

Abstract
Background: Hypertrophic cardiomyopathy (HCM) is an autosomal dominant disease caused by mutations in sarcomeric genes. However, extensive genetic screening failed to identify a mutation in about a third of cases. One possible explanation is that other diseases, caused by other genes, may mimic HCM.
Objective: To investigate the possible involvement of Danon’s disease, an X linked lysosomal disease, in a large population of patients with HCM.
Methods: A population of 197 index cases was considered; 124 were subsequently excluded because of a mutation in sarcomeric genes and 23 because of autosomal dominant inheritance. Fifty index cases were therefore included in molecular analysis (direct sequencing) of the lysosome associated membrane protein 2 (LAMP2) gene responsible for Danon’s disease.
Results: Two new mutations leading to premature stop codons were identified in patients who evolved towards severe heart failure (< 25 years old): 657C>T and 173_179del. The prevalence was therefore 1% of the total population (two of 197) or 4% of enrolled index cases (two of 50). Interestingly, Danon’s disease was responsible for half of the cases (two of four) with HCM and clinical skeletal myopathy but was not involved in isolated HCM (none of 41).
Conclusions: Danon’s disease may be involved in patients with previously diagnosed as HCM. A diagnosis strategy is proposed. To distinguish HCM from Danon’s disease is important because the clinical evolution, prognosis, mode of inheritance, and therefore genetic counselling are very different.
Keywords: gene, hypertrophic cardiomyopathy, Danon’s disease, LAMP2, lysosome associated membrane protein 2, skeletal myopathy
Hypertrophic cardiomyopathy (HCM) is a myocardial disease of genetic origin, usually with an autosomal dominant inheritance and caused by mutations affecting sarcomeric proteins.1,2 However, extensive screening of these genes failed to identify the mutation responsible in about one third of index cases.3 One possible explanation is that other cardiac diseases, caused by additional genes, may mimic HCM. This was recently shown for some diseases such as Fabry’s disease, responsible for 3% of HCM,4,5 and for HCM associated with the Wolff-Parkinson-White syndrome related to the γ2 subunit of AMP activated protein kinase (PRKAG2) gene.6
To test this hypothesis further, we analysed the possible involvement of Danon’s disease in consecutive index cases of idiopathic HCM. Danon’s disease is an X linked (dominant rather than recessive) lysosomal disease due to a primary deficiency of lysosome associated membrane protein 2 (LAMP2).7,8 The pathological hallmark of the disease is intracytoplasmic vacuoles containing autophagic material and glycogen in cardiac and skeletal muscle cells.7,9 The phenotype typically associated with mutations in the LAMP2 gene is characterised by cardiomyopathy and skeletal myopathy, sometimes conduction defect, Wolff-Parkinson-White syndrome, or mental retardation.7–9 The phenotypic expression of Danon’s disease is, however, variable. The involvement of the gene in common forms of HCM has not been examined.
We decided therefore to screen systematically the LAMP2 gene in consecutive patients previously diagnosed as HCM, after excluding a mutation in sarcomeric genes.
METHODS
Study population
We considered 197 independent index cases (including 172 with familial forms) of clinically diagnosed HCM (unexplained left ventricle hypertrophy with maximum wall thickness > 13 mm). Written informed consent was obtained from each patient in accordance with a study protocol approved by a local ethics committee. After mutational screening of nine sarcomeric genes, no mutation was found in 73 index cases.3 In these cases, the pedigrees were reviewed and families with certain autosomal dominant inheritance (male to male transmission) were excluded (n = 23). Fifty index cases were therefore included in the study of the LAMP2 gene (fig 1). Thirty two of the participants were male (64%) and familial forms accounted for 40 (80%). Among enrolled index cases, there were 41 patients with common and isolated HCM, four patients with HCM and clinical skeletal myopathy (with or without cardiac conduction abnormalities), two patients with HCM and increased serum creatine kinase (CK) concentration but normal skeletal muscle examination, and three patients with HCM and Wolff-Parkinson-White syndrome.
Figure 1.

Population evaluated in the study. HCM, hypertrophic cardiomyopathy.
