

Abstract
Objective: To see whether sudden unexpected death in chronic heart failure is preceded by intraindividual worsening in inflammation and in ECG criteria.
Design and setting: Prospective cohort study conducted in the community.
Patients: 34 patients with chronic heart failure were studied. Their mean (SD) age was 68 (8) years, 29 were men, mean (SD) left ventricular ejection fraction was 29 (9)%, and they were in New York Heart Association functional class II (n = 20), III (n = 11), and IV (n = 3). The patients were examined monthly over 24 months, with sequential measurement of C reactive protein and neutrophil counts and 24 hour ambulatory ECG monitoring measuring heart rate variability, mean heart rate, and arrhythmias. Intraindividual changes in these parameters were related to subsequent cardiac deaths.
Results: During follow up, nine patients died: five patients had a sudden unexpected death (SUD) and four died of progressive heart failure (PHF). There were significant intraindividual changes in neutrophil counts (p = 0.02), C reactive protein (p = 0.039), and heart rate variability (p ⩽ 0.018) in those who died of SUD and PHF. In contrast no significant changes were seen in ventricular extrasystoles, ventricular tachycardia episodes, brain natriuretic peptide, or aldosterone in the SUD group, but all of these parameters did increase as expected in those who died of PHF.
Conclusions: This is preliminary evidence that SUD may be preceded by intraindividual increases in both inflammation and autonomic dysfunction. Both may be causal in genesis but, even if they are not, intraindividual increases in either may be convenient markers to identify patients at high risk of impending SUD. Larger studies are needed to confirm the observation from this pilot study.
Keywords: chronic heart failure, heart rate variability, C reactive protein, sudden cardiac death
Despite advances in the management of chronic heart failure (CHF), patients with CHF still have a high mortality rate.1–4 These deaths appear to occur by different modes: sudden unexpected death (SUD) and death caused by progressive heart failure (PHF).5,6 PHF deaths are predictable by simple clinical measures such as worsening New York Heart Association (NYHA) class, lower left ventricular ejection fraction, or low exercise capacity.7–11 By contrast, SUD in CHF is not as easy to predict, with variable results in the literature.12–14 It is generally assumed that SUD is triggered either by an acute coronary syndrome or by a primary arrhythmia. This means that some measure of an impending coronary event or of “arrhythmogenicity” may increase just before the SUD.13,14
SUD in CHF may often be primarily caused by an acute coronary syndrome, which is in turn due to the rupture of an “inflamed” atherosclerotic plaque.15 If this were true, one may expect that intraindividual signs of inflammation such as neutrophilia or C reactive protein would increase just before the SUD. This hypothesis is made likely because of the landmark work of Ridker and colleagues,16 who showed clearly that C reactive protein is increased when measured years before the SUD. This obviously raises the question of whether the SUD event is triggered by a further, short term intraindividual increase in C reactive protein and inflammation just before the SUD event. This is made likely by the fact that sites of acute rupture in plaques have much more inflammation in them than stable plaques17 and by recent pathological data that plaque instability and thrombus formation exist for days to weeks before a SUD.18
On the other hand, if “arrhythmogenicity” contributes to SUD, then ambulatory electrocardiography may be useful, but it has so far proved variable in its ability to predict SUD.12–14 This variability may be because all current studies are cross sectional, where one sample is taken from a large number of patients and there is then a long time gap before the SUD occurs. The alternative study design would be a longitudinal study where many repeated ECGs are taken from the same patients to look for intraindividual changes before the SUD.19 Apart from the time gap problem, another problem with cross sectional studies is that most parameters have greater interindividual variability than they do intraindividual variability, and high interindividual variability severely limits the ability of cross sectional studies to identify individual risk. Two measures on the ambulatory ECG that may progressively worsen as SUD approaches are heart rate variability and spontaneous ventricular extrasystole activity.19,20 One previous report of two cases did suggest that heart rate variability worsened in patients before their SUD.19
We therefore performed a pilot study to see whether SUD in CHF was preceded by intraindividual increases in inflammation (C reactive protein/neutrophils) or in certain ECG measures of arrhythmogenicity. These are the most likely “trigger” events before SUD, which is why we focused on them in this study.
METHODS
Study group and data collection
We recruited 34 patients who met the following criteria: CHF for ⩾ 18 months, clinical stability for three months; NYHA class II to IV; CHF caused by coronary artery disease (94%) or idiopathic dilated cardiomyopathy; impaired left ventricular ejection fraction of < 40%; no acute coronary event within six months; and sinus rhythm. Exclusion criteria were planned coronary revascularisation; chronic obstructive pulmonary disease; significant renal dysfunction; diabetes mellitus; autonomic neuropathy; limiting peripheral arterial disease; implanted pacemaker; treatment with β blockers; and atrial fibrillation. The local Tayside ethics and research committee approved the study before recruitment. All patients gave written and informed consent to participate in the study.
