

Abstract
Objective: To test the hypothesis that identical twins show no inter-twin differences in cardiovascular structure or physiology in fetal life unless there has been twin-twin transfusion syndrome.
Design: Unselected prospective case–control observational study of fetoplacental haemodynamics including echocardiography at a median of 24 (16.7 to 32.3) weeks, with postnatal confirmation of congenital heart disease or normality.
Setting: Fetal medicine unit.
Patients: 136 women with monochorionic diamniotic twin pregnancies, of which 47 fetal twin pairs (35%) had twin-twin transfusion syndrome.
Results: There were no haemodynamic differences between the bigger fetus (twin 1) and the smaller co-twin (twin 2) in uncomplicated monochorionic diamniotic pairs. In twin-twin transfusion syndrome, recipient fetuses had increased aortic and pulmonary velocities compared with their donor co-twins (mean (SD): 0.73 (0.23) m/s and 0.63 (0.14) m/s), respectively, v 0.53 (0.16) m/s and 0.48 (0.10) m/s in donor twins; p = 0.003 (aortic) and < 0.0001 (pulmonary)), and also in comparison with twin 1 and twin 2. The overall prevalence of congenital heart disease was increased above that in singletons (3.8% v 0.56%; 6.9% in twin-twin transfusion v 2.3% in uncomplicated monochorionic diamniotic twins), with inter-twin discordance for defects. The prevalence in recipient twins was 11.9% (p = 0.014 v uncomplicated control twins).
Conclusions: Fetuses with an identical genome but no circulatory imbalance have similar cardiovascular physiology but discordant phenotypic expression of congenital heart disease. The high prevalence of congenital heart disease in monochorionic diamniotic twins merits detailed fetal echocardiography.
Keywords: monochorionic twins, twin-twin transfusion syndrome, congenital heart disease, fetal echocardiography
The incidence of spontaneous monozygotic (identical) twinning is about 4/1000 live births.1 Two thirds comprise monochorionic diamniotic twins, resulting from presumed division of the early embryonic mass between days 3 and 8 of gestation. This leads to the development of two fetuses with the same genome and therefore the same sex, who share a fused placental mass, with their gestational sacs divided by a thin translucent membrane composed of two layers of amnion. Vascular communications between the two placental territories typically comprise superficial bidirectional arterio-arterial and veno-venous anastomoses, located on the chorionic plate, and deep unidirectional arteriovenous anastomoses within shared cotyledons.
The twin-twin transfusion syndrome complicates around 15% of monochorionic diamniotic twin gestations.2 Unbalanced inter-twin transfusion secondary to unidirectional flow through deep arteriovenous anastomoses, with a paucity or absence of compensatory bidirectional superficial arterio-arterial anastomoses, is thought to be the cause.3 The diagnosis is made ultrasonographically, usually in the second trimester, by demonstrating the oligohydramnios/polyhydramnios sequence (where the deepest pool of amniotic fluid is less than 1 cm in the donor twin and more than 8 cm in the recipient).4 The donor becomes oliguric and volume depleted, while the recipient becomes polyuric, volume overloaded, and may develop hydrops fetalis. In the severe form of twin-twin transfusion syndrome, the so called “stuck twin phenomenon”, the donor lies devoid of amniotic fluid against the uterine wall shrouded by the inter-twin membrane, while the recipient’s sac becomes severely polyhydramniotic. Untreated, twin-twin transfusion has a perinatal mortality of 80%, with intrauterine demise or neonatal death from extreme prematurity.5 Several management options now available—including serial decompressive amnioreduction, amniotic septostomy, selective fetocide by bipolar cord occlusion, and laser photocoagulation of placental anastomoses—lead to an improvement in overall perinatal survival of 45–70%, with survival rates for an individual fetus of 65–90%.6–9
We first reported cardiac dysfunction, ventricular hypertrophy, and right ventricular outflow tract obstruction requiring postnatal valvotomy in a small series of infants who were recipient fetuses.10 This was confirmed by others.11–13 More recently we described abnormalities of vascular distensibility in survivors of chronic twin-twin transfusion syndrome in infancy.14 However, most published data are from small series without suitable controls, and thus might merely reflect the cardiovascular complications known to be increased in monochorionic diamniotic twins rather than the influence of the twin-twin transfusion syndrome.
We hypothesised that monochorionic diamniotic twins without twin-twin transfusion would show no inter-twin differences in cardiac structure or function. Our aim was to describe the prevalence of structural heart defects in a larger series of monochorionic diamniotic twins and to assess the possible influence on this of the twin-twin transfusion syndrome.
METHODS
Study population
Between February 1997 and December 2000, 159 monochorionic diamniotic twin gestations were managed at the Centre for Fetal Care (Queen Charlotte’s and Chelsea Hospital, London). Monochorionicity was diagnosed during the first trimester by the presence of two fetuses of the same sex, a common placental mass, and a thin dividing inter-twin membrane. All were followed on a two weekly basis to monitor fetal growth and wellbeing and to detect signs of twin-twin transfusion. Sixty one satisfied the criteria for twin-twin transfusion syndrome. Of the 159 monochorionic diamniotic twin pregnancies, 136 underwent detailed fetal echocardiography of both fetuses and were included in this study. Fetal echocardiography was not done in the remaining 23 monochorionic diamniotic twin pairs owing to very preterm delivery (n = 4), intrauterine death of both co-twins (n = 8), transfer of obstetric care (n = 6), or late referral just before delivery (n = 5).
