro-LVH genotype on phenotype in family members with the myosin binding protein C (MyBP-C) mutation
| Pro-LVH genotype | ||||
| Variable | Yes (n=16) | No (n=10) | p Value* | p Value† |
| Sex (male/female) | 5/11 | 2/8 | ns | ns |
| Age (years) | 47 (17) | 45 (17) | ns | ns |
| IVST (mm) | 16.4 (6.7) | 11.5 (2.0) | 0.012 | 0.09 |
| LV mass (g) | 319.5 (112.8) | 189.9 (47.6) | 0.002 | 0.02 |
| Pathological ECG | 10 (63%) | 0 | 0.005 | ns |
*Univariate analysis.
†General linear model or logistic regression analysis controlling for age, sex, and the presence of hypertension.
IVST, interventricular septal thickness; LV, left ventricular; ns, not significant.
Table 3.
Left ventricular (LV) mass and interventricular septal thickness (IVST) according to the number of pro-LVH polymorphisms
| Number of polymorphisms | ||||
| 0 | 1 | ≥2 | p Value | |
| LV mass (g) | 189.9 (47.6) | 295.9 (107.2) | 390.5 (137.1) | 0.02 |
| IVST (mm) | 11.5 (1.6) | 14.7 (6.1) | 21 (7) | 0.007 |
Analysis by individual polymorphisms
Comparison of individual mutations (for example, ACE DD v ACE ID/II) showed trends for associations between LVH (assessed either echocardiographically or by ECG) and the presence of the designated pro-LVH genotype but these were significant only for AGT, CMA, and CYP11B2. In contrast, compared with a control group that had none of the designated pro-LVH mutations (n = 10), carriers of each pro-LVH polymorphism had a highly significantly greater degree of LVH (table 4).
Table 4.
Influence of renin-angiotensin-aldosterone system (RAAS) genes on phenotype in family members with the MyBP-C mutation compared with the family group with no pro-LVH polymorphisms (n=10)
| RAAS gene | Genotype | p Value analysis | LV mass (g) | IVST (mm) | Pathological ECG |
| No pro-LVH genes present* (n=10) | 189.9 (47.5) | 11.5 (1.9) | 0 | ||
| ACE (n=6) | DD | 302.8 (58.7) | 15.2 (4.8) | 3 (50%) | |
| Univariate | 0.001 | 0.049 | 0.04 | ||
| Multivariate† | 0.04 | ns | ns | ||
| AGT (n=6) | CC | 334.5 (127.4) | 18.6 (6.8) | 3 (50%) | |
| Univariate | 0.005 | 0.007 | 0.04 | ||
| Multivariate† | 0.02 | 0.04 | ns | ||
| AGTR (n=1) | CC | 547.5 | 26 | 1 (100%) | |
| Univariate | – | – | 0.09 | ||
| Multivariate† | 0.001 | 0.001 | ns | ||
| CMA (n=6) | AA | 365.6 (142.0) | 20.3 (8.4) | 4 (67%) | |
| Univariate | 0.003 | 0.006 | 0.008 | ||
| Multivariate† | 0.06 | 0.06 | ns | ||
| CYP11B2 (n=4) | CC | 323.6 (110.8) | 15.5 (6.3) | 4 (100%) | |
| Univariate | 0.007 | 0.085 | 0.001 | ||
| Multivariate† | 0.01 | 0.007 | ns | ||
*Control group for statistical comparisons.
†General linear model (LV mass, ISVT) or logistic regression analysis (ECG abnormality) controlling for age, sex, and the presence of hypertension.
Influence of age, sex, and hypertension on expression of LVH
Male patients had a significantly greater LV mass than female patients (343.3 (121.8) v 242.2 (98.3), p = 0.04). Although LV mass and wall thickness tended to be greater in patients older than 40 years, there was no significant association between age and LVH, either echocardiographically or by ECG. Eight of 26 gene carriers had previously documented systemic hypertension. Cardiac hypertrophy was not greater in hypertensive (muscle mass 235 (63) g) than in normotensive gene carriers (muscle mass 287 (129) g; p = 0.27). Since RAAS polymorphisms may influence blood pressure the genotype–phenotype relation was analysed by excluding these eight gene carriers with hypertension. The phenotypic traits in those with no (n = 6) and those with pro-LVH (n = 12) genotypes were similar to the findings observed when hypertensive subjects were included.
