

Abstract
Objective: To examine whether 17-β-oestradiol (E2) may alter angiotensin II (Ang II) induced cell proliferation and to identify the putative underlying signalling pathways in rat cardiac fibroblasts.
Design: Cultured rat cardiac fibroblasts were preincubated with E2 then stimulated with Ang II. [3H]Thymidine incorporation and endothelin-1 (ET-1) gene expression were examined. The effect of E2 on Ang II induced NADPH oxidase activity, reactive oxygen species (ROS) formation, and extracellular signal regulated kinase (ERK) phosphorylation were tested to elucidate the intracellular mechanism of E2 in proliferation and ET-1 gene expression.
Results: Ang II increased DNA synthesis, which was inhibited with E2 (1–100 nmol/l). E2, but not 17-α-oestradiol, inhibited Ang II induced ET-1 gene expression as shown by northern blotting and promoter activity assay. This effect was prevented by co-incubation with the oestrogen receptor antagonist ICI 182 780 (1 µmol/l). E2 also inhibited Ang II increased NADPH oxidase activity, ROS formation, ERK phosphorylation, and activator protein-1 mediated reporter activity.
Conclusions: The results suggest that E2 inhibits Ang II induced cell proliferation and ET-1 gene expression, partially by interfering with the ERK pathway through attenuation of ROS generation. Thus, this study provides important new insight regarding the molecular pathways that may contribute to the proposed beneficial effects of oestrogen on the cardiovascular system.
Keywords: endothelin-1, 17-β-oestradiol, angiotensin II, cardiac fibroblasts, reactive oxygen species, extracellular signal regulated kinase
Oestrogen has been reported to have cardioprotective and antihypertensive effects in premenopausal women. 17-β-Oestradiol (E2) is the terminal biologically active natural oestrogen with the highest affinity to the oestrogen receptor. E2 has been reported to inhibit low density lipoproteins peroxidation in vitro,1 to an even larger extent than vitamin E does.2 Besides lipid modifying effects, E2 exerts its potentially vasoprotective effects through decreased vascular oxidative stress.3,4 Laufs and colleagues reported that E2 inhibits vascular reactive oxygen species (ROS) production by downregulating Rac1 GTPase in vascular smooth muscle cells.5 Furthermore, E2 is reported to exert radical scavenging effects through the regulation of radical scavenging enzyme activity and expression in the vascular cells.6 Thus, it is reasonable to propose that the cardioprotective effect of E2 may be related to its antioxidant properties; however, this hypothesis remains to be tested.
Angiotensin II (Ang II), the effector peptide of the renin–angiotensin system, is now known to have growth promoting properties in various cell types and a mitogenic effect on cardiac fibroblasts.7–9 Recent reports have shown that Ang II stimulates membrane bound NADPH oxidase, which generates ROS in cardiac fibroblasts.10 We recently reported that ROS are essential for Ang II induced proliferation and endothelin-1 (ET-1) gene expression in cardiac fibroblasts.11 However, whether E2 inhibits Ang II induced cell proliferation and ET-1 gene induction by attenuating ROS generation in cardiac fibroblasts remains unclear. This study was conducted to examine whether E2 inhibits Ang II induced ET-1 gene expression and to identify signalling protein kinase cascades that may be responsible for the putative effect of oestrogen.
METHODS
Materials
Dulbecco’s modified Eagle’s medium (DMEM), fetal calf serum, and tissue culture reagents were obtained from Life Technologies, Inc. A rat ET-1 cDNA probe (accession No M64711) was obtained as previously described.12 A full length of the ET-1 promoter region (4.4 kb) was fused to the chloramphenicol acetyltransferase (CAT) reporter gene.11 PBLCAT2 (containing CAT reporter gene with its promoter) and PBLCAT3 (containing the CAT gene only) were constructed as previously described.13 2′,7′-Dichlorofluorescein diacetate was obtained from Molecular Probes (Eugene, Oregon, USA). H2O2 was purchased from Acros Organics (Pittsburgh, Pennsylvania, USA). E2, N-acetylcysteine (NAC), and all other reagent grade chemicals were purchased from the Sigma Chemical Co (St Louis, Missouri, USA). ICI 182 780 was purchased from Tocris Cookson (Ballwin, Missouri, USA). The plasmid activator protein-1 (AP-1) luciferase containing the firefly luciferase reporter gene driven by a basic promoter element (TATA box) joined to tandem repeats of AP-1 binding element were obtained from Stratagene (La Jolla, California, USA).
Culture of cardiac fibroblasts
The investigation was conducted in accordance with the Guide for the care and use of laboratory animals published by the National Institutes of Health in the United States (NIH Publication No 85–23, revised 1996) and approved by the institutional animal care and use committee of Taipei Medical University. Primary cultures of neonatal rat cardiac fibroblasts were prepared as previously described.11 Cardiac fibroblasts grown in either 60 or 100 mm culture dishes from the second to fourth passage were used for the experiments. The purity of the fibroblasts was > 95% as determined by morphological characterisation and by immunostaining with antibodies to von Willebrand factor VIII. Cardiac fibroblasts were grown in DMEM without phenol red containing antibiotics and 10% fetal calf serum until 24 hours before experimentation, when cells were placed in a defined serum-free medium containing insulin (0.5 μmol/l) and transferrin (5 mg/ml) for all experiments. Cells were then preincubated with E2 for 12 hours and then with or without Ang II (100 nmol/l) for various incubation times as indicated, followed by harvesting. Cellular viability under all treatment conditions was determined by cell count, morphology, and trypan blue exclusion.