During the same period nine monochorionic monoamniotic pregnancies were managed at our institution and four pairs had fetal echocardiography. The remaining five pairs were not studied owing to early fetal death of both co-twins associated with umbilical cord entanglement (n = 3) or because of severe congenital anomalies. In view of the small numbers, we did not analyse monochorionic monoamniotic pregnancies.
Investigations
Fetal echocardiography was performed to detect structural abnormalities and to compare haemodynamic measurements between the bigger twin (twin 1 or recipient) and the smaller twin (twin 2 or donor). All cardiac studies were performed by the same investigator (HMG) and regular fetal biometry and placental studies were done by MJO and LW using an Acuson Sequoia Ultrasound system (Acuson Co, Mountain View, California, USA) with an 8C4, 6C2, or 5V2c transducer. These were stored on videotape for later analysis. Energy output was maintained at < 100 mW/cm2. Data were incomplete in 13 of the 136 pregnancies (8.8%) owing to severe oligohydramnios or polyhydramnios, frequent fetal movements, or maternal discomfort. However, cardiac morphological data and peripheral Doppler studies were obtained in all 136 pregnancies.
Serial fetal biometry and liquor volume, expressed as amniotic fluid index, were recorded. The estimated fetal weight discordance was calculated as follows:
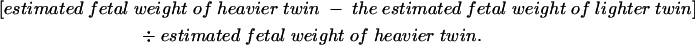 |
The pulsatility index of the umbilical artery was calculated from the formula:
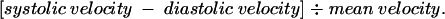 |
Twin-twin transfusion syndrome was diagnosed by the polyhydramnios–oligohydramnios sequence before 28 weeks. The presence or absence of arterio-arterial anastomoses was assessed with colour Doppler flow mapping and confirmed after delivery by placental perfusion studies. Chorionicity was confirmed postnatally by macroscopic and microscopic examination of the placenta.
Standard fetal echocardiographic views were recorded: fetal situs, four chamber view, and cardiothoracic ratio, the great arterial crossover, and the “three vessel view” to determine right-left symmetry of size, normal connections, and equal growth of both the transverse aortic and ductal arches. Additional views were used as appropriate to confirm or exclude septal defects. Colour flow mapping and Doppler were used to record flow through the atrioventricular and semilunar valves and in the arterial and venous ducts. The instantaneous fetal heart rate was calculated from two aortic Doppler waveforms, while all remaining measurements were performed on three consecutive cardiac cycles and the values averaged. Aortic and pulmonary valve diameters and right, left, and combined ventricular outputs were indexed to the estimated fetal weight, and ventricular outputs were calculated from the formula:
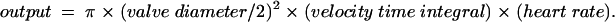 |
Ventricular hypertrophy was diagnosed if the septum or free wall thickness was more than the 95% confidence limits for estimated gestational age15; significant atrioventricular valve regurgitation was diagnosed if the colour flow map extended to the mid or back of the atrium with holosystolic Doppler profiles; and abnormalities of venous ductal or umbilical venous flow were diagnosed if there was absent or reversal of flow at end diastole.
Information on perinatal mortality was collected prospectively. Any suspected fetal structural cardiac abnormalities were confirmed by postnatal echocardiography in our centre (20 monochorionic diamniotic twins + 22 infants with twin-twin transfusion syndrome, comprising 15.4% of surviving infants) and the normality of the remainder was confirmed after correspondence with the subjects’ parents or the health professionals involved.
Statistical analysis
Results are expressed as mean (SD) or median (range), as appropriate. Analysis of variance (ANOVA) with Bonferroni or Dunnett T3 post hoc tests was used for intergroup comparisons. Students’ t test and the Mann–Whitney test were used for inter-twin comparisons, and categorical variables compared using Fisher’s exact test. Significance was set at p < 0.05. All analyses were performed using the statistical package SPSS 9.0 for Windows 1998.
RESULTS
We included 136 twin pregnancies (86% of the total monochorionic diamniotic twin cohort) in the study. These were evaluated by fetal echocardiography at a median gestational age of 24 weeks (range 16.7–32.3 weeks). Forty seven of these 136 pregnancies (35%) were complicated by twin-twin transfusion syndrome: 44 (94%) had the syndrome at the time of fetal echocardiography and three (6%) developed it later. Nine pregnancies had serial echocardiography because of chronic twin-twin transfusion; the haemodynamic progress of the recipient fetus of each pair is shown in table 5. The scan chosen for haemodynamic analysis was that nearest to the median study gestation (24 weeks) but the presence of atrioventricular valve regurgitation or ventricular dysfunction at any stage during the pregnancy was documented. Standard clinical variables were similar in twin-twin transfusion syndrome and control twin pregnancies, although as expected the former showed greater estimated fetal weight discordance, amniotic fluid index, and frequency of artery to artery anastomoses, along with earlier gestation at delivery and poorer perinatal outcome (table 1).
Table 5.
Functional changes following intervention and subsequently until delivery on haemodynamic abnormalities in recipient twins
| Postintervention | Subsequent | ||||
| Case | Dysfunction | AVVR | Dysfunction | AVVR | Intervention |
| 1 | ↑ | ↑ | ↓ | ↓ | Amnioreduction + digoxin |
| 2 | ↑ | ↑ | ↑ | ↑ | Amnioreduction |
| 3 | ↓ | ↓ | ↓ | ↓ | Amnioreduction |
| 4 | ∼ | ∼ | None | ||
| 5 | ↓ | ↓ | None | ||
| 6 | ↑ | ↑ | ↑ | ↑ | Amnioreduction + fetocide* |
| 7 | ↓ | ↓ | None | ||
| 8 | ↓ | ↓ | ↓ | ↓ | Two amnioreductions |
| 9 | ↓ | ↓ | ↓ | ↓ | Amnioreduction |
*Fetocide of donor.