Patients were seen at monthly intervals in their own home. A 24 hour ambulatory ECG monitor (Tracker2, Reynolds Medical Ltd, Hertford, UK) was applied and calibrated. Blood samples were taken after ⩾ 20 minutes supine rest. The 24 hour ECG results were processed with a Reynolds pathfinder 600 series workstation. Neutrophils were counted by our routine haematology laboratory by standard flow cytometry. Stored plasma was used for high sensitivity C reactive protein analysis with a standard commercial enzyme linked immunosorbent assay (ELISA) kit (Immuno-biological Laboratories, Hamburg, Germany). Chlamydia pneumoniae antibodies (IgG, IgA, IgM) and immune complexes were analysed on all samples as described before.21 All monthly samples were analysed for neutrophils, brain natriuretic peptide (BNP), and aldosterone. C reactive protein and chlamydiae were measured only on a single baseline sample and in the last three samples for each patient before death or at the end of study.
All tapes were subjected to standard Holter analysis with artefacts confirmed manually. Subsequently, time domain heart rate variability was analysed automatically from RR intervals that had normal morphology and cycle lengths between 80–120% of the preceding cycle duration. All tapes were analysed and their data files were reviewed and edited by an investigator (AMAS) blinded to the individual patient’s clinical status. On average, 97% of the data for each patient were available for further analysis after editing.
Death was classified as PHF or SUD based on the conventional expert committee view that SUD was a death within one hour of new symptoms without evidence of PHF before the new symptoms began.22
Heart rate variability was analysed according to standard guidelines.23
Statistical methods
The analyses were done with SPSS (version 10, SPSS Inc, Chicago, Illinois, USA). Most of the data were normally distributed. Data that were not normally distributed or were borderline in terms of normal distribution were analysed as they were and after a log transformation. Mean values at different time points were compared by repeated measurements analysis of variance and linear contrasts and when appropriate a one way analysis of variance followed with post hoc Bonferroni correction. Values of p < 0.05 were accepted as significant. The coefficients of variation of neutrophils, aldosterone, C reactive protein, BNP, standard deviation of all NN intervals (SDNN), and triangular index are 0.8%, 6%, 13%, 15%, 0.3%, and 1.2%, respectively.
RESULTS
Patient characteristics
No patient was treated with β blockade, as was standard practice at the time (mid 1990s) (table 1). The mean (SD) follow up period was 20 (5) months. Nine patients died during follow up, four of PHF (during months 7, 12, 21, and 24) and five of SUD (during months 6, 11, 15, 20, and 21). It is worth noting that all PHF deaths occurred within hospital and all SUD occurred in the community, which helps to validate that the deaths were correctly classified.22
Table 1.
Clinical characteristics of study subjects and baseline treatment
| Parameter total number | Mean (SEM) or number |
| Age (years) | 69 (1.3) |
| Diastolic blood pressure (mm Hg) | 72 (2.2) |
| Systolic blood pressure (mm Hg) | 126 (3.7) |
| Heart rate (beats/min) | 72 (2.2) |
| 24 hour SDANN | 142 (7.2) |
| 24 hour SDNNi | 70 (8.1) |
| Weight (kg) | 77 (2) |
| Body mass index | 30 (0.8) |
| Plasma urea (mmol/l) | 7 (0.8) |
| Plasma creatinine (μmol/l) | 131 (6.8) |
| Plasma sodium (mmol/l) | 136 (0.3) |
| Plasma ACE activity(U/l) | 12 (3.4) |
| NYHA class II/III/IV | 20/11/3 |
| LVEF (%) | 29 (1.3) |
| Previous myocardial infarction | 32 |
| Dilated cardiomyopathy* | 2 |
| Baseline treatment | |
| Furosemide (frusemide) | 29 |
| ACE inhibitor | 29 |
| Nitrate | 14 |
| Digoxin | 12 |
| Aspirin | 22 |
*Based on global left ventricular dilatation and lack of symptoms or signs of ischaemic heart disease at any time.
ACE, angiotensin converting enzyme; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association; SDANN, average of NN intervals for all five minute segments; SDNNi, mean (SD) of NN intervals for all five minute segments.