DNA synthesis
To measure synthesis of new DNA, cells (1 × 105/well) were plated in six-well (35 mm) dishes 24 hours before experiments as previously described.14 Cells were incubated with [3H]thymidine (5 μCi/ml). After the indicated treatment, cells were harvested by incubation at 4°C with trichloroacetic acid (5%) followed by solubilisation in a 1/10th normal NaOH solution. Radioactivity was determined by scintillation counting. Data are presented as the mean (SEM) of 9–12 determinations for three to four cell preparations and normalised to the untreated sample × 100 (that is, the percentage of control).
NADPH oxidase activity assay
NADPH oxidase was measured in cardiac fibroblasts as described previously.15
Detection of intracellular ROS
Intracellular ROS formation in cardiac fibroblasts was measured by monitoring changes in dichlorofluorescein fluorescence as described previously.16 Chemiluminescence of superoxide production was assayed as described previously.17
RNA isolation and northern blot analysis
Total RNA and northern blot analyses of ET-1 and 18S RNA were prepared as described previously.11
Transfection and CAT assays
For the transient transfections, cells were transfected with various expression vectors by the calcium phosphate method.13 For the transient transfections, cardiac fibroblasts were transfected with various expression vectors by the calcium phosphate method. DNA concentration for all samples was adjusted to an equal amount in each experiment. Briefly, cardiac fibroblasts were maintained in culture for 24 hours before transfection. The indicated expression vectors were mixed with calcium phosphate and immediately added to the cardiac fibroblast cell culture. After incubation for five hours, cells were washed three times with phosphate buffered saline and incubated with 10% serum DMEM. After 12 hours, cells were washed with serum-free media and further incubated for 48 hours in serum-free medium. To correct for variability in transfection efficiency, 5 μg of pSV-β′-galactosidase plasmid DNA was co-transfected in all the experiments. CAT and β-galactosidase were assayed as described previously.13
Western blot analysis
Rabbit polyclonal phospho-specific extracellular signal regulated kinase (ERK) antibodies were purchased from New England Biolabs (Beverly, Massachusetts, USA). ERK antibodies were purchased from Santa Cruz Biotechnology Inc (Santa Cruz, California, USA). Western blot analysis was performed as previously described.16
Luciferase assay
Cardiac fibroblasts plated on six-well (35 mm) dishes were transfected with the luciferase reporter construct possessing consensus AP-1 binding sites (AP-1-luciferase) (Stratagene). After incubation for 24 hours in serum-free DMEM, cardiac fibroblasts were cultured under various conditions as indicated for 48 hours and then assayed for luciferase activity with a luciferase reporter assay kit (Stratagene). As was the case for AP-1 transcriptional activity, the specific firefly luciferase activity was normalised for transfection efficiency to its respective β-galactosidase activity and expressed relative to the control.
Statistical analysis
Results are expressed as mean (SEM) for at least six experiments unless designated otherwise. Data were analysed with Student’s t test and analysis of variance followed by a Dunnett multiple comparison test with GraphPad Prism (GraphPad Software, San Diego, California, USA). A probability value of p < 0.05 was considered to be significant.
RESULTS
Effects of E2 on Ang II induced cell proliferation of cardiac fibroblasts
The effects of E2 on Ang II stimulated rat cardiac fibroblast proliferation was assessed by analysing DNA synthesis with [3H]thymidine incorporation. Preincubation of cardiac fibroblast s with E2 for 12 hours (1–100 nmol/l) followed by exposure to E2 with Ang II (100 nmol/l) for 24 hours resulted in a concentration dependent decrease in Ang II induced cell proliferation (fig 1A). Unlike E2, 17-α-oestradiol (100 nmol/l) had no effect on Ang II induced cell proliferation, whereas preincubation with the oestrogen receptor antagonist ICI 182 780 (1 µmol/l) prevented E2 mediated downregulation of Ang II induced cell proliferation (fig 1B). These data clearly suggest that E2 inhibits Ang II induced cell proliferation of cardiac fibroblasts.
Figure 1.