AVVR, atrioventricular valve regurgitation; ↑, improvement; ↓, worsening; ∼, no change.
Table 1.
Clinical data on monochorionic diamniotic twin pairs with and without twin-twin transfusion syndrome
| Variable | Non-TTTS pairs (n=89) | TTTS pairs (n=47) | p Value |
| Maternal age (years) | 33 (19 to 42) | 31 (17 to 42) | 0.08 |
| Parity | 1.0 (0 to 8) | 0.5 (0 to 5) | 0.54 |
| Sex (male/female)† | 46/40 | 19/28 | 0.20 |
| Gestation echo (weeks) | 23.8 (3.2) | 23.6 (3.0) | 0.85 |
| EFW (g) recipient or bigger twin‡ | 691 (338) | 699 (436) | 0.92 |
| EFW (g) donor or smaller twin‡ | 606 (311) | 491 (286) | 0.06* |
| EFW discordance (%) | 11.0 (9.6) | 29.6 (15.3) | <0.0001 |
| AFI (cm) | 19.3 (4.7) | 32.0 (7.7) | <0.0001 |
| AAA+ (Doppler) | 59/89 | 12/47 | <0.0001 |
| Gestation birth (weeks) | 36 (29.6 to 39.7) | 31.6 (23.6 to 40.3) | <0.0001 |
| Survival (%) | 166/178 (93%) | 60/94 (64%) | <0.0001 |
Values are mean (SD), range, or n.
*ANOVA between the four groups, p=0.021. Twin 1 v donor, p=0.026; all other Bonferroni multiple comparisons tests, p>0.05.
†The sex of three MCDA pairs unknown (early fetal death).
‡EFW analysis was paired (one twin 1, two recipients, and four donors were excluded owing to intrauterine demise of their co-twin before fetal echocardiography.
AAA+, arterio-arterial anastomoses present; AFI, amniotic fluid index; EFW, estimated fetal weight; MCDA, monochorionic, diamniotic twin; TTTS, twin-twin transfusion syndrome.
Thirty four of the 46 pregnancies with twin-twin transfusion syndrome (74%) required intervention at a median gestational age of 20.4 weeks (15.3–27.6 weeks). Nineteen of these 34 (56%) were managed using serial amnioreduction, five with amniotic septostomy and amnioreduction (15%), and 10 by selective fetocide (29%) of either the donor (n = 4) or the recipient twin (n = 6). Fetal echocardiography was undertaken before any intervention in 19 pregnancies (41%), although one donor and one recipient died in utero before a detailed cardiac scan was available. Decompressive amnioreduction preceded fetal echocardiography in 17 of the 46 cases (37%), amniotic septostomy in all five cases (11%), and selective fetocide in six cases (two donors and four recipients).
Forty six of the 272 fetuses did not survive the neonatal period. There were 19 intrauterine deaths, 12 selective terminations, and three neonatal deaths in the twin-twin transfusion syndrome group (survival rate = 64%). In the comparison group without twin-twin transfusion syndrome, there were 10 intrauterine deaths, one elective termination for severe abnormality, and one neonatal death (survival rate = 93%, p < 0.0001).
The haemodynamic data presented in tables 2 and 3 excluded 10 fetuses. The exclusions were because of congenital heart disease in two (severe pulmonary stenosis in a twin 1 and pulmonary atresia intact septum in a recipient at 17 weeks), while the remaining eight fetuses died in utero before detailed echocardiography could be performed, leaving a single survivor (one control MDCA and two twin-twin transfusion syndrome intrauterine deaths, and five selective terminations because of severe disease at presentation).16
Table 2.
Haemodynamic variables between monochorionic twin fetuses with and without twin-twin transfusion syndrome
| Variable | Twin 1 (n=87) | Twin 2 (n=89) | Recipient (n=41) | Donor (n=45) | ANOVA (p value; Bonferroni or Dunnett T3 post-hoc tests) |
| Cardiothoracic ratio (%) | 23.2 (3.9) | 23.1 (3.4) | 30.8 (9.1) | 23.9 (4.9) | <0.0001 |
| (R–T1) 95% CI, 2.89 to 12.4; p<0.0001 | |||||
| (R–T2) 95% CI, 3.02 to 12.5; p<0.0001 | |||||
| (R–D) 95% CI, 1.82 to 12.08; p=0.003 | |||||
| (T1–T2) 95% CI, −1.75 to 1.96; p>0.1 | |||||
| Aortic peak velocity (m/s) | 0.60 (0.13) | 0.59 (0.12) | 0.73 (0.23) | 0.53 (0.16) | <0.0001 |
| (R–T1) 95% CI, −0.002 to 0.26; p=0.05 | |||||
| (R–T2) 95% CI, 0.010 to 0.27; p=0.030 | |||||
| (R–D) 95% CI, 0.05 to 0.34; p=0.003 | |||||
| (T1–T2) 95% CI, −0.05 to 0.07; p=0.998 | |||||
| Pulmonary peak velocity (m/s) | 0.55 (0.13) | 0.53 (0.14) | 0.63 (0.14) | 0.48 (0.09) | <0.0001 |
| (R–T1) 95% CI, 0.013 to 0.159; p=0.012 | |||||
| (R–T2) 95% CI, 0.026 to 0.170; p=0.002 | |||||
| (R–D) 95% CI, 0.069 to 0.237; p<0.0001 | |||||
| (T1–T2) 95% CI, −0.048 to 0.071; p>0.1 | |||||
| Pulmonary valve diameter (cm) | 0.50 (0.13) | 0.48 (0.10) | 0.48 (0.14) | 0.42 (0.10) | 0.017 |
| (D–T1) 95% CI, −0.15 to −0.013; p=0.011 | |||||
| (D–T2) 95% CI, −0.14 to −0.0003; p=0.048 | |||||
| (D–R) 95% CI, −0.15 to 0.016; p=0.202 | |||||
| (T1–T2) 95% CI, −0.043 to 0.068; p>0.1 | |||||
| Umbilical artery pulsatility index | 1.27 (0.22) | 1.31 (0.31) | 1.33 (0.38) | 1.71 (0.46) | <0.0001 |
| (D–T1) 95% CI, 0.20 to 0.69; p<0.0001 | |||||
| (D–T2) 95% CI, 0.14 to 0.65; p=0.001 | |||||
| (D–R) 95% CI, 0.09 to 0.68; p=0.005 | |||||
| (T1–T2) 95% CI, −0.18 to 0.08; p=0.876 |
Values are mean (SD).