There were no significant differences at baseline except that C reactive protein and non-sustained ventricular tachycardia (NSVT) events were higher in the PHF group (table 2). On the other hand, intraindividual changes in several parameters were highly significantly different between the three groups. The patients who died (SUD and PHF) had significant intraindividual worsening in the sequential changes between baseline and the last three values of mean 24 hour heart rate variability (triangular index and SDNN) in contrast to the control survival group (fig 1). A similar pattern was seen for the white cell count, the neutrophil count, and C reactive protein in that they all increased progressively before death in both the PHF group and the SUD group (fig 2). Figures 3, 4, 5, and 6 show the individual data of those who died suddenly. A clear change was seen in most cases before SUD, which was not seen in the alive group as judged by the data in the lower part of table 2. Unlike others, we did not find that BNP predicted sudden death in CHF.
Table 2.
Baseline values and differences in means for the last three measurements compared with baseline values in various parameters
| Parameters | Alive (n = 25) | PHF (n = 4) | SUD (n = 5) | p Value (alive v PHF) | p Value (alive v SUD) |
| Single baseline value | |||||
| LVEF (%) | 29 (1.2) | 26 (5.5) | 27 (4) | 0.8 | 0.7 |
| Furosemide (mg/day) | 61 (10.8) | 56 (17.50 | 40 (14) | 0.8 | 0.3 |
| Triangular index | 38 (2.6) | 31 (6) | 46 (5.6) | 0.34 | 0.24 |
| SDNN (ms) | 142 (9) | 114 (17) | 173 (20) | 0.26 | 0.16 |
| MHR (beats/min) | 70.6 (2.9) | 64 (4.8) | 70.8 (5.3) | 0.4 | 0.98 |
| Ventricular extrasystoles | 1606 (356) | 2116 (1267) | 1008 (427) | 0.63 | 0.96 |
| NSVT events/patient | 0.4 (0.12) | 4.7 (2.8) | 0.2 (0.17) | 0.001 | 0.8 |
| Plasma potassium (mmol/l) | 5.01 (0.7) | 5.4 (0.3) | 5.06 (0.8) | 0.27 | 0.89 |
| Plasma creatinine (mmol/l) | 127 (8) | 145 (28) | 134 (17) | 0.48 | 0.71 |
| Plasma BNP (pg/ml) | 61 (9.9) | 48 (26) | 93 (20.9) | 0.38 | 0.14 |
| Plasma aldosterone (pg/ml) | 142 (26) | 102 (45) | 271 (116) | 0.68 | 0.22 |
| Plasma WBC (109/l) | 6.9 (0.42) | 7.9 (0.49) | 5.9 (0.49) | 0.25 | 0.32 |
| Plasma neutrophil (109/l) | 4.1 (0.3) | 5.6 (0.8) | 3.7 (0.79) | 0.07 | 0.59 |
| C reactive protein (μg/ml) | 4.2 (0.6) | 13 (5) | 7 (3.9) | 0.02 | 0.72 |
| Last three measures | Intraindividual changes from baseline between PHF and alive patients | Intraindividual changes from baseline between SUD and alive patients | |||
| Data are mean (SEM). | |||||
| Some data are borderline between being normally distributed and not being normally distributed. For those parameters, the data are presented and analysed in two ways (logarithmically transformed and not transformed). | |||||
| *Only one baseline value available for C reactive protein. | |||||
| BNP, brain natriuretic peptide; MHR, maximum heart rate; NSVT, non-sustained ventricular tachycardia; PHF, progressive heart failure; SDNN, standard deviation of all NN intervals; SUD, sudden unexpected death; WBC, white blood cells. | |||||
| 24 hour triangular index | −11.4 (3.9) | −11.3 (3.5) | 0.023 | 0.01 | |
| 24 hour SDNN (ms) | −51 (13.4) | −36.8 (11.8) | 0.002 | 0.013 | |
| 24 hour MHR (beats/min) | +14 (5.6) | −4.4 (4.9) | 0.04 | 0.25 | |
| 24 hour ventricular extrasystoles | +6293 (964) | +812 (850) | 0.0001 | 0.225 | |
| 24 hour log ventricular extrasystoles | +1 (0.36) | +0.17 (0.3) | 0.028 | 0.59 | |
| NSVT events/patient | +48 (14) | +0.04 (12) | 0.002 | 0.99 | |
| Plasma BNP (pg/ml) | +81.6 (22) | −13.5 (19.5) | 0.003 | 0.49 | |
| Log plasma BNP | +0.45 (0.17) | −0.03 (0.15) | 0.044 | 0.84 | |
| Plasma aldosterone (pg/ml) | +183 (50) | 7.7 (44) | 0.003 | 0.86 | |
| Log plasma aldosterone | +0.43 (0.12) | +0.02 (0.11) | 0.005 | 0.82 | |
| Plasma creatinine (mmol/l) | −1.8 (16) | −11 (13) | 0.339 | 0.91 | |
| Plasma neutrophil (109/l) | +2.3 (0.62) | +1.5 (0.55) | 0.003 | 0.026 | |
| C reactive protein (μg/ml)* | +9 (2.6) | +5 (2.3) | 0.001 | 0.04 | |
| Log C reactive protein* | +0.11 (0.13) | +0.26 (0.12) | 0.39 | 0.039 | |
Figure 1.
