

Abstract
Objective: To determine the effect of systemic arterial hypertension on the indices of aortic stenosis (AS) severity.
Methods: A severe supravalvar AS was created in 24 pigs. The maximum and mean pressure gradients across the stenosis were measured by Doppler echocardiography and by catheterisation. Both echocardiography and catheter data were used to calculate stenosis effective orifice area, energy loss coefficient, and peak systolic left ventricular wall stress. Measurements were taken both at normal aortic pressures and during hypertension induced by banding of the distal thoracic aorta in 14 pigs and by intravenous administration of phenylephrine in 10 pigs.
Results: During hypertension, systemic arterial resistance downstream from the stenosis increased greatly (all animals: 71 (40)%), whereas total systemic arterial compliance decreased significantly (−38 (21)%). Hypertension resulted in a moderate increase in effective orifice area (29 (14)%) and energy loss coefficient (25 (17)%) and substantial decreases in catheter gradients (maximum: −40 (20)%; mean: −43 (20)%; peak to peak: −70 (23)%) and Doppler gradients (maximum: −35 (17)%; mean: −37 (16)%). In multivariate analysis, peak to peak gradient was significantly (p < 0.001) related to the energy loss coefficient, mean flow rate, and arterial compliance, whereas maximum and mean catheter gradients were related only to the energy loss coefficient and flow rate. Of major importance, maximum systolic left ventricular wall stress increased greatly during hypertension (43 (23)%).
Conclusions: The severity of AS may be partially masked by the presence of coexisting hypertension. The markers of AS severity should thus be interpreted with caution in hypertensive patients and be re-evaluated when the patient is in a normotensive state.
Keywords: aortic stenosis, Doppler echocardiography, haemodynamic function, hypertension, left ventricular function
The transvalvar pressure gradient (TPG) is one of the major criteria used to evaluate aortic stenosis (AS) severity. As illustrated by the Gorlin equation,1 the TPG is physiologically determined by two factors: the effective orifice area (EOA) of the valve and transvalvar flow. Nonetheless, it has often been reported that the TPG may be reduced in patients with significant AS and concomitant systemic hypertension.2–4 This reduction was generally thought to depend on the simultaneous decrease in flow that may occur as a result of increased systemic arterial resistance (SAR). However, Laskey and colleagues3 suggested that the TPG may decrease irrespective of flow as a direct consequence of the increased SAR. As opposed to these results, Razzolini and colleagues5 found that, for each flow level, TPG increases linearly with SAR. Hence, controversy and uncertainty persist as to the eventual implication of systemic arterial hypertension in terms of interpretation of AS severity and ensuing clinical conduct.
Contrary to older assumptions, recent studies have shown that systemic hypertension is not rare in patients with AS. Antonini-Canterin and colleagues6 reported a prevalence of 32% of systolic hypertension in a series of 193 patients with severe symptomatic AS. Systemic arterial hypertension may result from an increase in SAR (systolodiastolic hypertension), a decrease in systemic arterial compliance (SAC) (systolic hypertension), or both abnormalities. Since it is difficult in AS patients to control and manipulate independently valvar (valve EOA and transvalvar flow) and arterial (SAR and SAC) haemodynamic factors, we elected to perform an animal study to examine the impact of systemic arterial hypertension on aortic valve haemodynamic function. The main objective of this study was to determine the effect of systemic arterial haemodynamic factors on the indices of AS severity.
MATERIALS AND METHODS
Animal care and experiments were conducted in accordance with the guidelines of the Canadian Council on Animal Care. The protocol was approved by the institutional animal care committee of Laval University, Sainte-Foy, Quebec, Canada.
Twenty four pigs weighing between 25–48 kg (body surface area 0.86–1.32 m2) were anaesthetised as previously described.7 After intramuscular premedication with ketamine, anaesthesia was induced by intravenous injection of fentanyl (5 μg/kg) and pentobarbital (6.5 mg/kg) and was maintained with a continuous infusion of fentanyl (4 μg/kg/h) and pentobarbital (10 mg/kg/h). Muscle relaxation was achieved by administration of pancuronium 0.30 mg/kg/h. A lateral thoracotomy was performed in the fourth left intercostal space. A severe supravalvar AS was created with an umbilical tape tightened around the aorta about 2 cm downstream from the aortic valve annulus.8
Systemic arterial hypertension was induced either by banding of the distal thoracic aorta (group A: 14 pigs) or by intravenous administration of phenylephrine (group B: 10 pigs). In group A, an umbilical tape was passed around the thoracic aorta at the level of the 9th–10th thoracic vertebra. The two extremities of the umbilical tape were then passed through the lumen of a piece of rigid rubber tube and clamped with a haemostatic clamp to allow for adjustment of banding circumference and thus of the severity of hypertension. In group B, the rate of the phenylephrine perfusion was adjusted between 2–7 µg/kg/min to produce mild systemic hypertension and then increased up to 8–15 µg/kg/min to produce severe systemic hypertension.
In both groups, the Doppler echocardiographic and catheter measurements described below were taken under the following experimental conditions: (1) normal condition; (2) severe AS; (3) severe AS + mild systemic hypertension; and (4) severe AS + severe systemic hypertension.
The ECG, pressure, and flow signals were simultaneously recorded and digitised (Digidata 1322, Axon Instruments, Foster City, California, USA). Pressure was measured with a multisensor Millar catheter (customised model, Millar Instruments, Houston, Texas, USA) about 1 cm upstream of the stenosis and 4 cm downstream from the stenosis to calculate maximum TPG (TPGmax), mean TPG (TPGmean), and peak to peak TPG (TPGptop). Cardiac output was measured with an ultrasonic flowmeter (T206, Transonic Systems, Ithaca, New York, USA) with the probe positioned around the main pulmonary artery. Left ventricular (LV) stroke volume was calculated by dividing cardiac output by heart rate. The mean systolic flow rate across the stenosis was calculated by dividing the stroke volume by the LV ejection time measured by Doppler echocardiography on the aortic jet velocity signal.9 The stenosis EOA was calculated as follows with the Gorlin formula:
 |
where Qmean is the mean systolic flow rate in ml/s. Previous studies have shown that the original Gorlin formula contains several errors that can be corrected by using a constant of 50 instead of 44.3.10
SAR was calculated as follows:
 |
where CO is the cardiac output in l/min and MAP and CVP are the mean aortic and central venous pressures, respectively.