One twin 1, five recipients, and two donors were excluded because of intrauterine demise before fetal echocardiography. One twin and one recipient were excluded because of structural heart defects (pulmonary valve stenosis and pulmonary atresia intact septum respectively).
ANOVA, analysis of variance; CI, confidence interval; D, donor; R, recipient; T1, twin one (larger); T2, twin two (smaller).
Table 3.
Non-significant haemodynamic variables in monochorionic diamniotic twin fetuses with and without twin-twin transfusion syndrome
| Variable | Twin 1 (n=87) | Twin 2 (n=89) | Recipient (n=41) | Donor (n=45) | ANOVA (p value) |
| AoI (cm/kg) | 0.69 (0.33) | 0.73 (0.31) | 0.65 (0.21) | 0.72 (0.30) | 0.09 |
| PVI (cm/kg) | 0.86 (0.39) | 0.92 (0.39) | 0.78 (0.25) | 0.99 (0.37) | 0.08 |
| Heart rate (beats/min) | 143 (9.6) | 145 (8.7) | 144 (9.5) | 146 (11.0) | 0.45 |
| Aortic ET (ms) | 184 (20) | 185 (21) | 189 (23) | 180 (21) | 0.46 |
| Aortic AT (ms) | 49 (13) | 51 (15) | 48 (12) | 45 (13) | 0.27 |
| Pulmonary ET(ms) | 197 (25) | 193 (24) | 201 (32) | 187 (26) | 0.20 |
| Pulmonary AT (ms) | 42 (15) | 45 (13) | 41 (12) | 39 (12) | 0.23 |
| LVOI (ml/min/kg) | 196 (84) | 196 (67) | 204 (100) | 183 (107) | 0.84 |
| RVOI (ml/min/kg) | 292 (124) | 327 (147) | 311 (142) | 268 (98) | 0.27 |
| CVOI (ml/min/kg) | 488 (164) | 532 (199) | 471 (151) | 440 (165) | 0.26 |
Values are mean (SD).
One twin 1, five recipients, and two donors were excluded because of intrauterine demise before fetal echocardiography. One twin and one recipient were excluded because of structural heart defects (pulmonary valve stenosis and pulmonary atresia with intact septum, respectively).
ANOVA, analysis of variance; AoI, aortic valve diameter indexed to estimated fetal weight; AT, acceleration time; CVOI, combined ventricular output index; ET, ejection time; LVOI, left ventricular output index; PVI, pulmonary valve diameter indexed to estimated fetal weight; RVOI, right ventricular output index.
No haemodynamic differences were seen between the heavier (twin 1) and lighter twins (twin 2) in control monochorionic diamniotic twins without twin-twin transfusion syndrome (tables 2 and 3). However, intergroup analysis showed that the mean cardiothoracic ratio and Doppler peak velocities at the pulmonary valve of the recipient twins were significantly higher than those of either the donor, or twin 1/twin 2 (ANOVA p < 0.0001 and p < 0.0001, respectively). Aortic Doppler peak velocities were significantly higher in the recipient twins than in donors or in twin 1 or twin 2 (p = 0.003, p = 0.05, and p = 0.03). The four groups did not differ significantly in heart rate, aortic valve diameter, left and right ventricular output indexed to estimated fetal weight, or aortic and pulmonary Doppler ejection and acceleration times. Inter-twin discordance in heart rate in pregnancies complicated by twin-twin transfusion syndrome is often recognised, with the donor twin having an increased heart rate, though cardiac performance—measured crudely using semilunar valve acceleration and ejection times—is maintained until significant fetal compromise is evident. This suggests that twins affected by twin-twin transfusion syndrome have physiological compensation for any circulatory imbalance present at the time of the scan. The donor pulmonary valve was significantly smaller than in either twin 1 or twin 2 (ANOVA p = 0.017; Bonferroni multiple comparisons tests, p = 0.011 and p = 0.048, respectively), but when aortic and pulmonary valve sizes were expressed in terms of estimated fetal weight there was no significant difference among the four groups of twins (p = 0.093 and p = 0.082, respectively).
Functional abnormalities were seen more commonly in the recipient twin, and rarely in monochorionic diamniotic twins without twin-twin transfusion syndrome (table 4). Two recipients (4.8%) had hydrops manifesting as ascites and pericardial effusion or generalised skin oedema.
