

Abstract
Nicotinic acetylcholine receptors (AChR) belong to a family of proteins that form ligand-gated transmembrane ion channels. They are involved in the fast transmission of signals between cells and the control of intercellular communication in the nervous system. A variety of therapeutic agents and abused drugs, including cocaine, inhibit the AChR and monoamine transporters and interfere with nervous system function. Here we describe a mechanism-based approach to prevent this inhibition. We had previously developed presteady-state kinetic (transient kinetic) techniques, with microsecond-to-millisecond time resolutions, for investigations of reactions on cell surfaces that allow one to determine the effects of inhibitors not only on the channel-opening probability but also on the opening and closing rates of the AChR channel. The transient kinetic measurements led to two predictions. (i) Ligands that bind to a regulatory site on the closed-channel conformation of the AChR with higher affinity than to the site on the open-channel form shift the equilibrium toward the closed-channel form, thereby inhibiting the receptor. (ii) Ligands that bind to a regulatory site with an affinity for the open conformation equal to or higher than their affinity for the closed conformations are expected not to inhibit the receptor and to displace inhibitors. The identification of such ligands in a combinatorial library of RNA ligands is reported. The implication of this approach to other protein-mediated reactions in which an inhibitor changes the equilibrium between active and inactive conformations is discussed.
The nicotinic acetylcholine receptor (AChR) is the prototypical member of a family of structurally related membrane proteins, the ligand-gated ion channels (1). These proteins regulate intercellular communication between the approximately 1012 cells of the mammalian nervous system, a process considered essential for brain function (2). Many therapeutic agents and abused drugs affect their function (3). For instance, the AChR is inhibited by the anticonvulsant MK-801 [(+)−dizocilpine] (4–6) and by several abused drugs, including cocaine (7–9). Cocaine affects more than three million people annually in the United States alone, at an estimated cost to society of more than 100 billion dollars.
Understanding the mechanism of the AChR and its inhibition is a longstanding and challenging problem (10) with major implications for medicine and drug addiction (11–12). Two decades ago, single-channel current-recording (13) measurements led to the proposal of a simple and generally accepted mechanism in which inhibitors enter the open channel and block it (14–17) (the channel-blocking mechanism, Mechanism A in Fig. 1). Although several variations of this open-channel-blocking mechanism have been proposed, including the conversion of an inhibitor-bound closed-channel conformation to a blocked open-channel form (18–21) (Mechanism B, Fig. 1), the open-channel-blocking mechanism, based mainly on single-channel current or other steady-state kinetic measurements (14–21), has met the test of time during the last 20 years. In the techniques used for those measurements, the channel-activating ligand is in quasi equilibrium (steady state) with the receptor. The question we asked was: Can additional information about the receptor-mediated reactions be obtained by using presteady-state kinetic techniques? Recently, presteady-state kinetic techniques that are suitable for measuring receptor-mediated reactions on cell surfaces in the millisecond-to-microsecond time region were developed (22–31). The time resolution of the laser-pulse photolysis technique (23–26) is sufficient to investigate the reaction before the channel has opened. One can, therefore, obtain information about the rate constants of channel opening (kop) and closing (kcl), the equilibrium constants for channel opening (Φ−1) and neurotransmitter binding to the site controlling channel opening (K1) (reviewed in ref. 25). The technique allowed us to ask questions, the answers to which are critical for understanding the mechanism of receptor-mediated reactions (reviewed in ref. 25). Moreover, as we shall demonstrate, the answers are critical for the discovery of ligands that prevent inhibition of the AChR. Is it possible that the receptor has regulatory (allosteric) sites on both the closed- and open-channel conformation? Can compounds bind to the regulatory site on the open conformation without blocking the channel? Does receptor inhibition arise when compounds bind with higher affinity to the regulatory site on the closed-channel conformation than to the site on the open-channel conformation, thereby shifting the equilibrium between the two forms to the closed-channel one (Mechanism C, Fig. 1)? Can compounds that bind to the allosteric site with equal or higher affinity for the open-channel form than for the closed-channel form displace inhibitors without themselves inhibiting the receptor?
Figure 1.