SAC was calculated by the following equation11:
 |
where SV is the stroke volume in ml and SAP and DAP are the systolic and diastolic aortic pressures, respectively. Previous studies have shown that SAC calculated with this equation accurately estimates the total SAC measured by other reference methods (area method or pressure decay method) and that it is a powerful predictor of clinical outcome in patients with hypertension or diabetes.11–14 Interestingly, this equation includes variables that can easily be measured non-invasively in the clinical setting.
The ratio of stroke volume to pulse pressure is considered a simple and accurate estimate of total SAC. However, it does not take into account the pulsatile characteristics of blood flow in arteries. A more precise and complete description of LV afterload imposed by the systemic arterial system is provided by the input impedance spectra of the systemic circulation15 but this complex approach is not feasible in practice. Other investigators have therefore suggested the use of effective arterial elastance (EAE), estimated as the steady state ratio of end systolic aortic pressure (ESAP) divided by stroke volume16–19:
 |
Doppler echocardiograms were recorded with a Sonos 5500 (Philips Medical Systems, Andover, Massachusetts, USA). An upper laparotomy was performed and the ultrasound probe (Model S3, Philips Medical Systems) was introduced into the abdominal cavity and positioned on the diaphragm at the level of the cardiac apex. This window allowed the visualisation of high quality apical five chamber images and optimal recording of the LV outflow tract pulsed wave velocity and aortic jet continuous wave velocity. The Doppler derived TPGmax and TPGmean were calculated with the modified Bernoulli equation and the Doppler derived EOA was calculated with the standard continuity equation. The Doppler derived mean systolic flow rate across the stenosis was calculated by dividing the stroke volume measured by Doppler echocardiography in the LV outflow tract by the LV ejection time.9 The diameter of the ascending aorta was measured at 2–3 cm downstream from the stenosis by epicardial two dimensional echocardiography with a 12 MHz probe (Model S12, Philips Medical Systems). The presence of aortic, mitral, or pulmonary valvar regurgitation was assessed by colour Doppler echocardiography. No valvar regurgitation > 0.5+ (trivial) was detected in this study.
Two dimensionally directed LV dimensions were measured in the left parasternal long axis with a 12 MHz probe positioned directly on the epicardium. LV minor axis internal dimension and posterior wall thickness were measured at end diastole and end systole. The peak systolic LV wall stress (PSWS) was estimated by the method of Grossman and colleagues20:
 |
where LVIDs and LVPWTs are the internal dimension and the posterior wall thickness of the LV at end systole, respectively, and LVPs is the LV systolic pressure measured by catheter.
Data analysis
Doppler measurements of aortic valve area rely on the peak velocity measured across the vena contracta and thus reflect the true EOA, whereas catheterisation measurements that use the pressure gradient after pressure recovery—that is, the net pressure gradient—yield larger values for EOA. Although the aortic valve area measured by catheterisation is a less accurate measure of the physiological EOA, it appears to be a better predictor of outcomes because it better reflects the true energy loss caused by the AS.21 We recently proposed a method for correcting the Doppler EOA for the effects of pressure recovery by calculating the energy loss coefficient (ELCo) as follows21,22:
 |
where EOAdop is the EOA measured by Doppler echocardiography and AA is the aortic cross sectional area of the ascending aorta.
In a previous study, we found that the energy loss coefficient measured by Doppler echocardiography accurately predicted the valve EOA measured by catheterisation.22 In fact, these two parameters are equivalent from a physiological stand point and they can be regarded as a “recovered EOA”—that is, an expression of EOA that takes into account the magnitude of pressure recovery that may occur downstream from the vena contracta.
Hence, the TPG measured by catheterisation corresponds to the net pressure gradient and, as suggested in our previous study, it is essentially determined by the energy loss coefficient and the mean transvalvar flow rate22:
 |
This equation can be rewritten as follows:
 |
In this study, we compared the TPG predicted by this equation with the TPG measured directly by catheterisation. In addition, we determined whether the systemic arterial factors (systolic aortic pressure, diastolic aortic pressure, pulse pressure, SAR, SAC, and effective arterial elastance) directly influence the TPGs irrespective of energy loss coefficient and flow rate as suggested in previous studies.3,5
Statistical analysis
Data were expressed as mean (SD) and compared in each group by a one way analysis of variance for repeated measurements to evaluate the effects of intervention (AS or systemic hypertension). A Holm-Sidak test was used for pairwise comparisons.
A stepwise forward regression analysis was performed to determine the independent effect of valvar and arterial haemodynamic variables on the TPGs and systolic LV wall stress. Variables with p < 0.1 in univariate analysis were entered in multivariate analysis. For the purpose of these multivariate analyses, the data of group A and of group B were pooled together.