Analysis by pedigree
The family was divided into three major branches (fig 1). Members of branch B had significantly more ventricular hypertrophy than either of the other branches and more pro-LVH polymorphisms (table 5). There were no sex differences or differences in the frequency of hypertension between the branches to account for these differences in mass. Similarly, there were no significant differences in age between the branches, although members of branch B tended to be older. However, in multivariate analysis the association of LV mass and of IVST with each branch of the family was independent of age (table 5).
Figure 1.

Pedigree of a family with hypertrophic cardiomyopathy (HCM). In generations III to V only those confirmed to have the myosin binding protein C (MyBP-C) mutation are represented (n = 26). Filled symbols represent family members fulfilling conventional diagnostic criteria for HCM, partially filled symbols represent those fulfilling criteria for the diagnosis of HCM in the context of a family history, and open symbols represent gene carriers with no clinical features of HCM. Numbers of pro-left ventricular hypertrophy (LVH) genotypes detected in the person are shown. –, no pro-LVH renin-angiotensin-aldosterone genotype.
Table 5.
LV wall thickness and pro-LVH genotypes in each branch of the family
| A | B | C | p Value | p Value* | |
| LV mass (g) | 221.7 (113.2) | 355.7 (122.4) | 241.0 (50.3) | 0.02 | 0.04 |
| IVST (mm) | 13.1 (4.8) | 19.4 (7.2) | 11.7 (1.7) | 0.01 | 0.04 |
| Pro-LVH mutations (yes/no) | 4/5 | 8/0 | 4/5 | 0.03 | – |
| Pro-LVH mutations/person | 0.7 | 1.5 | 0.4 | 0.02† | – |
| Age (years) | 45.1 (16.3) | 54.4 (16.3) | 39.2 (15.6) | 0.17 | – |
| Sex (male/female) | 3/6 | 2/6 | 2/7 | 0.86 | – |
| Hypertension (yes/no) | 4/5 | 1/7 | 3/6 | 0.36 | – |
*General linear model controlling for age.
†Kruskal-Wallis test.
Multivariate analysis
Multivariate analysis controlling for age, sex, and the presence of hypertension confirmed a significant independent association between the presence of pro-LVH mutations and LV mass (table 2). Each pro-LVH mutation considered to be individually relative to MyBP-C gene carriers who had no pro-LVH mutations was also independently associated with LV mass with the exception of the CMA AA allele, for which there was nonetheless a trend towards significance (p = 0.06) (table 4).
DISCUSSION
The influence of RAAS polymorphisms on LVH has been studied in a variety of settings, including hypertrophy caused by hypertension, endurance training, and aortic stenosis.9, 19, 30–35 In these contexts, ACE, AGT, and CYP11B2 genotypes have been shown to influence the degree of hypertrophy in many9, 13, 14, 30, 31, 35 but not all studies.32–34 Data on the role of RAAS polymorphisms specifically in HCM are also conflicting.6–8, 10–13 The variability of results in HCM may be partly accounted for by the heterogeneous patient populations in most studies since the effect of RAAS polymorphisms may be small relative to the effect of the underlying primary aetiological mutation. Few studies have involved individual families. One group of investigators has shown an association between LVH and ACE DD in 183 genetically independent patients with HCM, which contained a subgroup of 26 affected members of a single family.10 Another study evaluated the effect of ACE gene polymorphisms in patients with either MyBP-C (n = 33) or β myosin mutations (n = 81) and found an influence of ACE DD on hypertrophy in patients with a β myosin Arg403Leu mutation (n = 54) but not in those with other mutations, indicating that the influence of RAAS polymorphisms may depend on the underlying mutation.8 However, although subjects were matched for mutation, they were not all from the same family so that other genetic differences may have masked any influence of ACE DD.
In this study we have evaluated the influence of RAAS polymorphisms on the degree of hypertrophy in gene carriers from a single family with a MyBP-C mutation. Our study group consisted of 26 mutation carriers from a single family represented by a five generation pedigree (described in detail by Moolman and colleagues22). No information was available about generation 0, the “founder” generation. In generation I four affected siblings were known (all deceased; one patient died prematurely without having children). Gene carriers studied were descendants of three siblings in generation I. Patients and mutation carriers in generation II were first cousins. Thus, the probands investigated had many genes in common (first cousins share 25% of their genes), thereby facilitating the detection of any influence of RAAS polymorphisms.