Table 4.
Abnormal haemodynamic findings in monochorionic diamniotic twin fetuses with and without twin-twin transfusion syndrome
| Finding | Twin 1 (n=88) | Twin 2 (n=89) | Recipient (n=42) | Donor (n=45) | p Value (R v D) |
| Hypertrophy | 1 (1.1%) | 0 (0%) | 24 (57.1%) | 3 (6.7%) | <0.0001 |
| Abnormal function | 0 (0%) | 0 (0%) | 8 (19.0%) | 3 (6.7%) | 0.11 |
| Significant MR | 0 (0%) | 0 (0%) | 6 (14.3%) | 0 (0%) | 0.01 |
| Significant TR | 1 (1.1%) | 0 (0%) | 10 (23.8%) | 2 (4.4%) | 0.01 |
| UA AREDF | 0 (0%) | 3 (3.4%) | 3 (7.1%) | 10 (22.2%) | 0.07 |
| DV AREDF | 0 (0%) | 0 (0%) | 9 (21.4%) | 1 (2.2%) | 0.006 |
| Fetal hydrops | 0 (0%) | 0 (0%) | 2 (4.8%) | 0 (0%) | 0.23 |
| UV pulsations | 0 (0%) | 0 (0%) | 5 (11.9%) | 0 (0%) | 0.02 |
One twin 1, five recipients, and two donors were excluded owing to intrauterine demise before to fetal echocardiography. One twin and one recipient were excluded because of structural heart defects (pulmonary valve stenosis and pulmonary atresia with intact septum, respectively).
AREDF, absence or reversal of end diastolic flow; D, donor twin; DV, venous duct; MR, mitral regurgitation; R, recipient twin; TR, tricuspid regurgitation; UA, umbilical artery; UV, umbilical vein.
The effects of intervention for twin-twin transfusion syndrome on recipient cardiac function are presented in table 5. Three of nine recipients showed some improvement, which was transient in one case and sustained in another because selective fetocide of the donor halted the inter-twin transfusion process. Haemodynamic variables worsened in three despite intervention.
Cardiac and extracardiac malformations, including chromosomal abnormalities, are shown in table 6. The prevalence of congenital heart disease in twin-twin transfusion syndrome and in the control twins was 6.9% (6/87) and 2.2% (4/177), respectively. This difference is not significant (Fisher’s exact test: p = 0.086) but the prevalence of congenital heart disease in recipient twins was 11.9% (5/42), significantly higher than in the monochorionic diamniotic controls (Fisher’s exact test: p = 0.014). In twins without twin-twin transfusion syndrome there were two cases of pulmonary valve stenosis. One was diagnosed antenatally (Doppler velocity 3.1 m/s at 23 weeks gestation) and required balloon dilatation in the neonatal period. Pulmonary velocity in the second was only mildly elevated at 1 m/s at 19 weeks and remains mild, requiring no intervention at 3.4 years of age. In this same cohort, one tiny ventricular septal defect was identified antenatally which closed spontaneously after birth, and isolated aortic coarctation was diagnosed and repaired in the neonatal period in one twin 2.
Table 6.
Congenital anomalies and prevalence of congenital heart disease in monochorionic diamniotic twins with and without twin-twin transfusion syndrome
| Congenital anomaly | Twin 1 (n=88) | Twin 2 (n=89) | Recipient (n=42) | Donor (n=45) | Outcome |
| Heart | |||||
| Pulmonary valve stenosis | 2 | 2 | Balloon dilatation (in two) | ||
| Ventricular septal defect | 1 | 2 | 1 | Spontaneous closure | |
| Aortic coarctation | 1 | Surgical repair | |||
| PA/IVS | 1 | Selective termination | |||
| Other | |||||
| Single umbilical artery | 1 | 1 | |||
| Exomphalos | 1 | Surgical repair | |||
| Gastroschisis | 1 | Surgical repair | |||
| Chromosomal | |||||
| Turner’s mosaic | 1 | 1 | Intrauterine demise (both) | ||
| Trisomy 21 | 1 | 1 | Intrauterine demise, recipient | ||
| 46XX inv(9)(p11q13) | 1 | 1 | Alive and well | ||
| Prevalence of congenital heart disease | MCDA 2.26% | TTTS 6.90% | Total 3.79% | ||
One twin 1, five recipients, and two donors were excluded owing to intrauterine demise before fetal echocardiography.
PA/IVS, pulmonary atresia/intact ventricular septum; MCDA, monochorionic diamniotic twin; TTTS, twin-twin transfusion syndrome.
In the group with twin-twin transfusion syndrome, pulmonary valve stenosis was diagnosed in two recipients after birth, one of whom required balloon dilatation. This fetus had ventricular volume loading and hypertrophy, with moderate tricuspid and mitral regurgitation, but pulmonary Doppler velocity was normal (0.77 m/s) at 27.4 weeks. Fetal examination of the second case at 28 weeks was normal apart from a slightly increased cardiothoracic ratio (39%). Pulmonary Doppler velocity was 0.4 m/s, but mild pulmonary valve and bilateral peripheral pulmonary artery stenoses were present at the latest follow up at four months of age. Fetal ventricular septal defects were seen in one donor and one recipient which closed spontaneously after birth. One recipient had a complex structural cardiac defect (pulmonary atresia with intact ventricular septum). At 17 weeks of gestation this fetus had a dilated, poorly contractile heart, significant tricuspid regurgitation, reversal of flow in the venous duct, and pulsatile umbilical venous Doppler; the parents opted for selective termination of pregnancy. Fetuses with Turner’s mosaic or trisomy 21 all had structurally normal hearts.