Proposed mechanisms for the inhibition of AChR by MK-801 and cocaine. In each case, the upper line represents the minimum mechanism for the opening of the receptor-channel (10). Receptor R binds the neurotransmitter L (or another activating ligand, for instance carbamoylcholine). Katz and Thesleff (10) first suggested the binding of at least two ligand molecules to the receptor before the receptor-channel opens. R, RL, and RL2 represent the closed-channel conformations. RL2 represents the open-channel conformation of the receptor that allows inorganic cations to cross the cell membrane, thus initiating an electrical signal and intercellular communication. K1 is the observed dissociation constant for the activating ligand. kop and kcl are the rate constants for channel opening and closing, respectively; Φ−1 (=kop/kcl) is the channel-opening equilibrium constant (25, 43). The reactions shown occur in the microsecond-to-millisecond time region (13, 22–24, 44). For clarity, the desensitization reaction, which in the case of the AChR occurs in the 100- to 500-ms time region (44, 45), and the binding of the inhibitor I to the unliganded receptor form are not shown. The relatively slow transitions of receptor/inhibitor complexes to nonconductive forms (6, 9, 30) are also not shown. (A) Channel-blocking mechanism in which the inhibitor binds in the open channel and blocks it (14). (B) Extended channel-blocking mechanism. The inhibitor binds to the closed- and open-channel forms giving the nonconducting receptor forms IRL2 and IRL*2 (18). KI and KI are the observed inhibitor dissociation constants pertaining to the closed- and open-channel form, respectively. (C) Proposed cyclic inhibition mechanism involving a complex of the inhibitor with the open-channel conformation in which the open channel is not blocked by the inhibitor (i.e., it conducts ions). This minimum mechanism is based on chemical kinetic measurements and on predictions it makes regarding the properties of ligands that will inhibit the receptor and of those that will not inhibit the receptor but will prevent the binding of inhibitors. The principle of microscopic reversibility (37) requires that the ratio and KI/KI = Φ−1/ΦI0−1 where ΦI0−1 = k*op/k*cl. Therefore compounds that bind to a regulatory site with higher affinity for the closed-channel conformation than the open-channel form will shift the equilibrium toward the closed-channel form and inhibit the receptor. Compounds that bind to the open-channel conformation with equal or higher affinity than to the closed-channel form are expected to displace inhibitors from the regulatory sites without inhibiting receptor activity.
Here we demonstrate that by using transient kinetic techniques (22–26), one can obtain additional information about the inhibition mechanism.
Materials and Methods
Materials.
Carbamoylcholine was purchased from Sigma and (+)−dizocilpine (MK-801) from Research Biochemicals (Natick, MA). αCNB-caged carbamoylcholine (N-[2-α-carboxy]-2-nitrobenzyl-carbamoylcholine), synthesized according to Milburn et al. (23), was a gift from Molecular Probes. The chemicals for cell culture and the buffers were purchased from GIBCO and Sigma.
RNA Aptamers.
The preparation of the aptamers (RNA ligands) has been described (32). All aptamer solutions contained 0.11 units per microliter of a DTT-independent ribonuclease inhibitor anti-RNase (Ambion), a concentration that had no effect on receptor activity when tested in cell-flow experiments but that did ensure stability of the aptamer (32). Two types of RNA ligands with different consensus sequences were identified and designated as Class I or II aptamers (32). The number after the Roman numeral designates an aptamer within a class. The sequences of the aptamers in Class I are given in table 1 in ref. 32.
Cell Culture.
BC3H1 cells carrying the muscle type nicotinic AChR were cultured according to published methods (33).
Whole-Cell Current Recordings.
Currents were recorded by using the whole-cell configuration (34) for 2 s at ≈22°C, −60 mV, and pH 7.4. The solution in the recording pipet contained 140 mM KC1, 10 mM NaCl, 2 mM MgCl2, 1 mM EGTA, and 25 mM Hepes, balanced to pH 7.4. The bath buffer solution contained 145 mM NaCl, 5.3 mM KCl, 1.8 mM CaCl2, 1.2 mM MgCl2 and 25 mM Hepes, pH 7.4. Solutions were exchanged by using the cell-flow method as described (see below and refs. 22, 35, 36). The resistance of the recording electrode filled with buffer solution was typically 3–5 MΩ and the series resistance was 5–6 MΩ. The cells were held at a constant transmembrane voltage of −60 mV. Whole-cell currents were amplified by using an Axopatch 200B (Axon Instruments) amplifier and filtered by using a 4-pole, low-pass, Bessel filter incorporated in the amplifier, at 0.5–2 kHz (cell-flow) or 10 kHz (laser-pulse photolysis). The filtered signal was digitized by using a Labmaster DMA 100 kHz digitizing board (Scientific Solutions) controlled by Axon pclamp software. Typical digitizing frequencies were 1–5 kHz for cell-flow experiments and 20–40 kHz for laser-pulse photolysis experiments.
Cell-Flow Method.
The cell-flow method (22) was used for rapid solution exchange and to calibrate the amount of carbamoylcholine released with the laser-pulse photolysis technique, to test for cell damage by the laser pulse, and to monitor the recovery of the receptor after the inhibitor had been removed by washing the cells (using the cell-flow device) (22, 27, 28). The time resolution of this technique in experiments with the AChR-containing BC3H1 cells is only 10 ms (22), which is 100 times slower than the time resolution of the laser-pulse photolysis technique. The observed current must, therefore, be corrected for receptor desensitization that occurs during the time interval in which the neurotransmitter equilibrates with the cell surface receptors. The theory and practice have been developed to make this correction (22). The corrected current maximum observed in these experiments, a measure of the concentration of open receptor channels, agrees with the laser-pulse photolysis measurements (see below) which have a 100-fold better time resolution.
Laser-Pulse Photolysis Measurements.