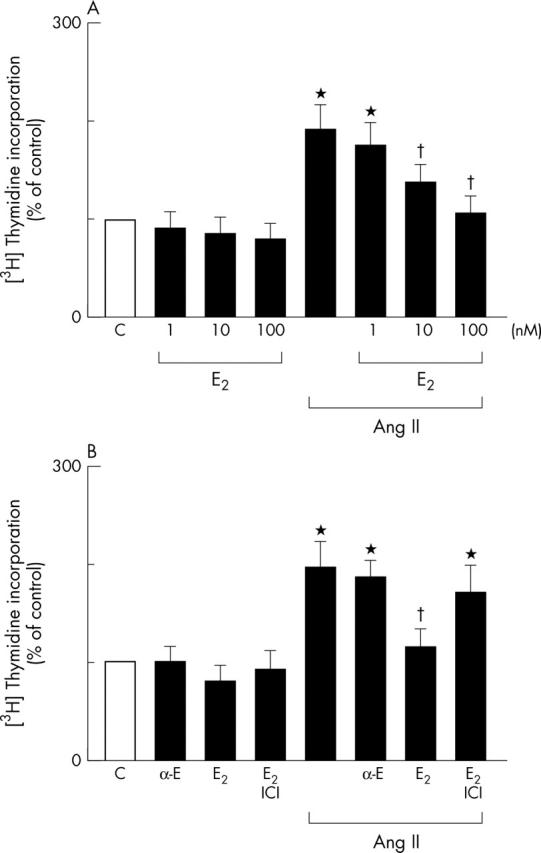
Effects of 17-β-oestradiol (E2) on angiotensin II (Ang II) induced cell proliferation in cardiac fibroblasts. All experiments were performed by incorporation of [3H]thymidine into DNA. Increases in [3H]thymidine incorporation are each expressed relative to the [3H] content (100%) in their respective drug-free controls (C). All data are shown as the mean (SEM) for triplicate determinations for six cell preparations. *p < 0.05 v C; †p < 0.05 v Ang II alone. (A) E2 inhibits Ang II induced DNA synthesis. Cells were preincubated with the indicated doses of E2 and treated with Ang II (100 nmol/l) for 24 hours. [3H]thymidine incorporation was then assayed. (B) Effects of the oestrogen receptor antagonist ICI 182 780 (ICI) on E2 mediated inhibition of Ang II induced DNA synthesis and lack of effect of 17-α-oestradiol (α-E) on Ang II induced DNA synthesis. Cells were preincubated with α-E (100 nmol/l), E2 (100 nmol/l), or the combination of E2 plus ICI (1 µmol/l) followed by either the application of Ang II (100 nmol/l) for 24 hours or its absence.
Effects of E2 on Ang II induced ET-1 gene expression in cardiac fibroblasts
Northern blot analysis was used to examine whether E2 inhibits Ang II increased ET-1 mRNA concentrations in cardiac fibroblasts (fig 2A). Cardiac fibroblasts were preincubated with E2 (100 nmol/l, 12 hours), treated with Ang II (100 nmol/l) for 30 minutes, then assayed for E2 inhibited Ang II induced ET-1 mRNA expression. Unlike E2, 17-α-oestradiol (100 nmol/l) had no effect on Ang II induced ET-1 mRNA, whereas preincubation with the oestrogen receptor antagonist ICI 182 780 (1 µmol/l) prevented E2 mediated downregulation of Ang II induced ET-1 mRNA (fig 2A). To determine whether E2 inhibition of Ang II induced ET-1 expression is regulated at the transcriptional level, an ET-1 promoter construct containing the ET-1 promoter region (−4.4 kb) and the reporter gene CAT was constructed and transiently transfected into cardiac fibroblasts. Exposure of cardiac fibroblasts to Ang II (100 nmol/l) for 24 hours significantly increased ET-1 promoter activity (fig 2B). Preincubation of cardiac fibroblasts with E2 (100 nmol/l) inhibited Ang II induced ET-1 promoter activity. Unlike E2, 17-α-oestradiol (100 nmol/l) had no effect on Ang II induced ET-1 promoter activity, whereas the preincubation of cardiac fibroblasts with the oestrogen receptor antagonist ICI 182 780 (1 µmol/l) prevented E2 mediated downregulation of Ang II induced ET-1 promoter activity (fig 2B). These data indicate that E2 inhibits Ang II induced ET-1 gene expression in cardiac fibroblasts.
Figure 2.