RESULTS
Tables 1 and 2 summarise the results of group A (systemic hypertension induced by banding of the aorta) and of group B (systemic hypertension induced by intravenous administration of phenylephrine), respectively. As expected, SAR increased greatly in both groups (group A: 70 (48)%; group B: 72 (31)%; all animals: 71 (40)%) during the protocol. Concomitantly, SAC decreased significantly (group A: −43 (23)%; group B: −31 (18)%; all animals: −38 (21)%) due to the dilatation of the aorta (group A: 16 (6)%; group B: 17 (10)%; all animals: 16 (9%)) that occurred as a consequence of the augmentation of aortic pressure. However, it should be noted that the reduction in SAC was more pronounced in group A than in group B, although the increase in the aortic diameter was similar in both groups. This difference is probably due to the distal banding that may have further compromised compliance of the aorta in group A. Interestingly, the use of aortic banding in group A tended to produce haemodynamic conditions similar to those observed in patients with systolic hypertension. Indeed, this intervention was associated with a major reduction in SAC and a major increase in pulse pressure (93 (62)%) (fig 1C). In contrast, the phenylephrine protocol used with group B produced systolodiastolic hypertension with only a moderate change in pulse pressure (23 (48)%) (fig 1D).
Table 1.
Changes in systemic arterial haemodynamic variables, aortic stenosis (AS) haemodynamic variables, and LV function in the 14 pigs of group A during induction of severe supravalvar AS and systemic hypertension (SH). In this group, SH was induced by aortic banding
| Normal | Severe AS | Severe AS + mild SH | Severe AS + severe SH | |
| Systemic arterial haemodynamic variables | ||||
| Systolic aortic pressure (mm Hg) | 92 (18) | 91 (14) | 103 (17)† | 146 (24)*† |
| Diastolic aortic pressure (mm Hg) | 61 (17) | 57 (9) | 58 (10) | 84 (13)*† |
| Pulse pressure (mm Hg) | 31 (6) | 33 (9) | 45 (10)*† | 62 (19)*† |
| Systemic arterial resistance (dyn.s/cm5) | 1705 (638) | 1615 (537) | 1837 (488) | 2759 (1159)*† |
| Systemic arterial compliance (ml/mm Hg) | 1.34 (0.37) | 1.29 (0.40) | 0.86 (0.21)*† | 0.67 (0.24)*† |
| Effective arterial elastance (mm Hg/ml) | 2.02 (0.57) | 2.07 (0.53) | 2.34 (0.67) | 3.54 (1.10)*† |
| Catheter derived AS haemodynamic variables | ||||
| Catheter EOA (cm2) | 2.16 (0.92) | 0.59 (0.20)* | 0.70 (0.31)* | 0.72 (0.31)* |
| Catheter maximum gradient (mm Hg) | 2 (1) | 49 (19)* | 40 (17)* | 29 (20)*† |
| Catheter mean gradient (mm Hg) | 1 (1) | 30 (14)* | 24 (12)* | 17 (13)*† |
| Catheter peak to peak gradient (mm Hg) | 0 (1) | 36 (19)* | 24 (15)*† | 10 (11)*† |
| Doppler derived AS haemodynamic variables | ||||
| Doppler EOA (cm2) | 1.74 (0.42) | 0.43 (0.13)* | 0.44 (0.14)* | 0.57 (0.14)*† |
| Aortic diameter (mm) | 17.9 (2.6) | 16.5 (1.5) | 17.7 (1.9) | 19.5 (2.0)† |
| Energy loss coefficient (cm2) | 2.20 (0.55) | 0.58 (0.21)* | 0.67 (0.28)* | 0.71 (0.26)* |
| Doppler maximum gradient (mm Hg) | 6 (2) | 61 (27)* | 54 (21)* | 37 (15)*† |
| Doppler mean gradient (mm Hg) | 3 (1) | 40 (18)* | 35 (15)* | 23 (11)*† |
| LV systolic function | ||||
| Heart rate (beats/min) | 88 (13) | 87 (11) | 87 (13) | 90 (15) |
| Stroke volume (ml/s) | 43 (12) | 42 (10) | 42 (12) | 40 (11) |
| Cardiac output (l/min) | 3.86 (1.19) | 3.54 (0.75) | 3.57 (0.80) | 3.38 (0.91) |
| Mean systolic flow rate (ml/s) | 150 (39) | 129 (29)* | 132 (31) | 120 (33)* |
| Peak systolic LV wall stress (kdyn/cm2) | 140 (41) | 191 (52)* | 216 (53)* | 278 (74)*† |
*Significant difference v normal stage; †significant difference between severe stenosis + SH stages and the severe stenosis stage.
EOA, effective orifice area; LV, left ventricular.
Table 2.
Changes in systemic arterial haemodynamic variables, AS haemodynamic variables, and LV function in the 10 pigs of group B during induction of severe supravalvar AS and SH. In this group, SH was induced by administration of phenylephrine
| Normal | Severe AS | Severe AS + mild SH | Severe AS + severe SH | |
| Systemic arterial haemodynamic variables | ||||
| Systolic aortic pressure (mm Hg) | 113 (13) | 107 (14) | 129 (15)*† | 162 (12)*† |
| Diastolic aortic pressure (mm Hg) | 82 (12) | 72 (10) | 90 (14)† | 121 (12)*† |
| Pulse pressure (mm Hg) | 32 (4) | 35 (9) | 39 (9) | 40 (10) |
| Systemic arterial resistance (dyn.s/cm5) | 1512 (306) | 1406 (283) | 1628 (322) | 2392 (534)*† |
| Systemic arterial compliance (ml/mm Hg) | 1.87 (0.32) | 1.69 (0.40) | 1.51 (0.43)* | 1.13 (0.24)*† |
| Effective arterial elastance (mm Hg/ml) | 1.94 (0.42) | 2.02 (0.37) | 2.31 (0.46) | 3.37 (0.77)*† |
| Catheter derived AS haemodynamic variables | ||||
| Catheter derived EOA (cm2) | 2.50 (0.27) | 0.64 (0.19)* | 0.71 (0.22)* | 0.82 (0.34)* |
| Catheter maximum gradient (mm Hg) | 2 (3) | 46 (19)* | 41 (14)* | 30 (12)*† |
| Catheter mean gradient (mm Hg) | 1 (1) | 31 (14)* | 27 (10)* | 20 (9)*† |
| Catheter peak to peak gradient (mm Hg) | 1 (1) | 37 (18)* | 29 (12)*† | 16 (11)*† |
| Doppler derived AS haemodynamic variables | ||||
| Doppler EOA (cm2) | 2.28 (0.27) | 0.51 (0.11)* | 0.56 (0.13)* | 0.66 (0.14)*† |
| Aortic diameter (mm) | 20.1 (2.1) | 18.0 (1.6)* | 19.4 (1.8) | 21.0 (2.1)† |
| Energy loss coefficient (cm2) | 2.47 (0.28) | 0.68 (0.27)* | 0.75 (0.27)* | 0.85 (0.29)* |
| Doppler maximum gradient (mm Hg) | 6 (1) | 68 (20)* | 64 (20)* | 46 (13)*† |
| Doppler mean gradient (mm Hg) | 3 (1) | 47 (15)* | 43 (12)* | 31 (9)*† |
| LV systolic function | ||||
| Heart rate (beats/min) | 90 (7) | 89 (7) | 95 (8) | 112 (16)*† |
| Stroke volume (ml) | 54 (8) | 49 (8) | 51 (9) | 45 (9) |
| Cardiac output (l/min) | 4.84 (0.86) | 4.37 (0.90) | 4.86 (1.16) | 4.85 (1.05) |
| Mean systolic flow rate (ml/s) | 198 (23) | 162 (25)* | 175 (31) | 173 (27)* |
| Peak LV systolic wall stress (kdyn/cm2) | 193 (45) | 268 (82) | 293 (81)* | 360 (66)*† |
*Significant difference v normal stage; †significant difference between severe stenosis + SH stages and the severe stenosis stage.