The laser-pulse photolysis method used has been described in detail (23–25, 27, 28). A cell in the whole-cell current-recording mode (34) is equilibrated with a photolabile biologically inert precursor of the neurotransmitter (caged neurotransmitter) (23, 26) specific for the receptor to be investigated. On photolysis, the precursor releases the neurotransmitter within 100 μs (23, 26–28). Photolysis is initiated with a pulse of light generated by a Lumonics (Ontario, Canada) nitrogen laser (λ = 337 nm, pulse duration 9 ns). The output of the laser was coupled into an optical fiber [Fiberguide Industries (Stirling, NJ), 200 or 300 μm in diameter], which delivered the light to the cell. The typical laser energies used were 2–8 mJ/mm2, depending on the required concentration of released carbamoylcholine. A standard concentration (100 μM) of carbamoylcholine was applied by using the cell-flow technique (22) before and after each photolysis experiment. The current amplitudes corrected for receptor desensitization (22, 35) were compared with the current amplitudes obtained in the laser-pulse photolysis experiments and with standard curves that allow one to relate current amplitudes to carbamoylcholine concentration (24). For each concentration of carbamoylcholine used, control experiments without inhibitor were performed, and the current amplitude corrected for receptor desensitization was taken as the 100% response.
Results and Discussion
In Mechanism A (Fig. 1), the inhibitor binds only to the open-channel form of the receptor. As can be seen from Eq. I-B (See Appendix), this mechanism requires that the apparent affinity of the receptor for the inhibitor increases [K̄I(obs) decreases] as the fraction of the receptor in the open-channel form (RL2)o increases. To test this prediction made by Mechanism A, we determined the effect of inhibitor concentration on the concentration of open channels at low- and high-neurotransmitter concentrations, when the fraction of receptors in the open-channel form is low and high, respectively. Cell-flow experiments with BC3H1 cells indicate that common AChR inhibitors, such as procaine (29), cocaine (9), MK-801 (6), and a synthetic analog (philanthotoxin-343) of the wasp toxin philanthotoxin 433 (30), bind with higher affinity to the closed-channel form than to the open-channel form. These results are inconsistent with Mechanism A in Fig. 1, in which inhibitors interact only with the open-channel conformation of the receptor. However, these experiments do not discriminate between Mechanisms B and C.
Mechanisms B and C (Fig. 1) differ in one important aspect. In Mechanism B, the complex of the inhibitor with the closed-channel form (IRL2) gives rise to an inhibitor-blocked open-channel form. In contrast, in Mechanism C (Fig. 1), IRL2 is in equilibrium with an unblocked open-channel form, IRL2 (open), to which the inhibitor is bound. As can be seen from Eq. II-C, Mechanism B requires that the observed rate constants for channel opening (kop) and closing (kcl) both decrease with increasing inhibitor concentration. In contrast, Mechanism C does not require this (see below). The laser-pulse photolysis technique (23–25, 27, 28) allows one to determine the effects of inhibitor concentration on both kop and kcl (Fig. 1). A typical current vs. time trace (Upper) obtained in a laser-pulse photolysis experiment done with the AChR in a BC3H1 cell, and a caged derivative of carbamoylcholine, a well characterized analog of acetylcholine, is shown in Fig. 2A. The current rises rapidly and then reaches a plateau. From the rate of the current rise measured over a wide range of ligand concentration, kop and kcl (Fig. 1), as well as the effects of inhibitors on them, were evaluated (6, 9, 29, 30; Eq. II-A–D).
Figure 2.

(A) Laser-pulse photolysis experiments (23, 24) with BC3H1 cells containing the muscle-type AChR (33). The laser-pulse photolysis method was used as has been described in detail (23, 24, 27, 28). Seventy micromolar carbamoylcholine was photolytically released from the 1-mM caged carbamoylcholine with which the cell was preequilibrated in the absence (Top) and presence (Bottom) of 500 μM MK-801 or in the presence of both 500 μM MK-801 and 2 μM aptamer II-3 (Middle). The cell was preequilibrated (using a device described in ref. 27) for 200 ms with the compounds used in the experiments, and then a 9-ns laser pulse at 337 nm released free carbamoylcholine. At the concentration used, MK-801 does not contribute to the absorption of light at 337 nm. The whole-cell current thus induced was recorded at ≈22°C, −60 mV, and pH 7.4. The desensitization reaction, which occurs on a slower time scale, is not shown. In the upper trace, the current amplitude is 2.6 nA and in the lowest trace, it is 0.5 nA. In the middle trace, 2 μM aptamer II-3 restores the current amplitude (2.3 nA) to about 90% of the noninhibited receptor amplitude (upper trace). The spikes seen in all three traces represent an instrument artifact. Nonlinear least-squares fitting (Marquardt algorithm) was performed by using Microcal origin. (B) Cell-flow experiments (22) with BC3H1 cells containing the muscle-type AChR (33). The whole-cell current was recorded for 2 s at ≈22°C, −60 mV, and pH 7.4. The upper traces parallel to the abscissa represent the current amplitudes corrected for receptor desensitization that occurs during the rising phase. For each concentration of carbamoylcholine used, control experiments without inhibitor were performed, and the current amplitude corrected for receptor desensitization was taken as the 100% response. All experiments were carried out in the presence of a constant concentration (0.5 μM) of aptamer I-14 and increasing concentrations of carbamoylcholine [100 μM, 250 μM, 400 μM, 500 μM corresponding to 0.30, 0.55, 0.65, and 0.75 fractions of open receptor channels as calculated from published cell-flow experiments relating the concentration of open receptor channels in BC3H1 cells to carbamoylcholine concentration (22, 24)]. The cell was preincubated for 2 s with the compounds used in the experiments. Similar results were obtained with a mixture of 12 Class I aptamers (Class I aptamers 1, 5, 6, 7, 9, 11, 13, 16, 18, 19, 20 and 22) (32). In the presence of a constant concentration (4 μM) of the aptamers, with which the cell was also preequilibrated for 2 s, over 50% of the receptors were inhibited in the presence of 100 μM carbamoylcholine. They were not inhibited in the presence of 500 μM carbamoylcholine, when the fraction of the receptors in the open-channel conformation is more than 2-fold larger than at 100 μM carbamoylcholine.