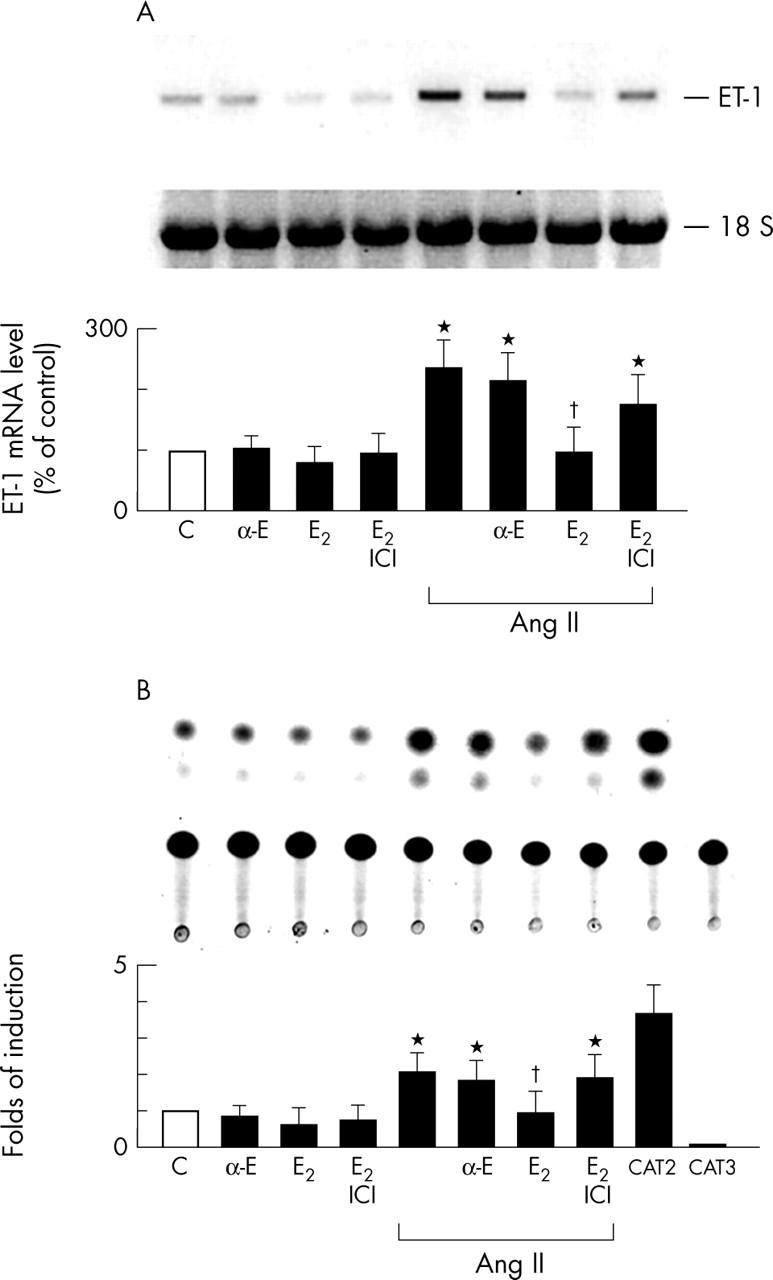
E2 down regulates Ang II induced endothelin-1 (ET-1) gene expression in cardiac fibroblasts. The results are shown as the mean (SEM) (n = 6). *p < 0.05 v C; †p < 0.05 v Ang II alone. (A) Downregulation of Ang II induced ET-1 mRNA by E2. Cells were preincubated with α-E (100 nmol/l), E2 (100 nmol/l), or the combination of E2 plus ICI (1 µmol/l) and then stimulated with Ang II (100 nmol/l) for 30 minutes or not. Total RNA was extracted and northern hybridisation was performed with 32P labelled ET-1 as a probe. 18S RNA was used to normalise the RNA applied in each lane. Data are presented as percentage changes of experimental groups compared with untreated C. (B) E2 inhibits Ang II induced ET-1 promoter activity. Cells were transfected with chimeric chloramphenicol acetyltransferase (CAT) fusion genes and preincubated with α-E (100 nmol/l), E2 (100 nmol/l), or the combination of E2 plus ICI (1 µmol/l) and then stimulated with Ang II (100 nmol/l) for 24 hours or not. Cells were harvested and CAT activities were measured. CAT activities after normalising to activities of β-galactosidase are shown as activity relative to C. CAT2 and CAT3 are positive and negative controls, respectively.
Effects of E2 on Ang II increased NADPH oxidase activity and ROS formation
We have shown that Ang II stimulates ROS production in cardiac fibroblasts.11 In this study, we further examined whether E2 prevents Ang II increased NADPH oxidase activity and ROS formation in cardiac fibroblasts. Cardiac fibroblasts were pretreated with E2 (1–100 nmol/l, 12 hours), then treated with Ang II (100 nmol/l). Preincubation of cultured cardiac fibroblasts with E2 (1–100 nmol/l) significantly inhibited Ang II induced NADPH oxidase activity and ROS formation as measured after Ang II treatment for 30 minutes (fig 3A, B, C). Pretreatment of cultured cardiac fibroblasts with E2 (100 nmol/l), NAC (10 mmol/l), or diphenyliodonium (DPI) (10 μmol/l) significantly inhibited Ang II induced ROS concentrations (fig 3D). Preincubation with the oestrogen receptor antagonist ICI 182 780 (1 µmol/l) also prevented the inhibitory effect of E2 (fig 3B). These findings support our earlier comments that E2 inhibits Ang II increased NADPH oxidase activity and intracellular ROS concentrations in cardiac fibroblasts.
Figure 3.