Figure 1.

Left ventricular (LV) and aortic pressure waveforms obtained in a pig of group A (A) at normal aortic pressure and (B) during mild and (C) severe systemic hypertension induced by banding of the aorta. (D) In this pig, in an additional stage after the removal of the aorta banding, phenylephrine was administered to produce severe hypertension (protocol similar to the one used in group B). Note the major decrease in pressure gradients during hypertension despite the presence of severe aortic stenosis (in this animal, effective orifice area (EOA) varies between 0.47–0.65 cm2 and EOA indexed for body surface area varies between 0.43–0.59 cm2/m2).
The average Doppler derived EOA at the level of the stenosis decreased greatly as a result of the supravalvar AS, whereas it increased again moderately after the induction of hypertension (group A: 29 (15)%; group B: 29 (12)%; all animals: 29 (14)%). Similar changes were observed for the energy loss coefficient (group A: 23 (17)%; group B: 26 (16)%; all animals: 25 (17)%) and the catheter EOA (group A: 24 (21)%; group B: 28 (20)%; all animals: 26 (21)%) but overall the values of these indices were higher than the average Doppler EOA due to the pressure recovery phenomenon as previously described.22 In multivariate analysis, the only independent determinants of the absolute change in Doppler EOA that occurred during hypertension were the absolute change in mean systolic flow rate (p = 0.01; ΔR2 = 0.24) and the absolute change in pulse pressure (p = 0.01; ΔR2 = 0.25).
There was a strong correlation and agreement (R = 0.82, slope = 0.95) between mean systolic flow rate measured by Doppler echocardiography in the LV outflow tract and mean systolic flow rate measured by ultrasonic flowmeter at the level of the main pulmonary artery. Given that the transvalvar flow rate is most often measured by Doppler echocardiography in the clinical setting, only the results of the Doppler flow measurements are presented hereafter. The average mean systolic flow rate decreased slightly (group A: −13 (9)%; group B: −18 (7)%; all animals: −15 (9)%) as a result of AS but did not change significantly after hypertension was induced (group A: −7 (12)%; group B: 8 (15)%; all animals: −1 (15)%).
Figure 1 shows the LV and aortic pressure waveforms obtained in one animal of group A. It is striking to note the dramatic reduction in the TPGs during hypertension despite the presence of a severe AS. Figure 2 shows the average changes in the catheter (fig 2A) and Doppler (fig 2B) TPGs in all animals of groups A and B. As expected, the induction of a severe AS was associated with a major increase in the baseline TPGs. On the other hand, when systemic hypertension was induced downstream from this severe AS, significant decreases in catheter derived TPGptop (group A: −76 (15)%; group B: −61 (29)%; all animals: −70 (23)%), TPGmax (group A: −43 (20)%; group B: −36 (20)%; all animals: −40 (20)%), and TPGmean (group A: −47 (22)%; group B: −37 (20)%; all animals: −43 (20)%) were observed. The Doppler derived TPGmax (group A: −39 (15)%; group B: −30 (19); all animals: −35 (17)%) and TPGmean (group A: −41 (14)%; group B: −30 (19); all animals: −37 (16)%) also decreased significantly during hypertension. These results confirm that the concomitant presence of systemic hypertension may cause the severity of AS to be underestimated.
Figure 2.