The kinetic measurements make it possible to distinguish between the three proposed mechanisms of inhibition. With increasing inhibitor concentration does only kcl decrease (Mechanism A, Fig. 1) or do both kop and kcl decrease (Mechanism B), or is it possible that one of these rate constants actually increases with increasing inhibitor concentration (Mechanism C)?
In the case of the inhibitors MK-801 (6), cocaine (9), and philanthotoxin-343 (30), the observed values obtained for kop or kcl do not simply decrease with increasing inhibitor concentration, as required by Mechanism B in Fig. 1 (Eq. II-C). For instance, in the case of MK801, kcl increases with increasing inhibitor concentration, whereas kop does not change (6). The simplest mechanism we can think of that is consistent with these results involves a cyclic equilibrium in which the channel is open with inhibitor bound (Mechanism C in Fig. 1). For this mechanism, the principle of microscopic reversibility (37) requires that the ratio of KI/KI (Fig. 1) is equal to the ratio of the channel-opening equilibrium constant in the absence (Φ−1 = kop/kcl) and the presence (ΦIO−1 = k*op/k*cl) of an inhibitor. This relationship predicts that ligands that bind to a regulatory site with higher affinity for the closed-channel conformation than for the open-channel form will shift the channel-opening equilibrium toward the closed-channel form, thus inhibiting the receptor. It further predicts that ligands that bind with equal or higher affinity to the regulatory site on the open-channel conformation will not affect the channel-opening equilibrium constant adversely. The latter type of ligand is expected to displace inhibitors from the regulatory site without itself inhibiting the receptor. Do such ligands exist?
To find such ligands, we explored the possibility of using combinatorially synthesized RNA ligands and the systematic evolution of ligands by exponential enrichment (SELEX) methodology (38–40) to isolate RNA ligands that bind to the cocaine site of the AChR in Torpedo californica electroplax membranes, which are rich in this receptor. From a very large pool (≈1013) of different RNA molecules, we obtained 24 RNA ligands that can be displaced from their binding site on the membrane-bound receptor by cocaine (32). On the basis of their consensus sequence and their ability to inhibit the AChR in BC3H1 cells, these ligands were placed into one of two classes. All 14 Class I aptamers inhibited the AChR in BC3H1 cells in the nanomolar concentration region. Ten Class II aptamers, in the micromolar concentration region, had no activity in this assay.
Here, we have used two transient kinetic methods (reviewed in ref. 25), the laser-pulse photolysis (23, 24, 27, 28) and cell-flow (22) techniques, to test the predictions of the inhibition Mechanism C of the AChR in BC3H1 cells (Fig. 1). Do aptamers that inhibit the receptor all bind with higher affinity to the closed-channel form than to the open form? Do Class II aptamers exist that not only do not inhibit the receptor but also protect it from inhibition?
In preliminary assays, Class I aptamer 14 (I-14) and Class II aptamer 3 (II-3) were shown to have the highest characteristic activity for each class of aptamer. Accordingly, these representative members of each aptamer class were tested in both laser-pulse photolysis (Fig. 2A) and rapid-mixing cell flow (Figs. 2B, 3, and 4) experiments.
Figure 3.