Molecular genetics
Genomic DNA was extracted from white blood cells of patients. We directly sequenced the entire coding regions of the LAMP2 gene, located on Xq24, including the nine exons (1 to 9a) and intron-exon junctions, with primers previously described.8 Sequencing reactions were performed with the ABI PRISM 3100 genetic analyser (Applied Biosystems, Courtaboeuf, France) and BigDye Terminator chemistry.
Muscle biopsies and immunohistochemistry
Skeletal muscle biopsies from patients with a mutation in the LAMP2 gene and from one control subject were analysed. Some transverse frozen sections were stained with the Gomori trichrome or periodic acid Schiff stain. Others were immunostained with a monoclonal antibody against the LAMP2 protein (monoclonal antibody anti-CD107b, clone H4B4, SouthernBiotech, Birmingham, Alabama, USA) or with a monoclonal antibody against the dystrophin protein (clone NCL-DYS1, Novocastra Laboratories, Newcastle, United Kingdom). In all cases a goat anti-mouse tetramethylrhodamine isothiocyanate conjugated antibody was used as a secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, Pennsylvania, USA).
RESULTS
Molecular genetics
Two new mutations in the LAMP2 gene were identified in two index cases (fig 2). They were a C/T657 transition in exon 4 (nucleotide position according to the GenBank accession number S79873) leading directly to a stop codon (Q174X) (patient 1); and a 7 bp deletion in exon 1 (173_179del), leading to a frameshift mutation and a premature stop codon (codon 17; patient 2). In the respective families, DNA was available for the mothers. The mother of patient 2 carried the mutation found in her son. The mother of patient 1 did not carry the mutation, which was therefore a de novo mutation.
Figure 2.

Results of genetic sequencing with two mutations identified in the lysosome associated membrane protein 2 (LAMP2) gene. Sequence electrophoregrams representative of a control DNA sample (wild type allele) or of patient 1, a C>T substitution in exon 4 at position 657 of the gene creating a premature stop codon (TAA, boxed); or of patient 2, a 7 bp deletion (boxed sequence) at position 173_179del in exon 1 leading to a change in reading frame and a premature stop codon.
Clinical features
Figure 3 shows family pedigrees.
Figure 3.

Pedigrees of families A and B. Black symbol, affected subject; white symbol, unaffected subject; shaded symbol, borderline status; slashed symbol, deceased member; 0 absence of a mutation; mut. + presence of a mutation; NA data not available.
In family A, HCM was diagnosed in index patient 1 (II-1, fig 3) at 14 years because of syncope. Sinus rhythm was present on the ECG, with a high QRS voltage and Wolff-Parkinson-White syndrome. Echocardiography exhibited increased left ventricle wall thickness (septum 18 mm, posterior wall 13 mm), no outflow tract gradient, and normal ejection fraction. Mild skeletal weakness and increased CK plasma concentration (four times the normal value) were observed. The evolution was characterised by a conduction defect (atrioventricular block and pause of five seconds) and atrial arrhythmia, leading to pacemaker implantation at 21 years. At the same time, myocardial contractility progressively decreased (ejection fraction 16%), ventricle diameter increased (79 mm), and heart failure developed. Genetic analysis and muscular biopsy were performed at that time. Diffuse limb muscle weakness and atrophy were observed, especially in the quadriceps and scapular muscles, without contractures. Plasma concentration of maltase acid was normal. There was no mental retardation (slight learning difficulties at school but intelligence quotient (IQ) of 91 at 14 years) and no ophthalmic abnormality. Heart transplantation was performed at 22 years because of severe heart failure but the patient died seven days later, following pulmonary complications. No additional cardiac or muscular disease was present (normal physical examination, echocardiography, and ECG) in relatives of the patient (in parents and the sister).