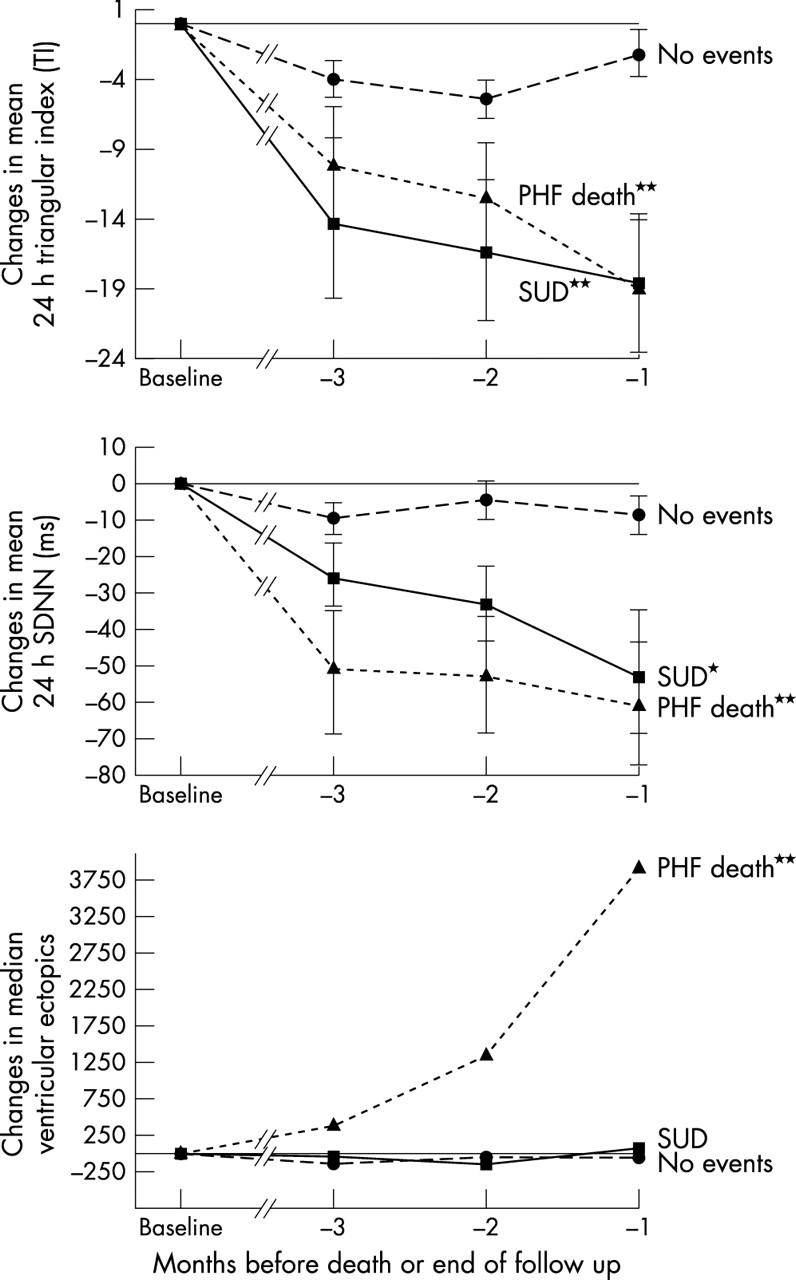
Changes (mean (SEM)) from baseline to the last three measurements (triangular index (TI), standard deviation of all NN intervals (SDNN), and ventricular extrasystole counts) before either death (sudden unexpected death (SUD) and progressive heart failure (PHF) groups) or the end of the study (for the alive group). // indicates multiple measurements. *p < 0.05; **p < 0.01.
Figure 2.
