

Abstract
Objective: To investigate cytokine gene expression in patients with aortic valve stenosis (AS) and with idiopathic dilated cardiomyopathy (DCM), and to correlate wall stress with myocardial proinflammatory cytokine gene expression.
Methods: Human left ventricular (LV) myocardial biopsies were obtained for subsequent reverse transcription polymerase chain reaction of tumour necrosis factor α (TNFα), interleukin (IL) -1β, and IL-6 mRNA. The study population consisted of 24 patients with AS and 10 patients with idiopathic DCM.
Results: Patients with AS had a larger ejection fraction (56 (5) v 37 (4)%, p < 0.01), smaller LV end diastolic volumes (146 (11) v 267 (21) ml, p < 0.01), and lower end systolic wall stress (44 (7) v 112 (11) kdyn/cm2, p < 0.001). Upregulation of TNFα, IL-1β, and IL-6 gene expression was detected in both groups. However, TNFα gene expression was significantly higher in AS than in DCM (p = 0.009). No correlation was found between haemodynamic parameters and TNFα gene expression. In patients with AS there was a strong inverse relation between circulating TNFα and TNFα gene expression (r = −0.685, p = 0.014), between circulating TNFα and IL-1β gene expression (r = −0.664, p = 0.018), and between soluble TNF receptor 2 and TNFα gene expression (r = −0.685, p = 0.020). Myocardial gene expression of TNFα was significantly higher in patients with well compensated AS than in patients with decompensated AS (p = 0.017). Similarly, patients with decompensated DCM were characterised by significantly lower TNFα gene expression than were patients with well compensated DCM (p = 0.011).
Conclusion: TNFα gene expression is significantly higher in patients with pressure overload than in normal hearts, in patients with idiopathic DCM, and in patients with compensated versus decompensated heart failure. Secondly, in patients with AS proinflammatory cytokine gene expression did not affect systolic performance. The higher TNFα gene expression in patients with compensated heart failure suggests that cytokine gene expression has an adaptive role in the early phase of LV remodelling.
Keywords: cardiomyopathy, cytokines, gene expression, heart failure
There is substantial evidence that myocardial cytokines such as tumour necrosis factor α (TNFα), interleukin (IL) -6,1 and IL-1β2 are generated after sustained haemodynamic overloading of the heart, whether it results from pressure3 or volume overload. Accordingly TNFα gene expression is upregulated in the ventricles of humans with idiopathic dilated and ischaemic cardiomyopathy.4 In these patients circulating TNFα protein concentrations correlate with the severity of the disease.5 Cytokines negatively affect cardiac contractility by modulating intracellular calcium fluxes during systole6 by inducing nitric oxide synthase, which can generate peroxynitrite in the presence of excess free radicals7 and by mediating apoptosis.8,9
The origin of these cytokines remains a matter of debate. Observational work has provided proof for endotoxin induced immune activation10,11 as well as for myocardial cytokine production.4,12 Under experimental conditions such as volume overload or passive muscle stretch, the myocardium generates biologically active TNFα13 and it has recently been shown that transaortic constriction results in myocardial upregulation of IL-1β, IL-6, and TNFα gene expression and their respective receptors.14 In addition unloading of the terminally failing heart by venting13 or by prolonged mechanical circulatory support15 results in a downregulation of myocardial TNFα gene expression.16 Taken together these data suggest that haemodynamic factors such as wall stress have a major role in the enhanced myocardial expression of relevant myocardial mediators such as cytokines. Nevertheless, recent clinical work showed that in transplant recipients with normal systolic function, without any histological evidence of rejection and without any arguments for pressure or volume overload, cytokine gene expression may be upregulated 10-fold to 100-fold higher than in patients with dilated cardiomyopathy (DCM).17 Furthermore, Clark and colleagues18 reported that the fall in serum TNFα and IL-6 concentrations observed after ventricular unloading was only temporary and that cytokine concentrations returned to preoperative concentrations at day 90. These observations suggest that factors other than haemodynamic load may interfere with cytokine gene expression.