A notable difference in penetrance and expressivity of HCM among mutation carriers characterised this family.22 Approximately 50% of them had ECG or echocardiographic abnormalities, or both, while the remainder had normal investigational results. Moreover, the expressivity of the disease mutation was strikingly different in three separate branches traced back to the three siblings of generation I. In one branch (branch B) all carriers with one exception had cardiovascular abnormalities diagnostic of HCM (albeit with late onset of disease). In the other branches (n = 10) only a minority of the carriers had features of HCM. Either environmental or inherited conditions may explain these phenotypic differences. Although environmental effects could not be excluded, the skewed distribution of phenotypes in the three branches is suggestive of genetic factors being involved. This study confirms RAAS polymorphisms as one of these genetic factors. These polymorphisms were significantly more frequent in branch B and members of this branch had significantly greater LV mass and IVST, which were independent of age differences.
In addition to the relatively homogeneous patient population, this study also differs from previous studies in that five non-allelic RAAS polymorphisms were analysed rather than only the smaller numbers characteristic of other studies. In this study, the pooling of these polymorphisms into a common unit of “pro-LVH” or “risk enhancing” genotypes showed a difference between carriers and non-carriers of pro-LVH genotypes. Gene carriers without such genotype had normal IVST, normal LV muscle mass, and no abnormal ECGs.
In addition, the measurement of five non-allelic RAAS polymorphisms increased the power to evaluate the genotype–phenotype association of single pro-LVH genotypes separately. There were only modest phenotypic differences between carriers and non-carriers of individual mutations. However, these non-carriers were frequently carriers of other pro-LVH genotypes. For instance, taking the ACE I/D polymorphism, six probands had the genotype ACE DD and 20 had ACE II or ID. However, 10 of the non-ACE DD genotypes had other pro-LVH genotypes. The situation was clarified by taking family members with no pro-LVH mutations as a control group. Four out of five RAAS pro-LVH polymorphisms were associated with LVH compared with this control group in both univariate and multivariate analyses. This result supports the concept of different polymorphic genotypes forming a “compound unit”, the components of which act in a related manner. The results of multivariate analysis confirm that each of these polymorphisms is associated with LVH independent of the influence of age, sex, and the presence of hypertension. It cannot be confirmed in this study that each polymorphism was associated with LVH independent of other pro-LVH polymorphisms since the sample size precluded the simultaneous inclusion of other polymorphisms as variables in multivariate analysis.
The role of chymase in the RAAS system has yet to be defined. There is evidence that it may account for a substantial proportion of angiotensin II produced within cardiac tissue36 but its importance may be greater in vitro than in vivo.37 However, it remains of interest in this era of direct angiotensin antagonists and other alternatives and adjuncts to ACE inhibition. It was included in this study because of a previously documented association with HCM and our data support the hypothesis that chymase polymorphisms may influence LVH.
In summary, the analysis of polymorphisms of five different RAAS genes in 26 MyBP-C mutation carriers of one family supports the conclusion that modifier genes contribute to the HCM disease phenotype. Our data also suggest that RAAS polymorphisms may form a pool of factors that individually or collectively contribute to LVH. Further investigations preferably of large families and of mutations in other HCM genes are required to decide whether the study of compound polymorphisms may better explain phenotypic variation in HCM than the analysis of single polymorphisms alone.
Acknowledgments
This study was supported by a grant of the START program of the University Hospital of Aachen and by the British Heart Foundation (NGM, WJM).