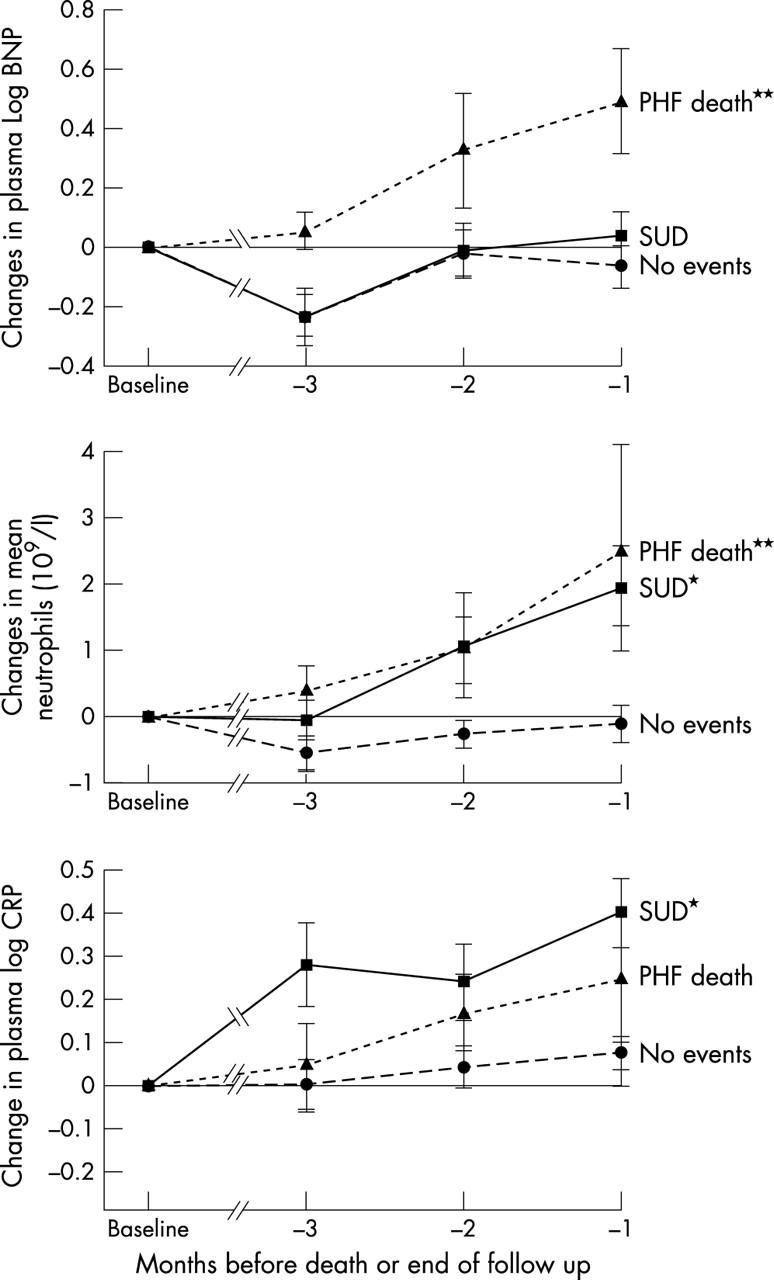
Changes (mean (SEM)) from baseline to the last three measurements of plasma brain natriuretic peptide (BNP), C reactive protein (CRP), and neutrophils before either death (SUD and PHF groups) or the end of the study (for the alive group). // indicates multiple measurements. *p < 0.05; **p < 0.01.
Figure 3.

Individual neutrophil changes from baseline until SUD in the five SUD patients. Each symbol represents a monthly value in each patient sequentially before their SUD.
Figure 4.

Individual C reactive protein (CRP) changes from baseline until SUD in the five SUD patients. Each symbol represents a monthly value in each patient sequentially before their SUD.
Figure 5.

Individual standard deviation of all NN intervals (SDNN) changes from baseline until SUD in the five SUD patients. Each symbol represents a monthly value in each patient sequentially before their SUD.
Figure 6.

Individual triangular index (TI) changes from baseline until SUD in the five SUD patients. Each symbol represents a monthly value in each patient sequentially before their SUD.
Patients who died in hospital of PHF also had significant increases in 24 hour mean ventricular extrasystoles, NSVT events, and 24 hour mean maximum heart rate in contrast to both SUD and control groups. In addition several parameters, which usually detect a worsening clinical state, followed the same pattern. These were 24 hour mean maximum heart rate, BNP, and aldosterone. All of these parameters behaved as expected in that they increased progressively in those who died of PHF but were unchanged in the SUD group. No significant changes were noted in any of the C pneumoniae titres or in immune complexes.