Effects of E2 on Ang II increased NADPH oxidase activity and reactive oxygen species (ROS) formation. Cells were preincubated with E2 (1–100 nmol/l) for 12 hours and then stimulated with Ang II (100 nmol/l) for 30 minutes or not. *p < 0.05 v C; †p < 0.05 v Ang II alone. (A) Effect of E2 (1–100 nmol/l) on Ang II increased NADPH oxidase activity. Cardiac fibroblasts after treatment were lysed and immediately assayed for NADPH oxidase activity. (B) Effect of E2 (1–100 nmol/l) on Ang II induced superoxide formation. Cardiac fibroblasts after treatment were lysed and immediately assayed for superoxide by the lucigenin method. (C) Effect of E2 (1–100 nmol/l) on Ang II induced ROS generation. Fluorescent intensities of dichlorofluorescein showed that intracellular ROS concentrations were increased by Ang II. (D) Effects of E2, E2 plus ICI, or antioxidants on Ang II induced ROS generation. Cells were preincubated with E2 (100 nmol/l), the combination of E2 plus ICI (1 µmol/l), or the antioxidant N-acetylcysteine (NAC) (10 mmol/l) or diphenyliodonium (DPI) (10 μmol/l) and then stimulated with Ang II (100 nmol/l) for 30 minutes or not. H2O2 (100 μmol/l) was used as a positive control.
Effects of E2 on Ang II activated ERK phosphorylation and AP-1 mediated reporter activity in cardiac fibroblasts
Ang II has previously been shown to activate ERK, and the activation of this pathway is redox sensitive.11 We recently reported that ROS were involved in the activation of the ERK pathway, which culminated in ET-1 gene expression.11,14 To gain insight into the mechanism of action of E2, we examined whether E2 affects the Ang II activated ERK pathway of cardiac fibroblasts. We examined the effect of E2 and antioxidants on Ang II induced ERK phosphorylation. As fig 4A shows, exposure of cardiac fibroblasts to Ang II (100 nmol/l) for 30 minutes rapidly activated phosphorylation of ERK. However, cardiac fibroblasts pretreated with E2 (100 nmol/l, 12 hours) and then with Ang II (100 nmol/l) for 30 minutes had significantly decreased levels of Ang II induced ERK phosphorylation. Pretreatment of cardiac fibroblasts with the antioxidant NAC (10 mmol/l, 30 minutes) or DPI (10 μmol/l, 30 minutes) also significantly decreased Ang II induced ERK phosphorylation. Moreover, Ang II increased AP-1 activation is involved in ET-1 gene induction.11 We further evaluated the effects of E2 on Ang II induced AP-1 functional activity with a reporter gene assay. E2 (100 nmol/l), NAC (10 mmol/l), or DPI (10 μmol/l) significantly attenuated Ang II induced AP-1 mediated reporter activation (fig 4B). These findings show that E2 inhibits the Ang II activated ERK signalling pathway and AP-1 activation, which is compatible with antioxidant action in cardiac fibroblasts.
Figure 4.
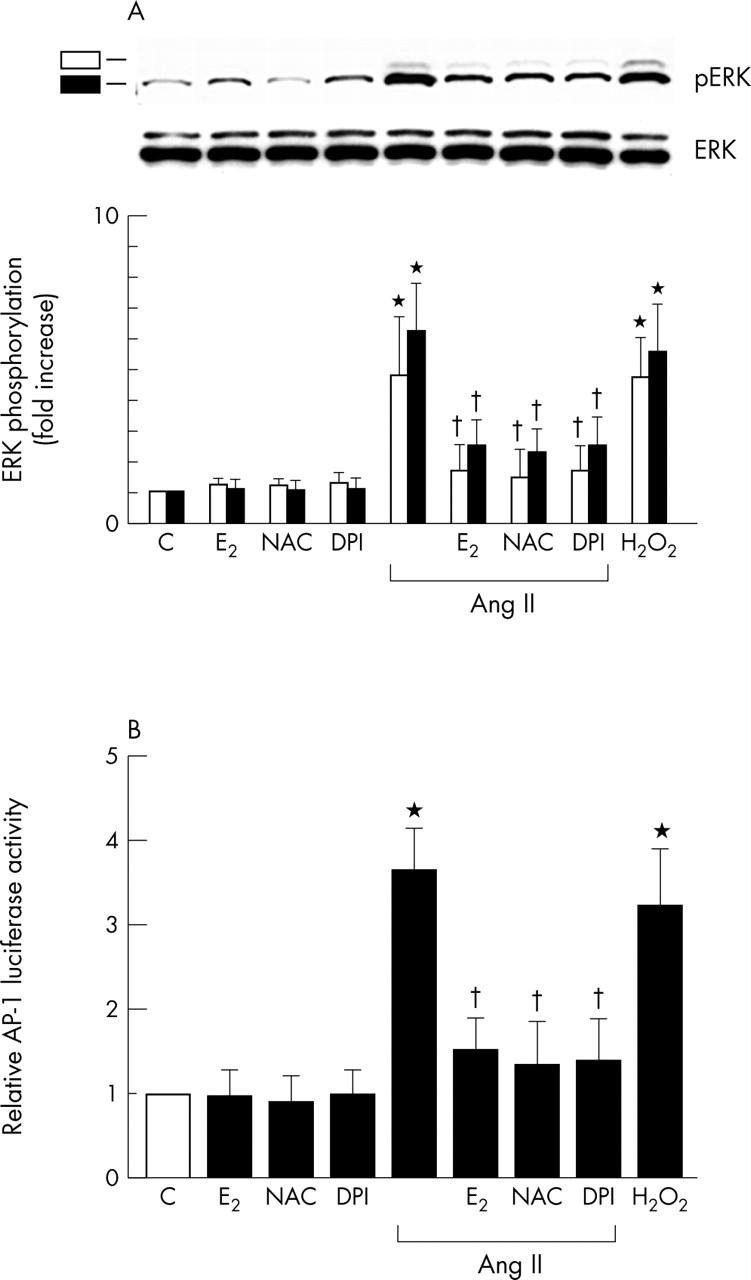
Inhibitory effect of E2 on Ang II increased extracellular signal regulated kinase (ERK) phosphorylation and activator protein 1 (AP-1) mediated reporter activity in cardiac fibroblasts. Results are shown as the mean (SEM) (n = 6). *p < 0.05 v control; †p < 0.05 v Ang II alone. (pERK, phosphorylated ERK.) (A) Effects of E2, E2 plus ICI, or various antioxidants on Ang II increased ERK phosphorylation. Cells were preincubated with E2 (100 nmol/l), NAC (10 mmol/l), or DPI (10 μmol/l) and then stimulated with Ang II (100 nmol/l) for 30 minutes. E2, NAC, or DPI inhibited Ang II induced phosphorylation of ERK. H2O2 (100 μmol/l) was used as a positive control. Phosphorylation of ERK was detected by western blotting with phospho-ERK antibody. Density was measured with a densitometer. Data are shown as fold increase relative to control groups. (B) Effects of E2, E2 plus ICI, or various antioxidants on Ang II increased AP-1 mediated reporter activity. Cardiac fibroblasts, transfected with AP-1-luciferase, were treated as indicated. Cells were preincubated with E2 (100 nmol/l), the combination of E2 plus ICI (1 µmol/l), or the antioxidant NAC (10 mmol/l) or DPI (10 μmol/l) and then stimulated with Ang II (100 nmol/l) for 24 hours or not. H2O2 (100 μmol/l) was used as a positive control. Luciferase activity was expressed as activity relative to untreated C.
DISCUSSION