DISCUSSION
We have described the fetal haemodynamics and prevalence of structural heart defects in the largest prospective case–control series of unselected monochorionic diamniotic twins with and without twin-twin transfusion syndrome reported to date. Our results show that identical twins without twin-twin transfusion syndrome have concordant fetal haemodynamics but that structural defects are sevenfold more common than in the general population 17 and usually occur in only one of a twin pair. In twin-twin transfusion syndrome, the associated circulatory imbalance is manifested by increased fetal aortic and pulmonary velocities in the recipient twin compared with either its co-twin or with uncomplicated monochorionic diamniotic twins. This reflects the active Frank–Starling mechanism in the recipient in response to an increased preload. Further responses to chronic volume loading in the recipient heart in this series were ventricular hypertrophy in 57% and significant right atrioventricular valve regurgitation in 24%.
While severe right and left sided cardiomyopathy has been described in some fetuses and neonates following twin-twin transfusion syndrome, an isolated increase in fetal aortic velocity has not been shown before in the recipient twin. The prevalence of significant haemodynamic abnormalities in the recipient twins in this study, while important, is less than the 80–100% quoted in other studies, reflecting possible bias associated with smaller observational series including our initial case series.10–13
Although the triggering mechanism for haemodynamic imbalance leading to twin-twin transfusion is not clearly understood, there is evidence that both twins experience a hostile intrauterine environment which may affect the regulatory control of their circulations. In utero calcification of the recipient aorta and pulmonary artery, with hyperplasia of the intima and media, has been reported in the absence of valvar disease and is likely to reflect the response of the arterial wall to chronic pressure overload and the increased sheer stress associated with twin-twin transfusion syndrome.18, 19 This was not seen in our series.
There have been case reports of neonatal hypertension in an “ex-donor” twin, possibly caused by reversal of twin-twin transfusion syndrome after the death of the recipient co-twin,20 and also of hypertension requiring treatment with calcium channel antagonists in neonates with left ventricular hypertrophic cardiomyopathy.12 One possible mechanism may be an increase in plasma endothelin-1 (our group found this to be increased 2.5-fold compared with donor co-twins). Endothelin-1 is a potent vasoconstrictive agent and was particularly high in five fetuses who required balloon dilatation for right ventricular outflow tract obstruction after birth.10 We speculate that increased endothelin-1, as documented on both fetal blood sampling and at delivery, may have contributed to recipient cardiac dysfunction.21
The donor fetus, in contrast, shows evidence of chronic hypovolaemia resulting in activation of the renin-angiotensin system. This has been confirmed by finding an overexpression of renin gene and protein in the donor kidney, with virtually absent expression in the recipient kidney.22, 23 This upregulation initially attempts to correct the volume depletion experienced by the donor, and transfusion of this increased renin to the recipient might further contribute to recipient hypertension. Raised concentrations of angiotensin II result in increased collagen synthesis, smooth muscle hypertrophy, and vascular medial hypertrophy in fetal sheep,24 and similar mechanisms may be responsible for the increased vascular stiffness we have shown in surviving donors in childhood.14 The increased umbilical artery pulsatility indices in the donor twins in our study compared with unaffected monochorionic diamniotic twins provides further evidence of a widespread feto-placental circulatory disorder in twin-twin transfusion syndrome. Umbilical artery Doppler abnormalities (absence of end diastolic flow) were seen more commonly in donor than in recipient twins, with significant inter-twin discordancy of the umbilical artery index. Previous studies of umbilical artery Doppler velocimetry in twin-twin transfusion syndrome have yielded inconsistent results25; our finding of altered umbilical artery flow in the smaller co-twin resembles that described in compromised fetuses with intrauterine growth restriction and suggests increased placental resistance.26
Twins are known to have an increased incidence of congenital malformations, but even in monochorionic twins only one twin is affected in more than 90% of cases.27, 28 The mechanism for the increase in malformations remains unknown, although several explanations have been proposed. One possibility is that the monozygotic twinning process itself increases the incidence of congenital heart disease, with postzygotic unequal division of the inner cell mass being responsible for discordant cardiovascular anatomy. Disturbance of laterality may also contribute to the development of heart defects in twins.29 Laterality is thought to be determined from the left side of the fetus. Inability of either of the co-twins to distinguish between left and right would have a major influence on the embryogenesis of the heart, the two sides of which are distinct in anatomy and function. Another contributor might be phenotypic variability of the same genome30 or possibly even genetic inter-twin differences caused by de novo mutations.
Existing data on the effects of the type of placentation on the incidence of congenital anomalies do not support the hypothesis that monochorionicity increases the rate of malformations in general.31 Nevertheless, congenital heart defects occur more commonly in monochorionic than in dichorionic monozygotic twins,32 and in terms of acquired congenital heart lesions monochorionic placentation and its complications may provide a less favourable environment for the developing fetus. In addition to the altered vascular responses described in childhood,14 fetal responses to circulatory imbalance in chronic twin-twin transfusion may provide the substrate for pulmonary valve stenosis. Despite haemodynamic similarity and an identical genome, the monochorionic diamniotic twin pairs in this study that were unaffected by twin-twin transfusion were discordant for congenital heart defects, with an incidence of congenital heart disease that was four times higher at 2.3% than in the general population.17 Superimposed on this high background prevalence, the presence of twin-twin transfusion syndrome increased the incidence of congenital heart disease in this study to 6.9%, 12-fold that of the general population.