Alleviation by aptamer II-3 of receptor inhibition by MK-801. The whole-cell current corrected for receptor desensitization was determined by the cell-flow technique (22). At a constant concentration of carbamoylcholine, the ratio of the maximum current amplitudes obtained in the absence, A, and presence, AI, of a constant concentration of MK-801 was determined as a function of the concentration of aptamer II-3 (22°C, −60 mV, and pH 7.4). The cell was preequilibrated with aptamer II-3 for 2 s. Each data point represents the average of two to three experiments by using on the average two cells per point. Eq. III-A was used to evaluate KI(obs) and KA(obs), the observed dissociation constants of the inhibitor and the aptamer, respectively. (A) Constant concentrations of 100 μM carbamoylcholine and 500 μM MK-801. The cells were preequilibrated with 500 μM MK-801 for 200 ms. KI(obs) = 180 ± 44 μM; KA(obs) = 0.32 ± 0.15 μM. (B) Constant concentrations of 500 μM carbamoylcholine and 300 μM MK-801. The cell was preequilibrated with 300 μM MK-801 for 200 ms. KI(obs) = 280 ± 50 μM; KA(obs) = 0.14 ± 0.07 μM. (C) Constant concentrations of 100 μM carbamoylcholine and 150 μM MK-801. The cell was preequilibrated with 150 μM MK-801 for 4 s. KI(obs) = 189 ± 74 μM; KII(obs) = 103 ± 23 μM; KA(obs) = 0.6 ± 0.1 μM, where KII(obs) is the inhibitor dissociation constant from the slowly equilibrating second inhibitory site. Experimental conditions were as described in Fig. 2 legend and in Materials and Methods (see also Eq. III-A, -B).
Figure 4.

Alleviation by aptamer II-3 of receptor inhibition by cocaine. The whole-cell current corrected for receptor desensitization was determined by using the cell-flow technique (22, 35). At a constant concentration of 100 μM carbamoylcholine, the ratio of the maximum current amplitude obtained in the absence, A, and presence, AI, of a constant concentration (150 μM) of cocaine was determined as a function of the concentration of aptamer II-3. The cells were preequilibrated with aptamer II-3 for 2 s. Each data point represents the average of two to three experiments by using on the average two cells per point. Eq. III-A was used to evaluate KI(obs) = 98 ± 23 μM; KII(obs) = 177 ± 40 μM; and KA(obs) = 0.7 ± 0.1 μM. The experimental conditions were as described in Fig. 2 legend and Materials and Methods.
The results of cell-flow experiments done with AChR-containing BC3H1 cells are shown in Fig. 2B. In the presence of 100 μM carbamoylcholine, when only 30% of the receptors are in the open-channel conformation [as determined from carbamoylcholine concentration vs. receptor activity curves (22, 24)], over 50% of the receptor activity is inhibited by 0.5 μM aptamer I-14. When the concentration of aptamer I-14 is kept constant but the carbamoylcholine concentration is progressively increased (and consequently the fraction of the receptors in the open-channel conformation increases), progressively less inhibition by the aptamer is observed (Fig. 2B). At 500 μM carbamoylcholine, 0.5 μM aptamer I-14 no longer inhibits the receptor (Fig. 2B Far Right). This is predicted by Mechanism C (Fig. 1). In this mechanism, ligands that bind to an allosteric site with higher affinity for the closed-channel conformation than for the open-channel one are expected to inhibit the receptor more strongly when the receptor is predominantly in the closed-channel conformation. Similar results were obtained with a mixture of 14 Class I aptamers (data not shown).
Mechanism C also predicts that ligands that bind to the same allosteric site as do inhibitors but with equal or higher affinity for the open-channel conformation than for the closed-channel form will not inhibit the receptor but will prevent the binding of these inhibitors. The laser-pulse photolysis experiment in Fig. 2A shows that 500 μM MK-801 is a powerful inhibitor of the receptor, decreasing the current amplitude by a factor of 5 (Bottom) compared with the control (Top). When 2-μM aptamer II-3 is also present, the inhibition by MK-801 is essentially abolished (Middle). As is expected from Mechanism C, aptamer II-3 is at least as effective in relieving receptor inhibition at low (Fig. 3A) concentrations of the open channel as it is at high (Fig. 3B) concentrations. The observed dissociation constant of the aptamer is ≈320 nM at 100 μM carbamoylcholine and ≈140 nM at 500 μM carbamoylcholine. All of the 10 Class II aptamers tested exhibit similar properties (data not shown).
The experiments in Fig. 3C differ from those in Fig. 3A in that the cell was preequilibrated with 150 μM MK-801 for 4 s before the measurements were made. The same carbamoylcholine concentration (100 μM) was used in both experiments (Fig. 3 A and C). After this preequilibration for 4 s (Fig. 3C), 33% of the receptors remained inhibited in the presence of 3 μM aptamer II-3. When, however, the preequilibration period was 200 ms (Fig. 3 A and B), all of the inhibition by MK-801 was relieved in the presence of 2 μM aptamer II-3. These results can be understood in the light of transient kinetic results obtained previously (6). The earlier results indicated that the muscle-type AChR in BC3H1 cells has two inhibitory sites for MK-801; one site equilibrates with MK-801 within 50 ms and the second site within 4 s (6). Additionally, at very high concentrations of carbamoylcholine, when the receptors are all in the open-channel form, a relatively slow transition of the open-channel form with inhibitor bound, IRL2 (open) (Mechanism C, Fig. 1) to a receptor form that does not conduct ions was observed (6, 9, 29, 30). Such a transition was first observed in single-channel current measurements (14).
The experiments with RNA aptamers give additional information about the two inhibitory sites. The selection of the aptamers was carried out (32) with membrane fragments in which both the plasma and cytosolic sides of the receptor are exposed to the solution. We therefore suggest that the second inhibitory site for MK-801, the slowly equilibrating site, is within the cell membrane rather than on the cytoplasmic surface of the receptor and is, therefore, not accessible to the negatively charged RNA aptamers.