DISCUSSION
The problem being addressed here is that, as stated in an editorial in Circulation, “we currently lack the tools to identify most people at risk of a sudden cardiac arrest with a degree of accuracy sufficient to warrant major therapeutic interventions”.24
The novel findings in this study relate to the SUD group only since our findings with the PHF group were seen in previous studies. Our main finding is that SUD is preceded by progressive intraindividual decreases in heart rate variability and intraindividual increases in markers of inflammation (neutrophils and C reactive protein), but not by any change in the traditional measures of disease progression such as BNP, aldosterone, heart rate, and extrasystoles. Indeed, the diagnostic accuracy of the changes in C reactive protein, neutrophils, and heart rate variability was fairly good with sensitivities of 60–100% and specificities of 84–92%, which meant that such changes were very seldom seen in the alive group. On the other hand, PHF deaths but not SUDs were preceded by intraindividual worsening not only in heart rate variability and inflammation but also by worsening in the traditional measures of disease severity: BNP, aldosterone, extrasystoles, and NSVT. We found no evidence to specifically implicate C pneumoniae in the intraindividual increases in inflammation.
The correct classification of those who died into PHF and SUD groups is clearly important. That all PHF deaths occurred during hospitalisation while all SUDs occurred in the community is some indication that our classification is correct. This can also be supported by the substantial increase in conventional measures of disease severity in the PHF death group but not in the SUD group. Our heart rate variability data support the recent large UK-HEART (UK heart failure evaluation and assessment of risk trial), where low SDNN predicted death caused by PHF.12 However, in contrast to our study, in UK-HEART no particular abnormality in heart rate variability was seen in the SUD group; however, UK-HEART was a cross sectional study where the time gap between the heart rate variability measure and the subsequent SUD may have been too long to detect such a relation. This may explain why a link between heart rate variability and SUD was more detectable in the present longitudinal study with frequent heart rate variability monitoring than in UK-HEART.
A key question is whether intraindividual increases in “inflammation” and intraindividual worsening in heart rate variability may be related. New information suggests that this is possible. Inflammation produces endothelial dysfunction,25,26 which in turn predicts coronary events.27,28 Endothelial dysfunction is also associated with reduced vascular nitric oxide, which is known to worsen autonomic function as indicated by heart rate variability.29,30 The final link in this hypothesis is that autonomic (vagal) dysfunction is probably one of many arrhythmogenic stimuli.31 Recent data are generally consistent with this in that SUD in CHF is often but not always associated with an identifiable acute coronary event at necropsy,32 which may be preceded by inflammation induced endothelial dysfunction.
These results have implications for risk stratification of patients with CHF, since impending SUD may be identifiable by sequential measuring of heart rate variability, neutrophils, or C reactive protein. Impending PHF death is usually easy to recognise at the bedside, whereas impending SUD is difficult to predict at the bedside. This means that it should be relatively easy clinically to ascertain whether an intraindividual increase in inflammation or autonomic dysfunction is heralding a SUD or a PHF death. A measure of heart rate or neurohormones may assist this differentiation, if doubt still exists. This may lead to better targeting of invasive procedures such as coronary revascularisation, insertion of coated stents or implantable defibrillators, or even transplantation towards those at high risk of imminent SUD. These measures may also help in the selection of patients for targeted anti-inflammatory treatments to prevent SUD. This may even involve short term administration of fairly “toxic” anti-inflammatories such as cyclosporine or steroids, which would be warranted if the patient is identified to be at risk of an imminent SUD but not warranted if the SUD risk is distant. Indeed the IMPRESS (immunosuppressive therapy for the prevention of restenosis after coronary artery stent implantation) study showed that short term high dose prednisolone can reduce cardiovascular events dramatically in a similar but different population.33
The ventricular extrasystole data are interesting in that worsening ventricular extrasystoles and increased NSVT occur only before PHF deaths but not before SUDs. This supports a previous review article that suggested that ventricular extrasystoles are a consequence of disease progression rather than a specific cause of future sudden death.34 The number of NSVT events and the number of patients in the SUD group are both small, which makes these NSVT results unreliable.
It is worth commenting that this study was done before the use of β blockers and implantable cardioverter-defibrillators in CHF, although both should defer deaths rather than prevent them entirely. The same intraindividual changes can still occur before the deferred death.