Because of these conflicting data, the present study was set up to investigate the stimulus for expression of TNFα, IL-1β, and IL-6 in a human setting and to correlate myocardial gene expression with haemodynamic parameters of left ventricular (LV) contractile performance and circulating cytokine concentrations. Therefore, LV endomyocardial biopsies were procured at the time of diagnostic cardiac catheterisation or at surgery from patients with pressure overload cardiomyopathy caused by symptomatic aortic valve stenosis (AS) and from patients with idiopathic DCM.
METHODS
Study population
Thirty four patients in stable condition (20 men, mean (SD) age 66 (11) years) with moderate to severe heart failure19 caused by idiopathic DCM (n = 10) and symptomatic AS (n = 24) referred for elective right left heart catheterisation were selected for the study. All patients provided informed consent and the protocol was approved by the local ethics committee. Idiopathic DCM was diagnosed on the basis of a normal coronary angiogram and evidence of abnormal LV function by means of contrast ventriculography (LV ejection fraction (LVEF) < 45% or indexed LV end diastolic volume > 102 ml/m2).20 Histological evaluation excluded an inflammatory cause of the DCM. The patients with AS were characterised by an LVEF > 50% and normal epicardial coronary arteries. AS was caused by degenerative aortic valve disease. Twelve patients with AS presented with effort induced syncope, six with exertional dyspnoea, and six with a history of angina. Patients with ischaemic heart disease or atrial fibrillation were excluded.
Cardiac catheterisation
The left and right sides of the heart were catheterised from the right femoral artery and vein. Right atrial pressure, pulmonary artery pressure, pulmonary capillary wedge pressure, and cardiac output were measured by use of a Swan-Ganz catheter. LV pressure was recorded with a catheter positioned in the LV cavity. LV angiograms were obtained in the right and left anterior oblique positions. Mean gradient across the aortic valve was measured and aortic valve area was calculated with the Gorlin formula.21
LV endomyocardial biopsies were procured during aortic valve replacement from patients with AS, whereas in patients with idiopathic DCM LV endomyocardial biopsies were obtained at the end of the diagnostic left to right heart catheterisation. All biopsies were procured at the level of the distal interventricular septum. They were immediately frozen in liquid nitrogen and stored at −80°C for subsequent detection of cytokine mRNA by reverse transcription (RT) polymerase chain reaction (PCR). In patients with DCM a histological diagnosis of myocarditis was excluded by routine endomyocardial biopsy. In patients with AS biopsies were procured immediately after the start of the pump run following excision of the stenotic aortic valve to avoid extracorporeal circulation mediated cytokine induction. As a control endomyocardial biopsies were obtained from four donor hearts at the time of transplantation. The mean (SD) age of the donors was 39 (3) years and there were three men and one woman. None of the donor hearts had ischaemic heart disease and the mean LVEF was 65 (12)%.
Echocardiographic analysis
LV meridional wall stress was estimated from M mode data in combination with pressure data, by the formula: wall stress = 0.334 × LV pressure × LVID/[PWT × (1 + PWT/LVID)], where PWT is posterior wall thickness and LVID the LV internal dimension.22 This was technically possible in 30 patients (10 DCM, 20 patients with AS) whereas the echocardiographic image quality was insufficient in four patients. Patients with AS were considered to have adequate compensation when end systolic wall stress was < 90 kdyn/cm2. When end systolic wall stress was ⩾ 90 kdyn/cm2 they were considered to have decompensated heart failure.23 Similarly patients with DCM with LV end diastolic pressure > 16 mm Hg were considered to have decompensated heart failure and reduced preload reserve.20
Cytokine protein plasma concentrations
In the patients with AS, plasma concentrations of TNFα, IL-6, soluble TNF receptor (sTNFR) 1, and sTNFR2 were sampled at the time of LV endomyocardial biopsy. Plasma concentrations were determined by a high sensitivity, commercially available enzyme linked immunosorbent assay (ELISA) kit (R&D Systems Inc, Minneapolis, Minnesota, USA). These assays have a high sensitivity with a lower limit of detection of 4.4 pg/ml for TNFα, 1.5 pg/ml for IL-6, 0.43 pg/ml for sTNFR1, and < 1 pg/ml for sTNFR2 with interassay and intra-assay variabilities lower than 7% according to the manufacturer. Plasma samples obtained from 10 age matched volunteers (mean age 55 (9) years) without evidence of systemic or cardiovascular disease were assayed to determine the reference plasma cytokine concentration.