In family B, HCM was diagnosed in index patient 2 (III-1, fig 3) in childhood during a systematic examination performed because of four cardiac deaths in the family (three uncles, 7 to 34 years, and the grandmother, at 32 years). ECG at 16 years showed sinus rhythm and large and deep Q waves in D1 and VL. Echocardiography showed increased wall thickness (inferior wall 20 mm, septal wall 12 mm, lateral wall 16 mm), no outflow tract gradient, normal left ventricular diameter (43 mm), and normal ejection fraction. There was no skeletal muscle abnormality. Atrial tachycardia and second degree atrioventricular block subsequently occurred. Ventricular tachycardia was induced during electrophysiological study leading to defibrillator cardioverter implantation at 22 years. At the time of genetic inquest (24 years), systolic function of the left ventricle was dramatically decreased (ejection fraction 20%, left ventricular diastolic diameter 64 mm). There was very mild distal muscle weakness, mild amyotrophy of the lower limbs with pes cavus, and increased CK concentration (five times the normal concentration). There was no mental retardation (despite slight scholarly difficulties in the past). Severe decrease of bilateral visual acuity was observed, related to choriocapillary ocular atrophy. The patient died of heart failure at 25 years while waiting for heart transplantation. Family DNA was not available from the four relatives who had died of cardiac disease. Cardiomyopathy was present in all four, skeletal myopathy in three of them, and conduction defect, pacemaker, and ophthalmic abnormalities in two of them. The mother (44 years) of the index patient also carried the mutation. She had no symptoms, a normal ECG, and very mild echocardiographic abnormalities (interventricular septum 12 mm, left atrial diameter 42 mm).
Skeletal muscle biopsies
The effects of the mutations on protein expression were analysed in skeletal muscle biopsies from the two index patients (fig 4). Abnormalities were similar for both. Light microscopy showed some sarcoplasmic clear vacuoles (fig 4B) that were periodic acid Schiff positive. Immunohistochemical study found dystrophin on vacuole membranes (fig 4F). In contrast, immunohistochemical analysis with antibodies against LAMP2 protein did not detect any material, showing that LAMP2 protein was absent in the skeletal muscles of the patients (fig 4D). All these features are typical of Danon’s disease.7–9
Figure 4.
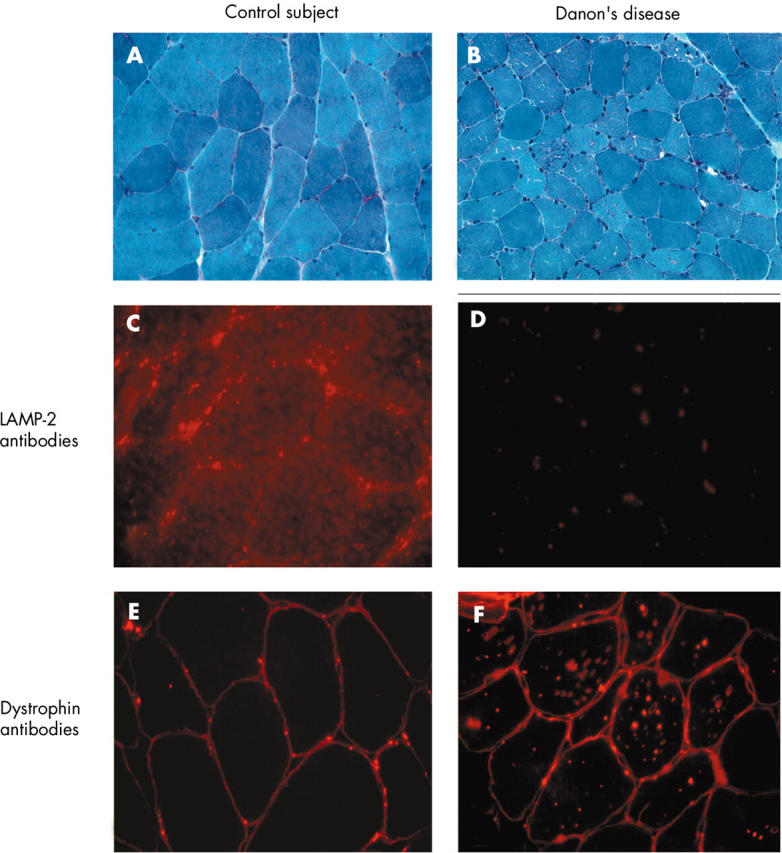
Skeletal muscle biopsy. (A, C, E) Normal sections; and (B, D, F) sections from the patient. All sections were analysed with a 60× oil objective lens, except image B, with 40×. (A, B) Light microscopy (Gomori trichrome stain). Presence of multiple small vacuoles in some fibres. (C, D) Immunohistochemistry with LAMP2 antibodies. (E, F) Immunohistochemistry with dystrophin antibodies.