Although the prevalence of congenital heart disease in twin-twin transfusion syndrome was higher than in control monochorionic diamniotic twins, this did not achieve significance. However, the prevalence of congenital heart disease in recipients alone (12%) was significantly higher than in monochorionic diamniotic controls.
In this study, three fetuses (two recipients) who subsequently developed neonatal pulmonary stenosis had normal fetal pulmonary velocities in the second trimester (range 0.4–1 m/s). This highlights the importance of caution in counselling in early pregnancy and the need for serial fetal echocardiograms, as right ventricular outflow tract obstruction is a dynamic process which may evolve until delivery, whereupon the haemodynamic insult ceases. Pulmonary stenosis in our recipients accounted for one third of the cases of congenital heart disease in twin-twin transfusion syndrome, with an incidence of 4.8%. This is considerably higher than the prevalence of pulmonary stenosis estimated in the population (0.5 to 1.5 per 1000).33, 34
Our findings support the suggestion that twins may not represent the ideal population for genetic studies of the cardiovascular system,35 as multifactorial and probably unknown antenatal factors influence the phenotypic outcome.
Conclusions
Monochorionic diamniotic twinning is associated with a 3.8% incidence of congenital heart disease. While uncomplicated monochorionic diamniotic twins show concordant haemodynamics, heart defects are usually only seen in one of a pair. The circulatory imbalance in twin-twin transfusion syndrome further augments this increased incidence of congenital heart disease in recipient twins.
With the increasing success of assisted reproduction techniques, the incidence of multifetal pregnancies, including monochorionic diamniotic twinning, is increasing.36 Monochorionic diamniotic pregnancies are recognised as posing a high risk and they require close antenatal surveillance. The prevalence of congenital heart disease in our cohort is comparable with that described in the pregnancies of parents who have congenital heart disease themselves, thus underscoring the importance of detailed fetal echocardiography in the care of all monochorionic diamniotic pregnancies.37, 38
Acknowledgments
Salary support was received from the British Heart Foundation (JLW), the Royal Brompton Hospital Clinical Research Committee (AAK), SPARKS (MJOT), and the Richard and Jack Wiseman Trust (LW and MJOT).
REFERENCES
- 1.Taylor MJO, Fisk NM. Prenatal diagnosis in multiple pregnancy. Baillieres Clin Obstet Gynaecol 2000;14:663–75. [DOI] [PubMed] [Google Scholar]
- 2.Duncan KR, Denbow ML, Fisk NM. The aetiology and management of twin–twin transfusion syndrome. Prenat Diagn 1997;17:1227–36. [DOI] [PubMed] [Google Scholar]
- 3.Denbow ML, Cox P, Talbert D, et al. Colour Doppler energy insonation of placental vasculature in monochorionic twins: absent arterio-arterial anastomoses in association with twin-to-twin transfusion syndrome. Br J Obstet Gynaecol 1998;105:760–5. [DOI] [PubMed] [Google Scholar]
- 4.Brennan JN, Diwan RV, Rosen MG, et al. Fetofetal transfusion syndrome: prenatal ultrasonographic diagnosis. Radiology 1982;143:535–6. [DOI] [PubMed] [Google Scholar]
- 5.Denbow ML, Fisk NM. The consequences of monochorionic placentation. Baillieres Clin Obstet Gynaecol 1998;12:37–51. [DOI] [PubMed] [Google Scholar]
- 6.Dennis LG, Winkler CL. Twin-to-twin transfusion syndrome: aggressive therapeutic amniocentesis. Am J Obstet Gynaecol 1997:177:342–9. [DOI] [PubMed] [Google Scholar]
- 7.Saade GR, Belfort MA, Berry DL, et al. Amniotic septostomy for the treatment of twin oligohydramnios–polyhydramnios sequence. Fetal Diagn Ther 1998;13:86–93. [DOI] [PubMed] [Google Scholar]
- 8.Deprest J, Audibert F, Van Schoubroeck D, et al. Bipolar coagulation of the umbilical cord in complicated monochorionic twin pregnancy. Am J Obstet Gynaecol 2000;182:340–5. [DOI] [PubMed] [Google Scholar]
- 9.Zikulnig L, Hecher K, Bregenzer T, et al. Prognostic factors in severe twin–twin transfusion syndrome treated by endoscopic laser surgery. Ultrasound Obstet Gynecol 1999;14:380–7. [DOI] [PubMed] [Google Scholar]
- 10.Zosmer N, Bajoria R, Weiner E, et al. Clinical and echocardiographic features of in utero cardiac dysfunction in the recipient twin in twin-to-twin transfusion syndrome. Br Heart J 1994;72:74–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cincotta R, Oldham J, Sampson A. Antepartum and postpartum complications of twin–twin transfusion. Aust NZ J Obstet Gynaecol 1996;36:303–8. [DOI] [PubMed] [Google Scholar]
- 12.Fesslova V, Villa L, Nava S, et al. Fetal and neonatal echocardiographic findings in twin–twin transfusion syndrome. Am J Obstet Gynecol 1998;179:1056–62. [DOI] [PubMed] [Google Scholar]