Myocardial cytokine gene expression
Because of the small size of the human heart tissue samples, a highly sensitive RT-PCR method was developed for cytokine mRNA quantification. As described previously in detail, this method allows quantification of tissue samples as low as 2–3 mg.24 The tissue samples were fresh frozen in liquid nitrogen and stored at −80°C. The frozen samples were thawed in 1 ml of Trizol solution and homogenised in a Polytron 3000 homogeniser at 30 000 rpm for 30 seconds. The total RNA was extracted and stored at −80°C according to the manufacturer’s protocol. Poly(A) RNA from the total RNA was captured with biotin labelled oligo-(dT) 20 according to the manufacturer’s protocol and immobilised by transferring the captured RNA to streptavidin coated microfuge tubes (mRNA Capture Kit, Roche Molecular Biochemicals, USA). After immobilisation the tubes were washed three times and stored at −80°C. First strand cDNA was synthesised directly on the immobilised poly(A) RNA according to the manufacturer’s protocol (1st Strand cDNA Synthesis Kit for RT-PCR, Roche Molecular Biochemicals).
PCR primer design and selection
PCR primers were designed so that there was no complementarity between the sense and antisense strands, thus avoiding the formation of primer dimers. Additionally, PCR sense and antisense primers were designed in such a way that they were positioned either on the intron-exon junction or on the exons of the respective target genes. With these criteria, the PCR primers for various oligonucleotides were designed with the DataMinder 3.2 computer program. To generate copy number cDNA standards, cDNA clones encoding the target genes for various cytokines and β actin were obtained (table 1).
Table 1.
Oligonucleotides
| Length | GC (%) | Tm (°C) | |
| TNFα | |||
| Primers | |||
| 5′-AAGAGTCCCCCAGGGACCTCM-3′ | 21 | 61 | 53 |
| 5′-CCTGGGAGTAGATGAGGTACA-3′ | 21 | 52 | 49 |
| Product | 230 | 88.2 | |
| IL-6 | |||
| Primers | |||
| 5′-CAGCTATGAACTCCTTCTCCACAAGC-3′ | 26 | 50 | 64.8 |
| 5′-CTGAGATGCCGTCGAGGATGTACCG-3′ | 25 | 60 | 67.9 |
| Product | 201 | 85.5 | |
| β Actin | |||
| Primers | |||
| 5′-AGCACGGCATCGTCACCAACT-3′ | 21 | 57.1 | 62.7 |
| 5′-TGGCTGGGGTGTTGAAGGTCT-3′ | 21 | 57.1 | 62.7 |
| Product | 180 | 86.0 |
Cytokine quantification
The numbers of mRNA copies for TNFα, IL-6, and IL-1β were determined by use of real time quantitative RT-PCR LightCycler technology. Because of the low quantity of the biopsy samples, β actin mRNA copy numbers were used to normalise the cytokine mRNA samples. The data are presented as copies of cytokine transcripts per 106 copies of β actin.
Statistical analysis
Parametric data are expressed as mean (SD). Student’s t test, a Mann-Whitney test, and a Spearman correlation coefficient were used for appropriate comparisons. A stepwise multiple regression analysis was used for multivariate re-evaluation of univariate correlations. Variables were the following: wall stress, aortic valve area, mean aortic gradient, LV end diastolic pressure, and LVEF. Significance was set at a two tailed probability level of p < 0.05.
RESULTS
Baseline characteristics
Table 2 reports clinical characteristics and table 3 reports echocardiographic and haemodynamic data.