Earlier presteady-state kinetic measurements with cocaine (9) had revealed that less inhibition occurred when BC3H1 cells were equilibrated with cocaine for 70 ms than for 200 ms. These time intervals are too close to decide between one or two cocaine-binding sites on the basis of kinetic experiments.
The displacement of cocaine from the receptor by aptamer II-3 is illustrated in Fig. 4. About 50% of the original receptor activity can be recovered when aptamer II-3 is used to displace cocaine. The observed KI value for cocaine that is not displaceable by this aptamer is 177 μM (Fig. 4 legend). By analogy with the results obtained with MK-801 and aptamer II-3, we suggest that cocaine, like MK-801, binds to more than one site on the receptor but competes with the aptamer for only one of these sites.
It is estimated that there are currently 14 million users of abused drugs in the United States alone [Substance Abuse and Mental Health Services Administration (1997) National Survey on Drug Abuse (Dept. Health and Human Services, Washington, D.C.)]. Many attempts have been made to find compounds, including the synthesis of hundreds of compounds, that will displace abused drugs, such as cocaine, from their sites of action on the monoamine transporters. So far, no compound has been found that is not also an inhibitor (11).
Our approach for finding compounds that displace cocaine from the inhibitory site of the nicotinic acetylcholine receptor differs in that we first investigated the mechanism of inhibition. For this purpose, we developed two transient kinetic methods. In the cell-flow technique, the observed current amplitude is corrected for receptor desensitization (22). The laser-pulse photolysis technique using caged neurotransmitters (23, 26; reviewed in ref. 25) allows elementary reaction steps of protein-mediated reactions on cell surfaces to be investigated with a microsecond-to-millisecond time resolution. It thus became possible to distinguish between alternative mechanisms of receptor inhibition. This allowed us to make a rational and successful search for ligands that bind to an inhibitory site on the AChR without affecting receptor activity but that can still displace inhibitors from the site.
The approach described can be applied not only to multisubunit proteins in which ligands can shift the equilibrium between existing protein conformations with different biological activities (41), like the AChR (42), but also to single-subunit proteins in which a ligand (for instance noncompetitive inhibitors) can induce a change (46) from an active to an inactive conformation. We hypothesize, therefore, that the mechanism-based approach described here will be applicable to the many proteins in which the equilibrium between active and inactive conformations is adversely affected by drugs (for instance, noncompetitive inhibitors) or by disease-causing mutations.
Acknowledgments
We thank Molecular Probes for the gift of caged carbamoylcholine. This work was supported by grants awarded by the National Institutes of Health (NS 08527 to G.P.H. and DA 11643A to Prof. Robert E. Oswald. G.P.H. is a co-Principal Investigator). H.U. was supported by a postdoctoral fellowship from the New York Chapter of the American Heart Association. H.-G.B. was supported by a postdoctoral fellowship from the Swiss Academy of Sciences. A.M.G. is supported by a postdoctoral fellowship from the New York Chapter of the American Heart Association. C.G. was supported by a Feodor–Lynen Postdoctoral Fellowship awarded by the Alexander von Humboldt Foundation. J.E.I. was a Cornell Hughes Scholar and Paul Schreurs Memorial Awardee. S.M.L. was a Cornell Hughes Scholar. V.J. was supported by a fellowship from the Cancer Research Fund of the Damon Runyon–Walter Winchell Foundation. S.E.C. was the recipient of a research grant from the President's Council of Cornell Women.
Abbreviation
- AChR
nicotinic acetylcholine receptor
Data Analysis.
Linear regression and nonlinear least-squares fitting (Marquardt algorithm) were performed using the Microcal origin program. The following assumptions have been made in developing the equations: L, I0 ≫ R0, and the rate-limiting steps are the opening and closing of the AChR channels.
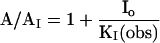 |
I-A |
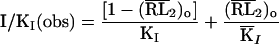 |
I-B |
Eq. I-A: To simplify the equations, we use a ratio method; in the ratio A/AI, A and AI represent the current maxima corrected for receptor desensitization in the absence and presence of inhibitor, respectively. I0 represents the inhibitor concentration and KI(obs), the observed dissociation constant of the inhibitor.
Eq. I-B: In Eq. I-B, KI and KI represent the dissociation constant of the inhibitor from the closed- and open-channel form, respectively. (RL2)0 represents the fraction of the receptors in the open-channel conformation. When the inhibitor binds only to the open channel, the first term in Eq. I-B is omitted.
Eq. II: From the rate of channel opening determined in transient kinetic experiments with a microsecond time resolution:
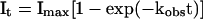 |
II-A |
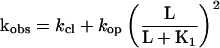 |
II-B |
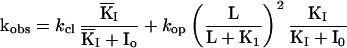 |
II-C |
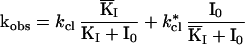 |
II-D |
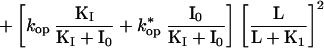 |
Eq. II-A: In the laser-pulse photolysis experiments with BC3H1 cells containing muscle-type AChRs, the current rise time was observed to follow a single exponential rate law over 85% of the reaction (23, 24). It represents the observed current at time t, and Imax represents the maximum current.