Neonatal cardiac fibroblasts were used in these experiments, and it is possible that different results might have been obtained had adult cardiac fibroblasts been used. However, we have previously shown that ROS mediates Ang II induced proliferation and ET-1 gene expression in neonatal rat cardiac fibroblasts.14 Furthermore, neonatal fibroblasts have properties (that is, [3H]proline incorporation and production of both matrix metalloproteinases and metalloproteinases) relevant to extracellular matrix turnover in the post-myocardial infarction heart.18
From several epidemiological studies, E2 is thought to have a protective effect against left ventricular hypertrophy, which is an important cardiovascular risk factor for morbidity and mortality.19–21 Premenopausal women have a lower prevalence of left ventricular hypertrophy than their age matched male counterparts.19 Left ventricular mass is significantly greater in men than in women even after indexing for body surface area.20,21 Experimental studies have shown cardioprotective roles of E222–25; however, the direct effect of E2 on cardiac cell growth remains controversial. Previous studies have shown that the exogenous administration of E2 alone either decreased,22 increased,26 or had no effect on DNA synthesis in cultured cardiac fibroblasts.27,28 In this study, we clearly showed that E2 modulates Ang II induced cell proliferation and ET-1 gene expression in cardiac fibroblasts. Increased ROS concentrations are involved in cell proliferation and ET-1 induction, which can be attenuated by antioxidant pretreatment of cells.11,14 In agreement with previous studies in other cell types,5,29 we have shown that E2 inhibits Ang II mediated ROS release in cardiac fibroblasts. More specifically, E2 prevented Ang II mediated NADPH oxidase activity but did not significantly reduce ROS after transfection with the active RacL61,5 pointing towards a role of Rac1 in the antioxidative effects of E2. Indeed, Laufs and colleagues5 also showed that E2 downregulated Rac1 protein and mRNA expression in a concentration and time dependent manner, both alone and in the presence of Ang II. Similarly, E2 inhibited basal and Ang II stimulated Rac1 activity.5 Downregulation of Rac1 expression by E2 was completely blocked in the presence of the non-selective oestrogen receptor antagonist ICI 182 780,5 indicating that the event was receptor mediated. In particular, it has been shown that activation of ERK is redox sensitive10,30,31 and that suppression of ROS inhibits ET-1 gene expression.11,14 One possible explanation for the inhibitory effect of E2 on Ang II induced cell proliferation and ET-1 gene expression may thus be the ability of E2 to attenuate ROS formation and then inhibit ERK phosphorylation and AP-1 mediated reporter activity in cardiac fibroblasts. Alternatively, E2 may inhibit Ang II induced cell proliferation and ET-1 gene expression by the increase of radical scavenging enzyme activity and expression.6 Although the effects of oestrogen in vitro on cardiac Ang II subtype 1 (AT1) receptor expression are not known, several studies have shown an alteration in Ang II receptor expression or binding with changes in circulating oestrogen concentrations in vivo.32 In vascular smooth muscle cells, oestrogen acts on the renin–angiotensin system at different points of the cascade: at the formation of Ang II, at the level of Ang II receptors, and on Ang II induced responses. Oestrogen has been shown to increase gene expression and plasma concentrations of angiotensinogen. Oestrogen deficiency has been shown to increase AT1 receptor mRNA concentrations, as well as the efficacy of Ang II in vasoconstriction (due to increased AT1 receptor density), whereas oestrogen replacement therapy in ovariectomised rats reversed AT1 receptor overexpression.29 Clarification of whether oestradiol affects the number of angiotensin receptors or angiotensin binding to its receptors may shed light on the mechanisms of oestradiol effects in fibroblasts. Thus, further experiments will be necessary to identify the detailed mechanisms of the inhibitory effects of E2 on Ang II induced ET-1 gene expression in cardiac fibroblasts.