Table 2.
Clinical characteristics
| AS (n = 24) | DCM (n = 10) | p Value | |
| Sex (men/women) | 14/10 | 6/4 | NS |
| Age (years) | 70 (8) | 56 (12) | 0.001 |
| NYHA class | 2.2 (0.9) | 2.6 (1.1) | 0.030 |
| BSA (m2) | 1.81 (0.05) | 1.94 (0.04) | NS |
| Serum creatinine (μmol/l) | 88 (18) | 106 (35) | NS |
| Serum blood urea (mmol/l) | 8.2 (1.8) | 14 (2.1) | NS |
| Serum sodium (mmol/l) | 142 (3) | 143 (2) | NS |
| White cell count (×109/l) | 8 (2) | 8 (3) | NS |
| CRP (mg/l) | 0.5 (0.1) | 0.4 (0.2) | NS |
| Medications | |||
| ACE-I or ARB | 14% | 27% | NS |
| Diuretics | 25% | 65% | 0.002 |
| β Blockers | 36% | 24% | NS |
| Digoxin | 0% | 0% | NS |
Data are mean (SD), number, or percentage.
ACE-I, angiotensin converting enzyme inhibitor; ARB, angiotensin I receptor blocker; AS, aortic valve stenosis; BSA, body surface area; CRP, C reactive protein; DCM, dilated cardiomyopathy; NS, not significant; NYHA, New York Heart Association.
Table 3.
Haemodynamic and echocardiographic characteristics
| AS (n = 24) | DCM (n = 10) | p Value | |
| LVEDV (ml) | 146 (11) | 267 (21) | <0.001 |
| LVESV (ml) | 84 (14) | 162 (18) | 0.005 |
| LVSP (mm Hg) | 196 (24) | 133 (23) | <0.001 |
| RAP (mm Hg) | 7 (1) | 8 (1) | NS |
| PAP mean (mm Hg) | 28 (3) | 26 (4) | 0.070 |
| PCWP (mm Hg) | 19 (2) | 17 (4) | NS |
| Cardiac output (l) | 4.94 (0.29) | 5.19 (0.64) | NS |
| LVEF (%) | 56 (5) | 37 (4) | 0.013 |
| ESWS (kdyn/cm2) | 44 (7) | 112 (11) | <0.001 |
| EDWS (kdyn/cm2) | 29 (17) | 43 (9) | 0.056 |
| LVEDD (mm) | 47 (6) | 68 (6) | 0.004 |
| IVSd (mm) | 13 (2) | 12 (3) | NS |
| AV area (cm2) | 0.64 (0.06) | NA | NA |
| AV gradient (mm Hg) | 51 (6) | NA | NA |
Data are mean (SD).
AV, aortic valve; EDWS, end diastolic wall stress; ESWS, end systolic wall stress; IVSd, interventricular septal thickness at end diastole; LVEDD, left ventricular end diastolic diameter; LVEDV, left ventricular end diastolic volume; LVEF, left ventricular ejection fraction; LVESV, left ventricular end systolic volume; LVSP, left ventricular systolic pressure; NA, not applicable; PAP, pulmonary artery pressure; PCWP, pulmonary capillary wedge pressure; RAP, right atrial pressure.
Patients with AS were older (p = 0.001) and were characterised by higher LVEF (p = 0.013) and smaller LV end diastolic (p < 0.001) and end systolic volumes (p = 0.005) than patients with DCM, despite similar pulmonary capillary wedge pressure (not significant) and right atrial pressure (not significant). End systolic wall stress (p < 0.001) was lower in patients with AS than in patients with DCM. There was a trend towards lower end diastolic wall stress in patients with AS, although it never became significant.