Eq. II-B: The relationship between the observed rate constant for the current rise kobs and kop, kcl, K1 of the reaction scheme (Fig. 1) and the concentration of activating ligand (carbamoylcholine) L.
Eq. II-C: kobs in the presence of an inhibitor that binds to both the open-channel form of the receptor RL2 (open) and to the closed-channel forms, and when the complex IRL*2 (Mechanism B, Fig. 1) is nonconducting. KI and KI are the dissociation constants of the inhibitor from the open-channel and closed-channel forms, respectively.
Eq. II-D: kobs in the presence of an inhibitor that binds to the closed- and open-channel forms of the receptor RL2 and the complex IRL2 (open) (Mechanism C, Fig. 1) is a conducting form of the receptor. k*op and k*cl are the rates for the interconversion between IRL2 and IRL2 (open) (Mechanism C, Fig. 1).
Eq. III: From the effect of inhibitor and RNA aptamer concentrations on A/AI:
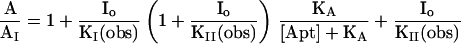 |
III-A |
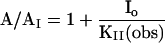 |
III-B |
Eq. III-A: A and AI, respectively, represent the current maxima, corrected for receptor desensitization, measured in the absence and the presence of both the inhibitor and the aptamer. KI(obs) and KII(obs) represent the dissociation constants of the inhibitor from receptor sites at which it does or does not compete with the aptamer respectively. [Apt] represents the concentration of the aptamer and KA(obs) is the dissociation constant of the aptamer. In the absence of aptamer, A/AI in Eq. III-A is a function of inhibitor concentration and KI(obs) and KII(obs). KII(obs) can be obtained from Eq. III-B.
Eq. III-B: In the presence of an inhibitory site with which the aptamer does not interact and when [Apt] ≫ KA, Eq. III-A becomes Eq III-B. KII(obs) can be calculated from Eq. III-B.
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.240459497.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.240459497
References
- 1.Kandel E R, Schwartz J H, Jessel T M. Essentials of Neural Science and Behavior. Stamford, CT: Appleton & Lange; 1995. p. 5. [Google Scholar]
- 2.Crick F H C. The Astonishing Hypothesis. The Scientific Search for the Soul. New York: Macmillan; 1994. [Google Scholar]
- 3.Hardman J G, Limbird L E, Molinoff P B, Ruddon R W, Gilman A G. Goodman & Gilman's The Pharmacological Basis of Therapeutics. 9th Ed. New York: McGraw–Hill; 1996. [Google Scholar]
- 4.Amador M, Dani J A. Synapse. 1991;7:207–215. doi: 10.1002/syn.890070305. [DOI] [PubMed] [Google Scholar]
- 5.Ramoa S, Alhoudon M, Aracava Y, Ivonis J, Lunt G G, Desphande S S, Wonnacott S, Aronstam R S, Albuquerque E X. J Pharmacol Exp Ther. 1990;254:71–82. [PubMed] [Google Scholar]
- 6.Grewer C, Hess G P. Biochemistry. 1999;38:7837–7846. doi: 10.1021/bi9827767. [DOI] [PubMed] [Google Scholar]
- 7.Karpen J W, Aoshima H, Abood L G, Hess G P. Proc Natl Acad Sci USA. 1982;79:2509–2513. doi: 10.1073/pnas.79.8.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swanson K L, Albuquerque E X. J Pharmacol Exp Ther. 1987;243:1202–1210. [PubMed] [Google Scholar]
- 9.Niu L, Abood L G, Hess G P. Proc Natl Acad Sci USA. 1995;92:12008–12012. doi: 10.1073/pnas.92.26.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katz B, Thesleff S. J Physiol (London) 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carroll F I, Howell L L, Kuhar M J. J Med Chem. 1999;42:2721–2736. doi: 10.1021/jm9706729. [DOI] [PubMed] [Google Scholar]
- 12.Javanmard S, Deutsch H M, Collard D M, Kuhar M J, Schweri M M. J Med Chem. 1999;42:4836–4843. doi: 10.1021/jm990306k. [DOI] [PubMed] [Google Scholar]
- 13.Neher E, Sakmann B. Nature (London) 1976;260:779–802. doi: 10.1038/260799a0. [DOI] [PubMed] [Google Scholar]
- 14.Neher E, Steinbach J H. J Physiol (London) 1978;277:153–176. doi: 10.1113/jphysiol.1978.sp012267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arias H R. Biochim Biophys Acta. 1998;1376:173–220. doi: 10.1016/s0304-4157(98)00004-5. [DOI] [PubMed] [Google Scholar]
- 16.Lester H. Annu Rev Biophys Biomol Struct. 1992;21:267–292. doi: 10.1146/annurev.bb.21.060192.001411. [DOI] [PubMed] [Google Scholar]