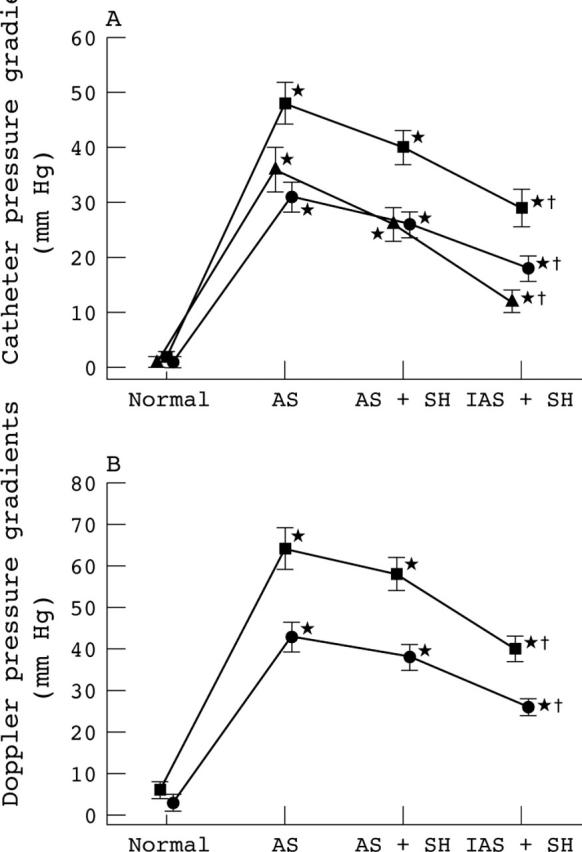
Changes in (A) catheter and (B) Doppler pressure gradients (maximum (▪); mean (•); peak to peak (▴)) in all animals of groups A and B (n = 24) during induction of aortic stenosis (AS) and systemic hypertension (SH) (mild: AS+SH I; severe: AS+SH II). *Significant difference v normal stage; †significant difference between severe stenosis + SH (AS+SH I or AS+SH II) stages and the severe stenosis (AS) stage. The error bars represent SEM.
In contrast to what was observed for TPGs, the peak systolic LV wall stress, which reflects the load imposed on the LV, increased greatly during hypertension. In fact, systemic hypertension produced an increase in wall stress (group A: 46 (19)%; group B: 40 (23)%; all animals: 43 (23)%) that was at least as important as the increase in wall stress caused by the severe AS (group A: 39 (33)%; group B: 41 (42)%; all animals: 41 (37)%) (fig 3).
Figure 3.

Changes in peak systolic LV wall stress in all animals of groups A and B (n = 24) during induction of AS and SH (mild: AS+SH I; severe: AS+SH II). *Significant difference v normal stage; †significant difference between severe stenosis + SH (AS+SH I or AS+SH II) stages and the severe stenosis (AS) stage. The error bars represent SEM.
Univariate and multivariate analyses were performed to determine the independent effect of each haemodynamic variable on the catheter derived TPGs as well as on the peak systolic LV wall stress. There was a strong correlation and agreement between the TPGs measured by catheter and TPGs predicted by equation 8 (fig 4). The two variables in this equation—the energy loss coefficient and the mean systolic flow rate measured by Doppler echocardiography—explain 89% of the variance of TPGmean and TPGmax (table 3). SAR has no independent effect on the TPGs, whereas SAC affects only TPGptop. The relative independent contribution of SAC to the variance of TPGptop is 20% (ΔR2 = 0.20) in the multivariate predictive model.
Figure 4.

Correlation between mean pressure gradient directly measured by catheterisation and mean pressure gradient predicted by equation 8 from the energy loss coefficient and the mean systolic flow rate measured by Doppler echocardiography. All data of group A and B are plotted (several data points are superimposed). The dashed line is the identity line.
Table 3.
Multivariate analysis of haemodynamic variables that independently determine transvalvar pressure gradients
| Variable | Transvalvar pressure gradient | ||
| Peak to peak | Peak | Mean | |
| Predicted gradient* | |||
| p Value | <0.001 | <0.001 | <0.001 |
| ΔR2 | 0.67 | 0.89 | 0.89 |
| Systolic arterial pressure | NS | NS | NS |
| Diastolic arterial pressure | NS | NS | NS |
| Pulse pressure | NS | NS | NS |
| Systemic arterial resistance | NS | NS | NS |
| Systemic arterial compliance | |||
| p Value | <0.001 | NS | NS |
| ΔR2 | 0.20 | NA | NA |
| Multivariate model | |||
| p Value | <0.001 | <0.001 | <0.001 |
| ΔR2 | 0.87 | 0.89 | 0.89 |
p Value and ΔR2 value for each independent variable in the multivariate predictive model are given. ΔR2 represents the respective contribution of the variable to the variance of TPG.
*Pressure gradient predicted by equation 8 from the energy loss coefficient and the mean systolic flow rate measured by Doppler echocardiography.
NA, not applicable; NS, not significant.
The energy loss coefficient, effective arterial elastance, and mean systolic flow rate were the only independent determinants of peak systolic LV wall stress with a relative contribution to its variance of 22% (ΔR2 = 0.22, p < 0.001), 32% (ΔR2 = 0.32, p < 0.001), and 9% (ΔR2 = 0.09, p < 0.001), respectively.
DISCUSSION
The assessment of patients with AS generally includes measurement of TPGs and valve EOA, as well as assessment of LV geometry and function. However, little attention is given to the anatomical and haemodynamic factors involved downstream from the stenosis. The results of this study suggest that, beyond stenosis severity, it is also important to consider the status of arterial haemodynamic function, since it may have important implications with regard to the markers of AS severity and the clinical management of these patients.
Effect of systemic hypertension on TPG
The results of this study suggest that the occurrence of systemic hypertension in patients with AS may result in an important reduction of TPGs. This phenomenon may be explained by several physiological mechanisms.
Firstly, the increase in LV afterload due to increased SAR and reduced SAC may cause a decrease in LV output and thus a reduction of TPGmax, TPGmean, and TPGptop. However, beyond the occurrence of AS, the induction of hypertension in our model did not result in any further significant decrease in the average mean flow rate. Nonetheless, there was a wide interindividual variation with regard to the change in flow rate that occurred with hypertension. Several animals had a large decrease in flow rate (see example in fig 1), whereas others had a substantial increase in flow rate. Most of the animals exhibiting an increase in flow rate during hypertension were in group B and this flow augmentation was mainly due to the shortening in LV ejection time that occurred as a consequence of phenylephrine induced tachycardia. It should, however, be noted that these findings are concordant with the clinical situation, where the direction and magnitude of the flow changes associated with systemic hypertension may vary widely from one patient to the other.