DISCUSSION
This study is the first attempt to evaluate the prevalence of Danon’s disease among a large population of patients with a previous diagnosis of HCM. The findings may therefore have implications for the diagnosis strategy of such patients. We found a mutation in two index patients. The prevalence in the total HCM population is 1% of all index cases (two of 197), or 3% of index cases without mutations in sarcomeric genes (two of 73) or 4% of index cases in the molecular analysis (two of 50). Interestingly, the frequency was closely related to the associated phenotype. Danon’s disease was responsible for half of the index cases (two of four) of HCM in patients who evolved towards clinical skeletal myopathy. In addition, Wolff-Parkinson-White syndrome was present in one and second degree atrioventricular block in both of these patients. In contrast, no mutation was found in isolated HCM (fig 1), in subgroups with HCM and Wolff-Parkinson-White syndrome (without skeletal myopathy), or in HCM and isolated CK increase.
A diagnostic strategy may be proposed to improve identification of patients with Danon’s disease, based on our results (in the specific context of patients with a previous diagnosis of HCM) and on results from previous studies (through the description of the clinical features of patients with Danon’s disease).8,9 As a first step, all patients with idiopathic HCM should be considered, whatever the familial context, except when it is a familial form with a male to male transmission (which excludes X linked disease). All of these patients should benefit from careful clinical and biological evaluation, including clinical muscular testing and determination of serum CK concentration, to search for additional features of Danon’s disease. Interestingly, in previous studies skeletal myopathy was mild in most cases (85%) but all male patients had increased serum CK concentration.9 Wolff-Parkinson-White syndrome was present in 35%, mild mental retardation was observed in 70%, and most patients had HCM and low ejection fraction.9 Women were less severely affected, with later onset cardiomyopathy. As a second step, and on the basis of our results, we propose that Danon’s disease can be reasonably excluded when HCM is isolated (without associated abnormalities). In contrast, in the presence of unusual clinical associations (especially HCM and clinical skeletal myopathy), the diagnosis of Danon’s disease should be discussed further. Other hereditary cardiac diseases should also be discussed (such as mitochondrial myopathy, glycogen storage disorders, and PRKAG2 gene related disease) because of overlapping clinical features (especially skeletal muscle disease and Wolff-Parkinson-White syndrome). As a third step, direct molecular screening of the LAMP2 gene should be performed to confirm or exclude Danon’s disease. An alternative would be to take a skeletal muscular biopsy to search for vacuolar myopathy, and if it is present, to analyse the biopsy immunohistochemically with specific LAMP2 antibodies. However, molecular genetic screening is less invasive, possibly more efficient or sensitive (if mutations do not lead to a stop codon and the absence of the protein), and remains the criterion of reference in women (where the protein may be present because of one normal X chromosome).