- 17.Lena V, Changeux J P. Trends Neurosci. 1993;16:181–186. doi: 10.1016/0166-2236(93)90150-k. [DOI] [PubMed] [Google Scholar]
- 18.Adams P R. J Physiol (London) 1976;260:531–552. doi: 10.1113/jphysiol.1976.sp011530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gage P W, Hamill O P, Wachtel R E. J Physiol (London) 1983;335:123–137. doi: 10.1113/jphysiol.1983.sp014524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogden D, Colquhoun S. Proc R Soc London Ser B. 1985;225:329–355. doi: 10.1098/rspb.1985.0065. [DOI] [PubMed] [Google Scholar]
- 21.Papke R L, Oswald R E. J Gen Physiol. 1989;93:785–811. doi: 10.1085/jgp.93.5.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Udgaonkar J B, Hess G P. Proc Natl Acad Sci USA. 1987;84:8758–8762. doi: 10.1073/pnas.84.24.8758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milburn T, Matsubara N, Billington A P, Udgaonkar J B, Walker J W, Carpenter B K, Webb W W, Marque J, Denk W, McCray J A, Hess G P. Biochemistry. 1989;29:49–55. doi: 10.1021/bi00427a008. [DOI] [PubMed] [Google Scholar]
- 24.Matsubara N, Billington A P, Hess G P. Biochemistry. 1992;31:5507–5514. doi: 10.1021/bi00139a012. [DOI] [PubMed] [Google Scholar]
- 25.Hess G P, Grewer C. Methods Enzymol. 1998;291:443–473. doi: 10.1016/s0076-6879(98)91028-x. [DOI] [PubMed] [Google Scholar]
- 26.Gee K R, Carpenter B K, Hess G P. Methods Enzymol. 1998;291:30–50. doi: 10.1016/s0076-6879(98)91005-9. [DOI] [PubMed] [Google Scholar]
- 27.Niu L, Grewer C, Hess G P. Techniques Protein Chem. 1996;VII:139–149. [Google Scholar]
- 28.Hess G P. In: Imaging Living Cells. Yuste R, Lanni F, Konnerth A, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1999. pp. 25.1–25.18. [Google Scholar]
- 29.Niu L, Hess G P. Biochemistry. 1993;32:3831–3855. doi: 10.1021/bi00066a001. [DOI] [PubMed] [Google Scholar]
- 30.Jayaraman V, Usherwood P N R, Hess G P. Biochemistry. 1999;38:11406–11414. doi: 10.1021/bi991219x. [DOI] [PubMed] [Google Scholar]
- 31.Jayaraman V, Thiran S, Hess G P. Biochemistry. 1999;38:11372–11378. doi: 10.1021/bi990454c. [DOI] [PubMed] [Google Scholar]
- 32.Ulrich H, Ippolito J E, Pagan O R, Eterovic V A, Hann R M, Shi H, Lis J T, Eldefrawi M E, Hess G P. Proc Natl Acad Sci USA. 1998;95:14051–14056. doi: 10.1073/pnas.95.24.14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schubert D, Harris A J, Devine E E, Heinemann S. J Cell Biol. 1974;61:398–402. doi: 10.1083/jcb.61.2.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamill O, Marty A, Neher E, Sakmann B, Sigworth F J. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 35.Hess G P, Udgaonkar J B, Olbricht W L. Annu Rev Biophys Biophys Chem. 1987;16:507–534. doi: 10.1146/annurev.bb.16.060187.002451. [DOI] [PubMed] [Google Scholar]
- 36.Krishtal O A S, Pidoplichko V I. Neuroscience. 1980;5:2325–2327. doi: 10.1016/0306-4522(80)90149-9. [DOI] [PubMed] [Google Scholar]
- 37.Hammes G. Thermodynamics and Kinetics for the Biological Sciences. New York: Wiley; 2000. [Google Scholar]
- 38.Tuerk C, Gold L. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 39.Ellington A D, Sostak J W. Nature (London) 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 40.Wilson D, Sostak J W. Annu Rev Biochem. 1999;68:611–647. doi: 10.1146/annurev.biochem.68.1.611. [DOI] [PubMed] [Google Scholar]
- 41.Monod J, Wyman J, Changeux J-P. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 42.Edelstein S J, Changeux J-P. Adv Prot Chem. 1998;51:121–184. doi: 10.1016/s0065-3233(08)60652-x. [DOI] [PubMed] [Google Scholar]
- 43.Cash D J, Aoshima H, Hess G P. Proc Natl Acad Sci USA. 1981;78:3318–3322. doi: 10.1073/pnas.78.6.3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hess G P, Cash D J, Aoshima H. Nature (London) 1979;282:329–331. doi: 10.1038/282329a0. [DOI] [PubMed] [Google Scholar]
- 45.Sakmann B, Patlack J, Neher E. Nature (London) 1980;286:71–73. doi: 10.1038/286071a0. [DOI] [PubMed] [Google Scholar]
- 46.Koshland D E, Jr, Nemethy G, Filmer D. Biochemistry. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]