Secondly, the rise in pressure regimen may result in a significant enlargement of EOA during hypertension, which may in turn contribute to reductions of TPGmax, TPGmean, and TPGptop. In our model, the EOA actually increased during the induction of hypertension, which may have been caused either by an increase in the contraction coefficient or by an actual increase in the anatomical orifice area of the stenosis. Previous studies in normal aortic valves have shown that the anatomical orifice area and thus the EOA of the valve are affected not only by flow but also by aortic pressure.23,24 Indeed, aortic pressure increase produces aortic root expansion, subsequent commissure separation, and stretching of the free edge of the valve leaflets, thus resulting in an increase in valve orifice area. In this context, it should be emphasised that the most common form of AS encountered nowadays is degenerative AS, which is sclerosis and rigidity of the aortic leaflets without commissural fusion. Hence, in patients with AS, the aortic root may have the potential to expand under increased pressure, thus resulting in an increase in valve EOA irrespective of the change in flow rate. In our animal model, the increase in EOA that occurred during hypertension may have been related to the expansion of the supravalvar stenotic orifice caused by the increased radial pressure applied against the aortic wall. Although the results of this animal study cannot be directly transposed to the clinical situation of valvar AS, these results as well as the results of previous studies on aortic valve dynamics23,24 suggest that this phenomenon may occur in patients with degenerative AS without commissural fusion.
Thirdly, our results also show that the SAC has an independent effect on TPGptop but not on TPGmax and TPGmean. Hence, the decrease in SAC associated with systemic hypertension further contributes to considerably reduce TPGptop. Indeed as opposed to TPGmax and TPGmean, TPGptop is highly sensitive to changes in the aortic pressure waveform that may occur with decreasing SAC (fig 1).
Thus, in summary, the changes in systemic arterial haemodynamic properties that are associated with systemic hypertension can cause a decrease in mean flow rate and an increase in valve EOA, which, in turn, may result in important reduction of Doppler and catheter TPGs.
It should also be pointed out that the net TPGs (that is, the TPGs measured by catheterisation) are determined by not only the valve EOA and flow rate but also the aortic diameter, which may influence the magnitude of pressure recovery downstream from the vena contracta.21,22,25–28 This may have a direct implication in patients with AS and concomitant hypertension, since the aortic diameter may increase as a result of aortic pressure increase. The results of this study show that the net TPGs estimated by catheterisation can be accurately predicted from indices measurable by Doppler echocardiography—that is, the energy loss coefficient and the mean systolic flow rate (fig 4).
Comparison with previous studies
Laskey and colleagues3 incorporated the TPG flow relation across the valve into the Windkessel model of systemic circulation. After mathematical elaboration, they eliminated the flow term and then obtained an equation that predicts a decrease in TPGmean when SAR is increased, irrespective of flow:
 |
where Pao is the instantaneous aortic pressure and A and B are coefficients that characterise the geometric and hydraulic properties of the stenotic aortic valve. This mathematical model was then applied to haemodynamic data obtained by catheterisation in 15 patients studied at rest and during mild exercise that produced a minimum change in SAR (−20 (9)%) and no significant change in SAC. From equation 9, it remains uncertain whether SAC and SAR have an independent effect on TPGmean. Indeed, if SAR increases while flow is constant, then Pao increases proportionally (see equation 2), so that the ratio of SAR to Pao is constant. Similarly SAC and dPao/dt vary in inverse directions so that their product tends to remain constant. This equation thus suggests that, in conditions where flow and valve EOA are held constant, TPGmean is relatively independent of systemic arterial properties, which would be in accordance with the results of the present study. Indeed, these results suggest that the impact of systemic hypertension on TPGmax and TPGmean is simply related to flow rate, valve EOA, and aortic diameter. To this effect, it should be emphasised that the influence of the aortic diameter, which may vary substantially during hypertension, was not analysed in previous studies3,5 and this could have also contributed to explain why, in these studies, SAR was found to have a direct effect on the catheter derived TPGs, independently of EOA and flow rate.
Effect of systemic hypertension on LV systolic wall stress
Contrary to appearances, the reduction in TPGs that may occur as a result of systemic hypertension in patients with AS should not be interpreted as reflecting a reduction of the load imposed on the LV. On the contrary, the LV of patients with AS and concomitant systemic hypertension is submitted to two additive loads: a valvar load and an arterial load. Indeed, this study shows that the increase in systemic arterial load (equivalent to severe hypertension in humans) distal to the valvar load (equivalent to severe AS in humans) results in a dramatic increase in peak systolic LV wall stress despite a concomitant decrease in TPGs. In fact, the strongest independent determinant of systolic LV wall stress was the effective arterial elastance, which reflects the LV afterload imposed by the systemic arterial system. These results are consistent with the study of Khot and colleagues,29 who reported that nitroprusside rapidly and greatly improves cardiac function in patients with LV systolic dysfunction and severe AS. In this context, it should be emphasised that a failing ventricle is extremely sensitive to an increase in afterload. Hence, the reduction of the arterial component of the LV afterload by nitroprusside resulted in a major reduction of systolic wall stress and a major improvement of cardiac output in these patients with low flow AS.
Clinical implications
This study shows that the severity of AS may be partially masked by the presence of coexisting hypertension. The association of these two entities is highly insidious, since it increases the burden on the LV as shown by the major increase in LV wall stress despite a significant decrease in TPGs.
Evidently, it can be argued that the EOA did not change proportionately to the TPGs and that its calculation would still have reflected the severity of the stenosis. However, in patients with thickened valve leaflets and no commissural fusion, EOA may increase substantially in response to an increase in pressure or flow and would then no further reflect the severity of the stenosis. Nonetheless, systolic wall stress would remain increased in this situation and this would certainly not improve the patient’s clinical condition. On the other hand, treating the patient for hypertension would reverse a normotensive state and the markers of AS severity would reappear and indicate that there is still an increased burden on the LV.