- 13.Simpson LL, Marx GR, Elkadry EA, et al. Cardiac dysfunction in twin–twin transfusion syndrome: a prospective, longitudinal study. Obstet Gynecol 1998;92:557–62. [DOI] [PubMed] [Google Scholar]
- 14.Cheung YF, Taylor MJO, Fisk NM, et al. Fetal origins of reduced arterial distensibility in the donor twin in twin–twin transfusion syndrome. Lancet 2000;355:1157–8. [DOI] [PubMed] [Google Scholar]
- 15.Allan LD, Joseph MC, Boyd EG, et al. M-mode echocardiography in the developing human fetus. Br Heart J 1982;47:573–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taylor MJO, Shalev E, Tanawattanacharoen S, et al. Ultrasound guided umbilical cord occlusion using bipolar diathermy for stage III/IV twin–twin transfusion. Prenat Diagn 2002;22:70–6. [DOI] [PubMed] [Google Scholar]
- 17.Wren C, Richmond S, Donaldson L. Temporal variability in birth prevalence of cardiovascular malformations. Heart 2000;83:414–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Popek EJ, Strain JD, Neumann A, et al. In utero development of pulmonary artery calcification in monochorionic twins: a report of three cases and discussion of the possible etiology. Pediatr Pathol 1993;13:597–611. [DOI] [PubMed] [Google Scholar]
- 19.Nicosia RF, Krouse TB, Mobini J. Congenital aortic intimal thickening. Its occurrence in a case of twin-transfusion syndrome. Arch Pathol Lab Med 1981;105:247–9. [PubMed] [Google Scholar]
- 20.Baud O, Lebidois J, Van Peborgh P, et al. Fetal and neonatal hypertension in twin–twin transfusion syndrome: a case report. Fetal Diagn Ther 1998;13:223–6. [DOI] [PubMed] [Google Scholar]
- 21.Bajoria R, Sullivan M, Fisk NM. Endothelin concentrations in monochorionic twins with severe twin–twin transfusion syndrome. Hum Reprod 1999;14:1614–18. [DOI] [PubMed] [Google Scholar]
- 22.Mahieu-Caputo D, Dommergues M, Delezoide A-L, et al. Twin-to-twin transfusion syndrome. Role of the fetal renin-angiotensin system. Am J Pathol 2000;156:629–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kilby MD, Platt C, Whittle MJ, et al. Renin expression in fetal kidneys of pregnancies complicated by twin–twin transfusion syndrome. Pediatr Dev Pathol 2001;4:175–9. [DOI] [PubMed] [Google Scholar]
- 24.Robillard JE, Gomez RA, Meernik JG, et al. Role of angiotensin II on the adrenal and vascular responses to hemorrhage during development in fetal lambs. Circ Res 1982;50:645–50. [DOI] [PubMed] [Google Scholar]
- 25.Giles WB. Doppler ultrasound in multiple pregnancies. Baillieres Clin Obstet Gynaecol 1998;12:77–89. [DOI] [PubMed] [Google Scholar]
- 26.Hecher K, Campell S, Doyle P, et al. Assessment of fetal compromise by Doppler ultrasound investigation of the fetal circulation. Arterial, intracardiac, and venous blood flow velocity studies. Circulation 1995;91:129–38. [DOI] [PubMed] [Google Scholar]
- 27.Bryan E, Little J, Burn J. Congenital anomalies in twins. Baillieres Clin Obstet Gynaecol 1987;1:697–721. [DOI] [PubMed] [Google Scholar]
- 28.Nakayama D, Masuzaki H, Yoshimura S, et al. Monozygotic twins discordant for single umbilical artery and congenital heart disease. Am J Obstet Gynecol 1998;179:256–7. [DOI] [PubMed] [Google Scholar]
- 29.Burn J. Disturbance of morphological laterality in humans. Ciba Found Symp 1991;162:282–96. [DOI] [PubMed] [Google Scholar]
- 30.Goodship J, Cross I, Scambler P, et al. Monozygotic twins with chromosome 22q11 deletion and discordant phenotype. J Med Genet 1995;32:746–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corney G, MacGillivray I, Campbell DM, et al. Congenital anomalies in twins in Aberdeen and Northeast Scotland. Acta Genet Med Gemellol 1983;32:31–5. [DOI] [PubMed] [Google Scholar]
- 32.Cameron AH, Edwards JH, Derom R, et al. The value of twin surveys in the study of malformations. Eur J Obstet Gynecol Reprod Biol 1983;14:347–56. [DOI] [PubMed] [Google Scholar]
- 33.Samanek M, Voriskova M. Congenital heart disease among 815,569 children born between 1980 and 1990 and their 15-year survival: a prospective Bohemia survival study. Pediatr Cardiol 1999;20:411–17. [DOI] [PubMed] [Google Scholar]
- 34.Grech V. History, diagnosis, surgery and epidemiology of pulmonary stenosis in Malta. Cardiol Young 1998;8:337–43. [DOI] [PubMed] [Google Scholar]
- 35.Luft FC. Twins in cardiovascular genetic research. Hypertension 2001;37:350–6. [DOI] [PubMed] [Google Scholar]
- 36.Derome C, Deron R, Vlietinck R et al. Iatrogenic multiple pregnancies at east Flanders, Belgium. Fertil Steril 1993;60:493–6. [PubMed] [Google Scholar]
- 37.Whittemore R, Wells JA, Castellsague X. A second-generation study of 427 probands with congenital heart disease and their 837 children. J Am Coll Cardiol 1994;23:1459–67. [DOI] [PubMed] [Google Scholar]
- 38.Burn J, Brennan P, Little J, et al. Recurrence risks in offspring of adults with major heart defects: results from first cohort of British collaborative study. Lancet 1998;351:311–16. [DOI] [PubMed] [Google Scholar]