To distinguish HCM from Danon’s disease may be important, as the pathophysiology, clinical evolution, prognosis, mode of inheritance, and therefore genetic counselling are very different. Firstly, the natural history of Danon’s disease seems to be characterised in male patients by an early onset and a very poor prognosis. This was the case in our two patients with heart failure and death at 22 and 25 years. In the largest population studied so far (20 male patients), the mean age at onset was less than 20 years in all cases and all patients except one died before 30 years.9 Deaths were related to left ventricular dysfunction and congestive heart failure or sudden death. This is in contrast with the natural history of HCM, where cardiac death is evaluated to be about 1–2% a year and only 10% evolve towards heart failure.10,11 Secondly, and contrary to HCM, the clinical spectrum of Danon’s disease is broad with additional cardiac or non-cardiac features. Until now, ophthalmic abnormality has not been regarded as a manifestation of Danon’s disease. However, we suggest that it may be, as it was observed in one of our patients (patient 2 and his family) as well as in a previously published case.12 Overall, this specific spectrum and evolution justifies a specific medical follow up. Thirdly, there is no specific treatment for Danon’s disease. Management with inotrope negative or chronotrope negative agents, used for HCM, should be used with caution in Danon’s disease because of the frequent evolution towards systolic left ventricular dysfunction and atrioventricular block. Fourthly, Danon’s disease is an X linked dominant disorder and this inheritance may be difficult to recognise. As mothers of patients may be clinically healthy and because de novo mutations are possible (reported by Arad et al6 and present in our patient 1), some patients present as “sporadic” cases without a familial context. In contrast, when mothers of patients are clinically affected, the mode of inheritance may mimic the autosomal mode. Only male to male transmission allows the recognition of autosomal dominant inheritance and excludes X linked inheritance. These difficulties underline the usefulness of an accurate cardiac and molecular diagnosis. The information and genetic counselling subsequently given to patients and their relatives will obviously be different for HCM13 and Danon’s disease, especially as regards the risk of transmission. The discussion about prenatal diagnosis is also different since the severity of the disease appears obvious in all patients with Danon’s disease, whereas the prognosis is more variable and quite good for most patients with HCM related to sarcomeric genes.
In conclusion, physicians must be aware of Danon’s disease, which may mimic HCM but results in a very different clinical evolution, prognosis, mode of inheritance, and subsequently different genetic counselling. A careful clinical evaluation for this disease should therefore be considered for all patients with idiopathic HCM, especially for male patients with a diagnosis at less than 30 years but also for women with later onset. The presence of unusual clinical associations should subsequently lead to molecular screening to confirm or rule out the disease.
Abbreviations
CK, creatine kinase
HCM, hypertrophic cardiomyopathy
IQ, intelligence quotient
LAMP2, lysosome associated membrane protein 2
PRKAG2, γ2 subunit of AMP activated protein kinase
REFERENCES
- 1.Marian AJ, Roberts R. The molecular genetic basis for hypertrophic cardiomyopathy. J Mol Cell Cardiol 2001;33:655–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 2001;104:557–67. [DOI] [PubMed] [Google Scholar]
- 3.Richard P , Charron P, Carrier L, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations and implications for molecular diagnosis strategy. Circulation 2003;107:2227–32. [DOI] [PubMed] [Google Scholar]
- 4.Nakao S , Takenaka T, Maeda M, et al. An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. N Engl J Med 1995;333:288–93. [DOI] [PubMed] [Google Scholar]
- 5.Sachdev B , Takenaka T, Teragichi H, et al. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 2002;105:1407–11. [DOI] [PubMed] [Google Scholar]
- 6.Arad M , Benson DW, Perez-Atayed AR, et al. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest 2002;109:357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danon MJ, Oh SJ, DiMauro S, et al. Lysosomal glycogen storage disease with normal acid maltase. Neurology 1981;31:51–7. [DOI] [PubMed] [Google Scholar]
- 8.Nishino I , Fu J, Tanji K, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000;406:906–9. [DOI] [PubMed] [Google Scholar]
- 9.Sugie K , Yamamoto A, Murayama K, et al. Clinicopathological features of genetically confirmed Danon disease. Neurology 2002;58:1773–8. [DOI] [PubMed] [Google Scholar]
- 10.Maron BJ. Hypertrophic cardiomyopathy, a systematic review. JAMA 2002;287:1308–20. [DOI] [PubMed] [Google Scholar]
- 11.Wigle ED, Rakowski H, Kimball BP, et al. Hypertrophic cardiomyopathy: clinical spectrum and treatment. Circulation 1995;92:1680–92. [DOI] [PubMed] [Google Scholar]
- 12.Lacoste-Collin L , Garcia V, Uro-Coste E, et al. Danon’s disease: a case with a novel LAMP-2 gene mutation. Neuromuscul Disord 2002;12:882–5. [DOI] [PubMed] [Google Scholar]
- 13.Charron P , Heron D, Gargiulo M, et al. Genetic testing and genetic counselling in hypertrophic cardiomyopathy: the French experience. J Med Genet 2002;39:741–6. [DOI] [PMC free article] [PubMed] [Google Scholar]