In addition, it should be emphasised that TPGs are an integral part of the algorithm underlying the evaluation of AS severity. Hence, the occurrence of low TPGs in the presence of normal flow would certainly suggest to the unseasoned observer that the stenosis is possibly not as severe as it may seem from the EOA measurement and that there may be an error in the calculation of EOA. These considerations may become even more prevalent in the catheterisation laboratory, where TPGptop is often the parameter relied on most heavily to evaluate AS severity. Also, patients often have a higher blood pressure due to anxiety when they undergo catheterisation.
Given the evidence that AS severity can be significantly masked by hypertension and that no parameter can be meaningfully used to reconcile this discrepancy, a logical extension of the present findings with regard to clinical practice appear to be the following. Firstly, blood pressure by sphygmomanometry should be measured systematically when evaluating patients for AS in the echocardiographic laboratory. Secondly, if the findings are ambiguous and patients are hypertensive, the examination should be repeated once they have returned to a normotensive state. Thirdly, likewise, if patients are hypertensive when evaluated in the catheterisation laboratory, an attempt should be made at lowering their blood pressure and repeating the measurements when patients are in a normotensive state. Fourthly, the TPGptop should not be used to assess stenosis severity in patients with systolic hypertension and high differential pressure because it is highly dependent on SAC independently of AS severity. This recommendation may also be extended to patients with dyslipidaemia, diabetes, atherosclerosis, or coronary artery disease, since these conditions have also been associated with reduced SAC.30–33. Lastly, serial evaluations should take into account whether the patient’s blood pressure is the same from one examination to the other.
Beyond its potential usefulness in diagnosing AS severity, the normalisation of systemic arterial pressure may also have a beneficial effect from a treatment standpoint.29 Further studies are needed to evaluate the impact of antihypertensive medication on the prevention of LV dysfunction and associated clinical outcomes in patients with AS.
Study limitations
The study was conducted in an animal model not necessarily reflecting all the conditions present in the clinical situation. The biomechanics and haemodynamic function of aortic valve stenosis are more complex than supravalvar aortic banding. Hence, this study could not assess the impact of systemic arterial factors on the aortic valve opening and closing dynamics. Also, the methods used to produce systemic hypertension in this animal model do not necessarily reflect the actual physiopathology of hypertension in patients with AS. Further studies are thus needed to determine whether the results obtained in this acute model of supravalvar AS are also applicable to patients with valvar AS. Nonetheless, the study does provide a physiological basis for previous observations reporting that TPGs measured during cardiac catheterisation are often reduced compared with Doppler TPGs in patients with significant AS and concomitant systemic hypertension.2–4 Also, it strongly emphasises that blood pressure measurements should become an integral part of the examination of these patients and that AS severity should ideally be evaluated when the patient is in a normotensive state.
Acknowledgments
This work was supported by an operating grant from the Canadian Institutes of Health Research (MOP-10929). Dr Pibarot is the director of the Canada Research Chair in Valvular Heart Disease, Canadian Institutes of Health Research, Ottawa, Canada. We thank Lynn Atton, Guy Noël, Justin Robillard, and Guy Rossignol for their technical assistance.
Abbreviations
AS, aortic stenosis
EOA, effective orifice area
LV, left ventricular
Pao, instantaneous aortic pressure
SAC, systemic arterial compliance
SAR, systemic arterial resistance
TPG, transvalvar pressure gradient
TPGmax, maximum transvalvar pressure gradient
TPGmean, mean transvalvar pressure gradient
TPGptop, peak to peak transvalvar pressure gradient
Presented at the 75th Scientific Sessions of the American Heart Association, 17–20 November 2002, Chicago, Illinois, USA.
REFERENCES
- 1.Gorlin R, Gorlin SG. Hydraulic formula for calculation of the area of the stenotic mitral valve, other cardiac valves, and central circulatory shunts. Am Heart J 1951;41:1. [DOI] [PubMed] [Google Scholar]
- 2.Roberts WC, Perloff JK, Costantino T. Severe valvular aortic stenosis in patients over 65 years of age: a clinicopathologic study. Am J Cardiol 1971;27:497–506. [DOI] [PubMed] [Google Scholar]
- 3.Laskey WK, Kussmaul WG, Noordergraaf A. Valvular and systemic arterial hemodynamics in aortic valve stenosis. Circulation 1995;92:1473–8. [DOI] [PubMed] [Google Scholar]
- 4.Chambers J . Can high blood pressure mask severe aortic stenosis? J Heart Valve Dis 1998;7:277–8. [PubMed] [Google Scholar]
- 5.Razzolini R, Gerosa G, Leoni L, et al. Transaortic gradient is pressure-dependent in a pulsatile model of the circulation. J Heart Valve Dis 1999;8:279–83. [PubMed] [Google Scholar]
- 6.Antonini-Canterin F, Huang G, Cervesato E, et al. Symptomatic aortic stenosis: does systemic hypertension play an additional role? Hypertension 2003;41:1268–72. [DOI] [PubMed] [Google Scholar]
- 7.Xu J, Durand LG, Pibarot P. A new, simple, and accurate method for noninvasive estimation of pulmonary arterial pressure. Heart 2002;88:76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith MD, Dawson PL, Elion JL, et al. Correlation of continuous wave Doppler velocities with cardiac catheterization gradients: an experimental model of aortic stenosis. J Am Coll Cardiol 1985;6:1306–14. [DOI] [PubMed] [Google Scholar]
- 9.Kadem L, Pibarot P, Dumesnil JG, et al. Independent contribution of the left ventricular ejection time to the mean gradient in aortic stenosis. J Heart Valve Dis 2002;11:615–23. [PubMed] [Google Scholar]
- 10.Dumesnil JG, Yoganathan AP. Theoretical and practical differences between the Gorlin formula and the continuity equation for calculating aortic and mitral valve areas. Am J Cardiol 1991;67:1268–72. [DOI] [PubMed] [Google Scholar]
- 11.Chemla D, Hébert J-L, Coirault C, et al. Total arterial compliance estimated by stroke volume-to-aortic pulse pressure ratio in humans. Am J Physiol (Heart Circ Physiol) 1998;274:H500–5. [DOI] [PubMed] [Google Scholar]
- 12.Segers P, Verdonck P, Deryck Y, et al. Pulse pressure method and the area method for the estimation of total arterial compliance in dogs: sensitivity to wave reflection intensity. Ann Biomed Eng 1999;27:480–5. [DOI] [PubMed] [Google Scholar]
- 13.De Simone G, Roman MJ, Koren MJ, et al. Stroke volume/pulse pressure ratio and cardiovascular risk in arterial hypertension. Hypertension 1999;33:800–5. [DOI] [PubMed] [Google Scholar]
- 14.Palmieri V, Bella JN, Roman MJ, et al. Pulse pressure/stroke index and left ventricular geometry and function: the LIFE study. J Hypertens 2003;21:781–7. [DOI] [PubMed] [Google Scholar]
- 15.Nichols W, O’Rourke MF. Input impedance as ventricular load. In: Nichols W, O’Rourke MF, eds. McDonald blood flow in the arteries. theoretical, experimental and clinical principles. London: Arnold, 1998:285–93.