The present study delivers important new insight into the molecular mechanisms of action of E2 in cardiac fibroblasts. Moreover, our results show that E2 suppresses the ERK pathway and reduces Ang II induced cell proliferation and ET-1 gene expression. It appears plausible that the Ang II activated signalling pathway consists of a number of redox sensitive steps and that E2 treatment may modulate the redox state of the cell.
In summary, this study has shown that E2 inhibits Ang II induced ROS formation, ERK phosphorylation, AP-1 mediated reporter activity, ET-1 gene expression, and cell proliferation in cardiac fibroblasts. These findings support the proposed beneficial effects of oestrogen in the cardiovascular system.
Acknowledgments
This study was supported by grants from Shin Kong Wu Ho-Su Memorial Hospital (SKH-TMU-92-15 to T-H Cheng and H-H Chao; SKH-TMU-93-03 to J-C Liu and J-Y Liou).
Abbreviations
Ang II, angiotensin II
AP-1, activator protein-1
AT1, angiotensin II subtype 1
CAT, chloramphenicol acetyltransferase
DMEM, Dulbecco’s modified Eagle’s medium
DPI, diphenyliodonium
E2, 17-β-oestradiol
ERK, extracellular signal regulated kinase
ET-1, endothelin-1
NAC, N-acetylcysteine
ROS, reactive oxygen species
REFERENCES
- 1.Rifici VA, Khachadurian AK. The inhibition of low-density lipoprotein oxidation by 17-beta estradiol. Metabolism 1992;41:1110–4. [DOI] [PubMed] [Google Scholar]
- 2.Ayres S, Tang M, Subbiah MT. Estradiol-17beta as an antioxidant: some distinct features when compared with common fat-soluble antioxidants. J Lab Clin Med 1996;128:367–75. [DOI] [PubMed] [Google Scholar]
- 3.Hong MK, Romm PA, Reagan K, et al. Effects of estrogen replacement therapy on serum lipid values and angiographically defined coronary artery disease in postmenopausal women. Am J Cardiol 1992;69:176–8. [DOI] [PubMed] [Google Scholar]
- 4.Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med 1999;340:1801–11. [DOI] [PubMed] [Google Scholar]
- 5.Laufs U, Adam O, Strehlow K, et al. Down-regulation of Rac-1 GTPase by estrogen. J Biol Chem 2003;278:5956–62. [DOI] [PubMed] [Google Scholar]
- 6.Strehlow K, Rotter S, Wassmann S, et al. Modulation of antioxidant enzyme expression and function by estrogen. Circ Res 2003;93:170–7. [DOI] [PubMed] [Google Scholar]
- 7.Fujisaki H, Ito H, Hirata Y, et al. Natriuretic peptides inhibit angiotensin II-induced proliferation of rat cardiac fibroblasts by blocking endothelin-1 gene expression. J Clin Invest 1995;96:1059–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gray MO, Long CS, Kalinyak JE, et al. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovasc Res 1998;40:352–63. [DOI] [PubMed] [Google Scholar]
- 9.Crabos M, Roth M, Hahn AW, et al. Characterization of angiotensin II receptors in cultured adult rat cardiac fibroblasts: coupling to signaling systems and gene expression. J Clin Invest 1994;93:2372–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sano M, Fukuda K, Sato T, et al. ERK and p38 MAPK, but not NF-kappaB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ Res 2001;89:661–9. [DOI] [PubMed] [Google Scholar]
- 11.Mervaala EM, Cheng ZJ, Tikkanen I, et al. Endothelial dysfunction and xanthine oxidoreductase activity in rats with human renin and angiotensinogen genes. Hypertension 2001;37:414–8. [DOI] [PubMed] [Google Scholar]
- 12.Deng AY, Dene H, Pravenec M, et al. Genetic mapping of two new blood pressure quantitative trait loci in the rat by genotyping endothelin system genes. J Clin Invest 1994;93:2701–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng TH, Shih NL, Chen SY, et al. Reactive oxygen species modulate endothelin-I-induced c-fos gene expression in cardiomyocytes. Cardiovasc Res 1999;41:654–62. [DOI] [PubMed] [Google Scholar]
- 14.Cheng TH, Cheng PY, Shih NL, et al. Involvement of reactive oxygen species in angiotensin II-induced endothelin-1 gene expression in rat cardiac fibroblasts. J Am Coll Cardiol 2003;42:1845–54. [DOI] [PubMed] [Google Scholar]