Abbreviations
ACE, angiotensin converting enzyme
AGT, angiotensinogen
AGTR1, angiotensin II receptor type 1
CMA, cardiac chymase A
D, deletion
HCM, hypertrophic cardiomyopathy
IVST, interventricular septal thickness
I, insertion
LV, left ventricular
LVDD, left ventricular diastolic dimension
LVH, left ventricular hypertrophy
MyBP-C, myosin binding protein C
PWT, posterior wall thickness
RAAS, renin-angiotensin-aldosterone system
REFERENCES
- 1.Marian AJ, Roberts R. Recent advances in the molecular genetics of hypertrophic cardiomyopathy. Circulation 1995;92:1336–47. [DOI] [PubMed] [Google Scholar]
- 2.Niimura H, Bachinski LL, Sangwatanaroj S, et al. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med 1998;338:1248–57. [DOI] [PubMed] [Google Scholar]
- 3.Spirito P, Seidman CE, McKenna WJ, et al. The management of hypertrophic cardiomyopathy. N Engl J Med 1997;336:775–85. [DOI] [PubMed] [Google Scholar]
- 4.Griendling KK, Murphy TJ, Alexander RW. Molecular biology of the renin-angiotensin system. Circulation 1993;87:1816–28. [DOI] [PubMed] [Google Scholar]
- 5.Sadoshima J, Izumo S. Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts: critical role of the AT1 receptor subtype. Circ Res 1993;73:413–23. [DOI] [PubMed] [Google Scholar]
- 6.Marian AJ, Yu QT, Workman R, et al. Angiotensin-converting enzyme polymorphism in hypertrophic cardiomyopathy and sudden cardiac death. Lancet 1993;342:1085–6. [DOI] [PubMed] [Google Scholar]
- 7.Yoneya K, Okamoto H, Machida M, et al. Angiotensin-converting enzyme gene polymorphism in Japanese patients with hypertrophic cardiomyopathy. Am Heart J 1995;130:1089–93. [DOI] [PubMed] [Google Scholar]
- 8.Tesson F, Dufour C, Moolman JC, et al. The influence of the angiotensin I converting enzyme genotype in familial hypertrophic cardiomyopathy varies with the disease gene mutation. J Mol Cell Cardiol 1997;29:831–8. [DOI] [PubMed] [Google Scholar]
- 9.Schunkert H, Hense HW, Holmer SR, et al. Association between a deletion polymorphism of the angiotensin-converting-enzyme gene and left ventricular hypertrophy. N Engl J Med 1994;330:1634–8. [DOI] [PubMed] [Google Scholar]
- 10.Lechin M, Quinones MA, Omran A, et al. Angiotensin-I converting enzyme genotypes and left ventricular hypertrophy in patients with hypertrophic cardiomyopathy. Circulation 1995;92:1808–12. [DOI] [PubMed] [Google Scholar]
- 11.Ishanov A, Okamoto H, Yoneya K, et al. Angiotensinogen gene polymorphism in Japanese patients with hypertrophic cardiomyopathy. Am Heart J 1997;133:184–9. [DOI] [PubMed] [Google Scholar]
- 12.Brugada R, Kelsey W, Lechin M, et al. Role of candidate modifier genes on the phenotypic expression of hypertrophy in patients with hypertrophic cardiomyopathy. J Investig Med 1997;45:542–51. [PubMed] [Google Scholar]
- 13.Osterop AP, Kofflard MJ, Sandkuijl LA, et al. AT1 receptor A/C1166 polymorphism contributes to cardiac hypertrophy in subjects with hypertrophic cardiomyopathy. Hypertension 1998;32:825–30. [DOI] [PubMed] [Google Scholar]
- 14.Kupari M, Hautanen A, Lankinen L, et al. Associations between human aldosterone synthase (CYP11B2) gene polymorphisms and left ventricular size, mass, and function. Circulation 1998;97:569–75. [DOI] [PubMed] [Google Scholar]
- 15.Pfeufer A, Osterziel KJ, Urata H, et al. Angiotensin-converting enzyme and heart chymase gene polymorphisms in hypertrophic cardiomyopathy. Am J Cardiol 1996;78:362–4. [DOI] [PubMed] [Google Scholar]
- 16.Jeunemaitre X, Soubrier F, Kotelevtsev IV, et al. Molecular basis of human hypertension: role of angiotensinogen. Cell 1992;71:169–80. [DOI] [PubMed] [Google Scholar]
- 17.Rigat B, Hubert C, Alhenc-Gelas F, et al. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest 1990;86:1343–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amant C, Hamon M, Bauters C, et al. The angiotensin II type 1 receptor gene polymorphism is associated with coronary artery vasoconstriction. J Am Coll Cardiol 1997;29:486–90. [DOI] [PubMed] [Google Scholar]
- 19.Van Geel PP, Pinto YM, Buikema H, et al. Is the A1166C polymorphism of the angiotensin II type 1 receptor involved in cardiovascular disease? Eur Heart J 1998;19(suppl G):G13–7. [PubMed] [Google Scholar]