- 16.Sunagawa K, Maughan WL, Burkhoff D, et al. Left ventricular interaction with arterial load studied in isolated canine ventricle. Am J Physiol (Heart Circ Physiol) 1983;245:H773–80. [DOI] [PubMed] [Google Scholar]
- 17.Sunagawa K, Sagawa K, Maughan WL. Ventricular interaction with the loading system. Ann Biomed Eng 1984;12:163–89. [DOI] [PubMed] [Google Scholar]
- 18.Segers P, Stergiopulos N, Westerhof N. Relation of effective arterial elastance to arterial system properties. Am J Physiol (Heart Circ Physiol) 2002;282:H1041–6. [DOI] [PubMed] [Google Scholar]
- 19.Chemla D, Antony I, Lecarpentier Y, et al. Contribution of systemic vascular resistance and total arterial compliance to effective arterial elastance in humans. Am J Physiol (Heart Circ Physiol) 2003;285:H614–20. [DOI] [PubMed] [Google Scholar]
- 20.Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest 1975;56:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia D, Pibarot P, Dumesnil JG, et al. Assessment of aortic valve stenosis severity: a new index based on the energy loss concept. Circulation 2000;101:765–71. [DOI] [PubMed] [Google Scholar]
- 22.Garcia D, Dumesnil JG, Durand LG, et al. Discrepancies between catheter and Doppler estimates of valve effective orifice area can be predicted from the pressure recovery phenomenon: practical implications with regard to quantification of aortic stenosis severity. J Am Coll Cardiol 2003;41:435–42. [DOI] [PubMed] [Google Scholar]
- 23.Thubrikar M, Bosher LP, Nolan SP. The mechanism of opening of the aortic valve. J Thorac Cardiovasc Surg 1979;77:863–70. [PubMed] [Google Scholar]
- 24.Higashidate M, Tamiya K, Beppu T, et al. Regulation of the aortic valve opening: in vitro dynamic measurement of aortic valve orifice area. J Thorac Cardiovasc Surg 1995;110:496–503. [DOI] [PubMed] [Google Scholar]
- 25.Niederberger J, Schima H, Maurer G, et al. Importance of pressure recovery for the assessment of aortic stenosis by Doppler ultrasound: role of aortic size, aortic valve area, and direction of the stenotic jet in vitro. Circulation 1996;94:1934–40. [DOI] [PubMed] [Google Scholar]
- 26.Baumgartner H, Steffenelli T, Niederberger J, et al. “Overestimation” of catheter gradients by Doppler ultrasound in patients with aortic stenosis: a predictable manifestation of pressure recovery. J Am Coll Cardiol 1999;33:1655–61. [DOI] [PubMed] [Google Scholar]
- 27.Heinrich RS, Marcus RH, Ensley AE, et al. Valve orifice area alone is an insufficient index of aortic stenosis severity: effects of the proximal and distal geometry on transaortic energy loss. J Heart Valve Dis 1999;8:509–15. [PubMed] [Google Scholar]
- 28.Schöbel WA, Voelker W, Haase KK, et al. Extent, determinants and clinical importance of pressure recovery in patients with aortic valve stenosis. Eur Heart J 1999;20:1355–63. [DOI] [PubMed] [Google Scholar]
- 29.Khot UN, Novaro GM, Popovic ZB, et al. Nitroprusside in critically ill patients with left ventricular dysfunction and aortic stenosis. N Engl J Med 2003;348:1756–63. [DOI] [PubMed] [Google Scholar]
- 30.Airaksinen KE, Salmela PI, Linnaluoto MK, et al. Diminished arterial elasticity in diabetes: association with fluorescent advanced glycosylation end products in collagen. Cardiovasc Res 1993;27:942–5. [DOI] [PubMed] [Google Scholar]
- 31.Toto-Moukouo JJ, Achimastos A, Asmar RG, et al. Pulse wave velocity in patients with obesity and hypertension. Am Heart J 1986;112:136–40. [DOI] [PubMed] [Google Scholar]
- 32.Sutton-Tyrrell K, Newman A, Simonsick EM, et al. Aortic stiffness is associated with visceral adiposity in older adults enrolled in the study of health, aging, and body composition. Hypertension 2001;38:429–33. [DOI] [PubMed] [Google Scholar]
- 33.O’Rourke MF, Staessen JA. Clinical applications of arterial stiffness: definitions and references values. Am J Hypertens 2002;15:426–44. [DOI] [PubMed] [Google Scholar]