- 15.Cheng JJ, Chao YJ, Wang DL. Cyclic strain activates redox-sensitive proline-rich tyrosine kinase 2 (PYK2) in endothelial cells. J Biol Chem 2002;277:48152–7. [DOI] [PubMed] [Google Scholar]
- 16.Cheng TH, Shih NL, Chen SY, et al. Reactive oxygen species mediate cyclic strain-induced endothelin-1 gene expression via Ras/Raf/extracellular signal-regulated kinase pathway in endothelial cells. J Mol Cell Cardiol 2001;33:1805–14. [DOI] [PubMed] [Google Scholar]
- 17.Wung BS, Cheng JJ, Hsieh HJ, et al. Cyclic strain-induced monocyte chemotactic protein-1 gene expression in endothelial cells involves reactive oxygen species activation of activator protein 1. Circ Res 1997;81:1–7. [DOI] [PubMed] [Google Scholar]
- 18.Peng J, Gurantz D, Tran V, et al. Tumor necrosis factor-alpha-induced AT1 receptor upregulation enhances angiotensin II-mediated cardiac fibroblast responses that favor fibrosis. Circ Res 2002;91:1119–26. [DOI] [PubMed] [Google Scholar]
- 19.Gardin JM, Wagenknecht LE, Anton-Culver H, et al. Relationship of cardiovascular risk factors to echocardiographic left ventricular mass in healthy young black and white adult men and women. The CARDIA study. Coronary artery risk development in young adults. Circulation 1995;92:380–7. [DOI] [PubMed] [Google Scholar]
- 20.Levy D, Garrison RJ, Savage DD, et al. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham heart study. N Engl J Med 1990;322:1561–6. [DOI] [PubMed] [Google Scholar]
- 21.Hayward CS, Webb CM, Collins P. Effect of sex hormones on cardiac mass. Lancet 2001;357:1354–6. [DOI] [PubMed] [Google Scholar]
- 22.Dubey RK, Gillespie DG, Jackson EK, et al. 17Beta-estradiol, its metabolites, and progesterone inhibit cardiac fibroblast growth. Hypertension 1998;31:522–8. [DOI] [PubMed] [Google Scholar]
- 23.Nuedling S, Kahlert S, Loebbert K, et al. 17 Beta-estradiol stimulates expression of endothelial and inducible NO synthase in rat myocardium in-vitro and in-vivo. Cardiovasc Res 1999;43:666–74. [DOI] [PubMed] [Google Scholar]
- 24.Van Eickels M, Grohe C, Cleutjens JP, et al. 17beta-estradiol attenuates the development of pressure-overload hypertrophy. Circulation 2001;104:1419–23. [DOI] [PubMed] [Google Scholar]
- 25.Xin HB, Senbonmatsu T, Cheng DS, et al. Oestrogen protects FKBP12.6 null mice from cardiac hypertrophy. Nature 2002;416:334–8. [DOI] [PubMed] [Google Scholar]
- 26.Lee HW, Eghbali-Webb M. Estrogen enhances proliferative capacity of cardiac fibroblasts by estrogen receptor- and mitogen-activated protein kinase-dependent pathways. J Mol Cell Cardiol 1998;30:1359–68. [DOI] [PubMed] [Google Scholar]
- 27.Grohe C, Kahlert S, Lobbert K, et al. Effects of moexiprilat on oestrogen-stimulated cardiac fibroblast growth. Br J Pharmacol 1997;121:1350–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mercier I, Colombo F, Mader S, et al. Ovarian hormones induce TGF-beta(3) and fibronectin mRNAs but exhibit a disparate action on cardiac fibroblast proliferation. Cardiovasc Res 2002;53:728–39. [DOI] [PubMed] [Google Scholar]
- 29.Nickenig G, Strehlow K, Wassmann S, et al. Differential effects of estrogen and progesterone on AT(1) receptor gene expression in vascular smooth muscle cells. Circulation 2000;102:1828–33. [DOI] [PubMed] [Google Scholar]
- 30.Shih NL, Cheng TH, Loh SH, et al. Reactive oxygen species modulate angiotensin II-induced beta-myosin heavy chain gene expression via Ras/Raf/extracellular signal-regulated kinase pathway in neonatal rat cardiomyocytes. Biochem Biophys Res Commun 2001;283:143–8. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka K, Honda M, Takabatake T. Redox regulation of MAPK pathways and cardiac hypertrophy in adult rat cardiac myocyte. J Am Coll Cardiol 2001;37:676–85. [DOI] [PubMed] [Google Scholar]
- 32.Krishnamurthi K, Verbalis JG, Zheng W, et al. Estrogen regulates angiotensin AT1 receptor expression via cytosolic proteins that bind to the 5′ leader sequence of the receptor mRNA. Endocrinology 1999;140:5435–8. [DOI] [PubMed] [Google Scholar]