- 20.Azizi M, Hallouin MC, Jeunemaitre X, et al. Influence of the M235T polymorphism of human angiotensinogen (AGT) on plasma AGT and renin concentrations after ethinylestradiol administration. J Clin Endocrinol Metab 2000;85:4331–7. [DOI] [PubMed] [Google Scholar]
- 21.Danser AH, Derkx FH, Hense HW, et al. Angiotensinogen (M235T) and angiotensin-converting enzyme (I/D) polymorphisms in association with plasma renin and prorenin levels. J Hypertens 1998;16:1879–83. [DOI] [PubMed] [Google Scholar]
- 22.Moolman JA, Reith S, Uhl K, et al. A newly created splice donor site in exon 25 of the MyBP-C gene is responsible for inherited hypertrophic cardiomyopathy with incomplete disease penetrance. Circulation 2000;101:1396–402. [DOI] [PubMed] [Google Scholar]
- 23.Klues HG, Schiffers A, Maron BJ. Phenotypic spectrum and patterns of left ventricular hypertrophy in hypertrophic cardiomyopathy: morphologic observations and significance as assessed by two-dimensional echocardiography in 600 patients. J Am Coll Cardiol 1995;26:1699–708. [DOI] [PubMed] [Google Scholar]
- 24.Maron BJ, Gottdiener JS, Epstein SE. Patterns and significance of distribution of left ventricular hypertrophy in hypertrophic cardiomyopathy: a wide angle, two dimensional echocardiographic study of 125 patients. Am J Cardiol 1981;48:418–28. [DOI] [PubMed] [Google Scholar]
- 25.Sahn DJ, DeMaria A, Kisslo J, et al. Recommendations regarding quantitation in M-mode echocardiography: results of a survey of echocardiographic measurements. Circulation 1978;58:1072–83. [DOI] [PubMed] [Google Scholar]
- 26.McKenna WJ, Spirito P, Desnos M, et al. Experience from clinical genetics in hypertrophic cardiomyopathy: proposal for new diagnostic criteria in adult members of affected families. Heart 1997;77:130–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Surawicz B, Uhley H, Borun R, et al. The quest for optimal electrocardiography. Task Force I: standardization of terminology and interpretation. Am J Cardiol 1978;41:130–45. [DOI] [PubMed] [Google Scholar]
- 28.Bonnardeaux A, Davies E, Jeunemaitre X, et al. Angiotensin II type 1 receptor gene polymorphisms in human essential hypertension. Hypertension 1994;24:63–9. [DOI] [PubMed] [Google Scholar]
- 29.Lindpaintner K, Pfeffer MA, Kreutz R, et al. A prospective evaluation of an angiotensin-converting-enzyme gene polymorphism and the risk of ischemic heart disease. N Engl J Med 1995;332:706–11. [DOI] [PubMed] [Google Scholar]
- 30.Montgomery HE, Clarkson P, Dollery CM, et al. Association of angiotensin-converting enzyme gene I/D polymorphism with change in left ventricular mass in response to physical training. Circulation 1997;96:741–7. [DOI] [PubMed] [Google Scholar]
- 31.Dellgren G, Eriksson MJ, Blange I, et al. Angiotensin-converting enzyme gene polymorphism influences degree of left ventricular hypertrophy and its regression in patients undergoing operation for aortic stenosis. Am J Cardiol 1999;84:909–13. [DOI] [PubMed] [Google Scholar]
- 32.Schunkert H, Hengstenberg C, Holmer SR, et al. Lack of association between a polymorphism of the aldosterone synthase gene and left ventricular structure. Circulation 1999;99:2255–60. [DOI] [PubMed] [Google Scholar]
- 33.Lindpaintner K, Lee M, Larson MG, et al. Absence of association or genetic linkage between the angiotensin-converting-enzyme gene and left ventricular mass. N Engl J Med 1996;334:1023–8. [DOI] [PubMed] [Google Scholar]
- 34.Hamon M, Amant C, Bauters C, et al. Association of angiotensin converting enzyme and angiotensin II type 1 receptor genotypes with left ventricular function and mass in patients with angiographically normal coronary arteries. Heart 1997;77:502–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karjalainen J, Kujala UM, Stolt A, et al. Angiotensinogen gene M235T polymorphism predicts left ventricular hypertrophy in endurance athletes. J Am Coll Cardiol 1999;34:494–9. [DOI] [PubMed] [Google Scholar]
- 36.Urata H, Boehm KD, Philip A, et al. Cellular localization and regional distribution of an angiotensin II-forming chymase in the heart. J Clin Invest 1993;91:1269–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei CC, Meng QC, Palmer R, et al. Evidence for angiotensin-converting enzyme- and chymase-mediated angiotensin II formation in the interstitial fluid space of the dog heart in vivo. Circulation 1999;99:2583–9. [DOI] [PubMed] [Google Scholar]