In summary, our data raise the possibility that SUD may be predictable if we concentrate on intraindividual changes in neutrophils, C reactive protein, or heart rate variability rather than using absolute across the board interindividual values. This was a relatively small pilot study, although the work involved was large—540 ambulatory ECG tapes and 540 assays of most analytes. However, the credibility of our results is increased by the finding that changes were the same in two independent measures of inflammation (C reactive protein and neutrophils). Nevertheless, larger studies will be needed to confirm this intriguing observation. Monthly neutrophil counts may ultimately prove to be a cheap and effective way of targeting novel treatments to patients with CHF at high risk of impending SUD.35 Furthermore, these data may be relevant to a wider group of patients, since half of all SUDs are said to occur in patients with at least a history of CHF.36
Acknowledgments
The study was supported in part by both the British Heart Foundation and the Scottish Office Department of Health.
Abbreviations
BNP, brain natriuretic peptide
CHF, chronic heart failure
ELISA, enzyme linked immunosorbent assay
IMPRESS, mmunosuppressive therapy for the prevention of restenosis after coronary artery stent implantation
NSVT, non-sustained ventricular tachycardia
NYHA, New York Heart Association
PHF, progressive heart failure
SDNN, standard deviation of all NN intervals
SUD, sudden unexpected death
TI, triangular index
UK-HEART, UK heart failure evaluation and assessment of risk trial
REFERENCES
- 1.Ho KKL, Pinsky JL, Kannel WB, et al. The epidemiology of heart failure: the Framingham study. J Am Coll Cardiol 1993;22 (suppl) :6–13. [DOI] [PubMed] [Google Scholar]
- 2.Massie BM, Shah NB. The heart failure epidemic: magnitude of the problem and potential mitigating approaches. Curr Opin Cardiol 1996;11:221–6. [DOI] [PubMed] [Google Scholar]
- 3.Mcdonagh TA, Morrison CE, Lawrence A, et al. Symptomatic and asymptomatic left ventricular systolic dysfunction in an urban population. Lancet 1997;350:829–33. [DOI] [PubMed] [Google Scholar]
- 4.Cowie MR, Mosterd A, Wood DA, et al. The epidemiology of heart failure. Eur Heart J 1997;18:208–25. [DOI] [PubMed] [Google Scholar]
- 5.Goldman S , Johnson G, Cohn JN, et al. Mechanism of death in heart failure: the vasodilator heart failure trials. Circulation 1993;87 (suppl) :3124–33. [PubMed] [Google Scholar]
- 6.Hinkle LE, Thaler JT. Clinical classification of cardiac deaths. Circulation 1982;65:457–64. [DOI] [PubMed] [Google Scholar]
- 7.Szlachcic J , Massie BM, Kramer BL, et al. Correlates and prognostic indication of exercise capacity in chronic heart failure. Am J Cardiol 1985;55:1037–42. [DOI] [PubMed] [Google Scholar]
- 8.Mancini DM, Eisen H, Kussmaul W, et al. Value of peak oxygen consumption for optimal timing of cardiac transplantation in ambulatory patients with heart failure. Circulation 1991;83:778–86. [DOI] [PubMed] [Google Scholar]
- 9.Roul G , Moulichon M-E, Bareiss P, et al. Prognostic factors of chronic heart failure in NYHA class II or III: value of invasive exercise haemodynamic data. Eur Heart J 1995;16:1387–98. [DOI] [PubMed] [Google Scholar]
- 10.Cohn JN, Levine TB, Olivari MT, et al. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med 1984;311:819–23. [DOI] [PubMed] [Google Scholar]
- 11.Cohn JN, Johnson GR, Shabetai R, et al. Ejection fraction, peak oxygen consumption, cardio-thoracic ratio, ventricular arrhythmias and plasma norepinephrine as determinants of prognosis in heart failure. Circulation 1993;87 (suppl) :5–16. [PubMed] [Google Scholar]
- 12.Nolan J , Batin PD, Andrews R, et al. Prospective study of heart rate variability and mortality in chronic heart failure: results of the United Kingdom heart failure evaluation and assessment of risk trial (UK-Heart). Circulation 1998;98:1510–6. [DOI] [PubMed] [Google Scholar]
- 13.La Rovere MT, Pinna GD, Maestri R, et al. Short term HRV strongly predicts sudden cardiac death in chronic heart failure patients. Circulation 2003;107:565–70. [DOI] [PubMed] [Google Scholar]
- 14.Galiner M , Pathak A, Fourcade J, et al. Depressed low frequency power of HRV as an independent predictor of sudden death in chronic heart failure. Eur Heart J 2000;21:475–481. [DOI] [PubMed] [Google Scholar]
- 15.Davies MJ, Thomas A. Thrombosis and acute coronary lesions in sudden cardiac ischaemic death. N Engl J Med 1984;310:1137–40. [DOI] [PubMed] [Google Scholar]
- 16.Ridker PM, Hennekens CH, Buring JE, et al. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 2000;342:836–43. [DOI] [PubMed] [Google Scholar]
- 17.van der Wal AC, Becker AE, van der Loos CM, et al. Sites of internal rupture of erosion of thrombosed coronary atherosclerotic plaques is characterised by an inflammatory process irrespective of the dominant plaque morphology. Circulation 1994;89:36–44. [DOI] [PubMed] [Google Scholar]
- 18.de Gouveia RH, van der Wal AC, van der Loos CM, et al. Sudden unexpected death in young adults: discrepancies between initiation of acute plaque complications and the onset of acute coronary death. Eur Heart J 2002;23:1433–40. [DOI] [PubMed] [Google Scholar]
- 19.Nakagawa M , Saikawa T, Ito M. Progressive reduction of heart rate variability with eventual sudden death in two patients. Br Heart J 1994;71:87–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bigger T . Why patients with congestive heart failure die: arrhythmias and sudden cardiac death. Circulation 1987;75 (suppl) :28–35. [PubMed] [Google Scholar]
- 21.Tavendale R , Parratt D, Pringle SD, et al. Serological markers of Chlamydia pneumoniae infection in men and women and subsequent coronary events. Eur Heart J 2002;23:301–7. [DOI] [PubMed] [Google Scholar]
- 22.Narang R , Cleland JGF, Erhardt L, et al. Mode of death in chronic heart failure: a request and proposition for more accurate classification. Eur Heart J 1996;17:1390–403. [DOI] [PubMed] [Google Scholar]
- 23.Anon. Heart rate variability: standards of measurements, physiological interpretation, and clinical use. Task force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Circulation 1996;93:1043–65. [PubMed] [Google Scholar]
- 24.Zipes DP. Saving time saves lives. Circulation 2001;104:2506–8. [PubMed] [Google Scholar]
- 25.Bhagat K , Moss R, Collier J, et al. Endothelial stunning following a brief exposure to endotoxin: a mechanism to link infection to infarction. Cardiovasc Res 1996;53:481–2. [PubMed] [Google Scholar]
- 26.Bhagat K , Vallance P. Inflammatory cytokines impair endothelium-dependent dilatation in human veins in vivo. Circulation 1997;96:3042–7. [DOI] [PubMed] [Google Scholar]
- 27.Al Suwaidi JA, Hamasaki S, Higano ST, et al. Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circulation 2000;101:948–54. [DOI] [PubMed] [Google Scholar]
- 28.Schachinger V , Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation 2000;101:1899–906. [DOI] [PubMed] [Google Scholar]
- 29.Chowdhary S , Townend JN. Role of nitric oxide in regulation of cardiovascular autonomic control. Clin Sci 1999;97:5–17. [PubMed] [Google Scholar]
- 30.Spieker LE, Corti R, Binggeli C, et al. Baroreceptor dysfunction induced by nitric oxide synthase inhibition in humans. J Am Coll Cardiol 2000;36:213–8. [DOI] [PubMed] [Google Scholar]
- 31.Zuanetti G , De Ferrari GM, Priori SG, et al. Protective effect of vagal stimulation on reperfusion arrhythmias in cats. Circ Res 1987;61:429–35. [DOI] [PubMed] [Google Scholar]
- 32.Uretsky BF, Thygesen K, Armstrong PW, et al. Acute coronary findings at autopsy in heart failure patients with sudden death. Circulation 2000;102:611–6. [DOI] [PubMed] [Google Scholar]
- 33.Versaci F , Gaspardone A, Tomai F, et al. Immunosuppressive therapy for the prevention of restenosis after coronary artery stent implantation (IMPRESS). J Am Coll Cardiol 2002;40:1935–42. [DOI] [PubMed] [Google Scholar]
- 34.Packer M . Lack of relation between ventricular arrhythmias and sudden death in patients with chronic heart failure. Circulation 1992;85:50–6. [PubMed] [Google Scholar]
- 35.Miller LW, Kubo SH, Young JB, et al. Report of the consensus conference on candidate selection for heart transplantation–1993. J Heart Lung Transplant 1995;14:1143–53. [PubMed] [Google Scholar]
- 36.Hinkle LE, Thaler HT. Clinical classification of sudden cardiac deaths. Circulation 1982;65:457–64. [DOI] [PubMed] [Google Scholar]