Myocardial cytokine gene expression in AS, DCM, and normal hearts
As fig 1 shows, myocardial cytokine gene expression was barely detectable in healthy controls. In patients with DCM (p < 0.001) and in patients with AS (p < 0.001) myocardial expression of TNFα mRNA copies corrected for β actin mRNA copies was increased compared with healthy donor hearts. TNFα gene expression was significantly higher in patients with aortic valve disease than in patients with idiopathic DCM (p = 0.009). There were no differences between IL-1β and IL-6 gene expression in both groups (fig 1). Myocardial TNFα gene expression correlated significantly with IL-6 gene expression (r = 0.619, p < 0.001) as well as with IL-1β gene expression (r = 0.407, p = 0.017). Lastly, IL-1β correlated with IL-6 gene expression (r = 0.487, p = 0.016).
Figure 1.
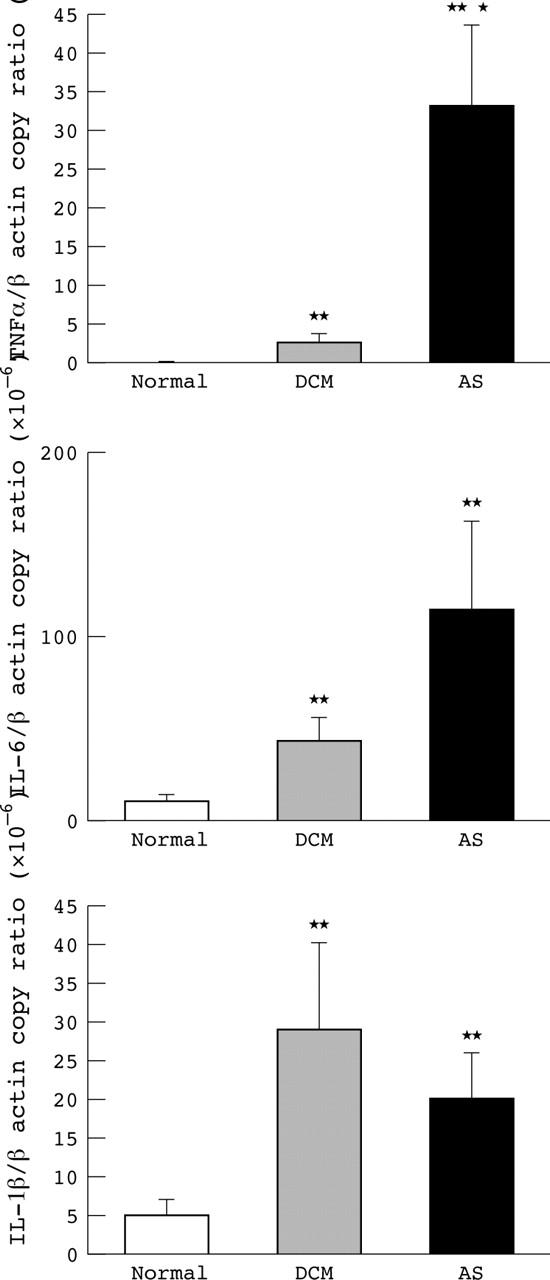
Level of gene expression of tumour necrosis factor α (TNFα), interleukin (IL) -6, and IL-1β in normal hearts, patients with dilated cardiomyopathy (DCM), and patients with aortic valve stenosis (AS). Numbers of copies of TNFα, IL-6, and IL-1β are indexed to a million copies of β actin. *p < 0.05 v AS; **p < 0.001 v normal.
Haemodynamic parameters and cytokine gene expression
No correlation was noted between parameters of LV contractile function such as LVEF, pulmonary capillary wedge pressure, mean pulmonary artery pressure, right atrial pressure, cardiac output, end diastolic wall stress, end systolic wall stress, aortic valve area, and myocardial cytokine gene expression of TNFα, IL-1β, and IL-6. Multivariate stepwise analysis found that no variable was independently related to TNFα formation in either the AS or in the DCM group. However, myocardial gene expression of TNFα was significantly higher in patients with well compensated AS than in patients with decompensated AS (p = 0.017). Similarly, patients with decompensated DCM were characterised by significantly lower TNFα gene expression than were patients with well compensated DCM (p = 0.011) (fig 2).
Figure 2.
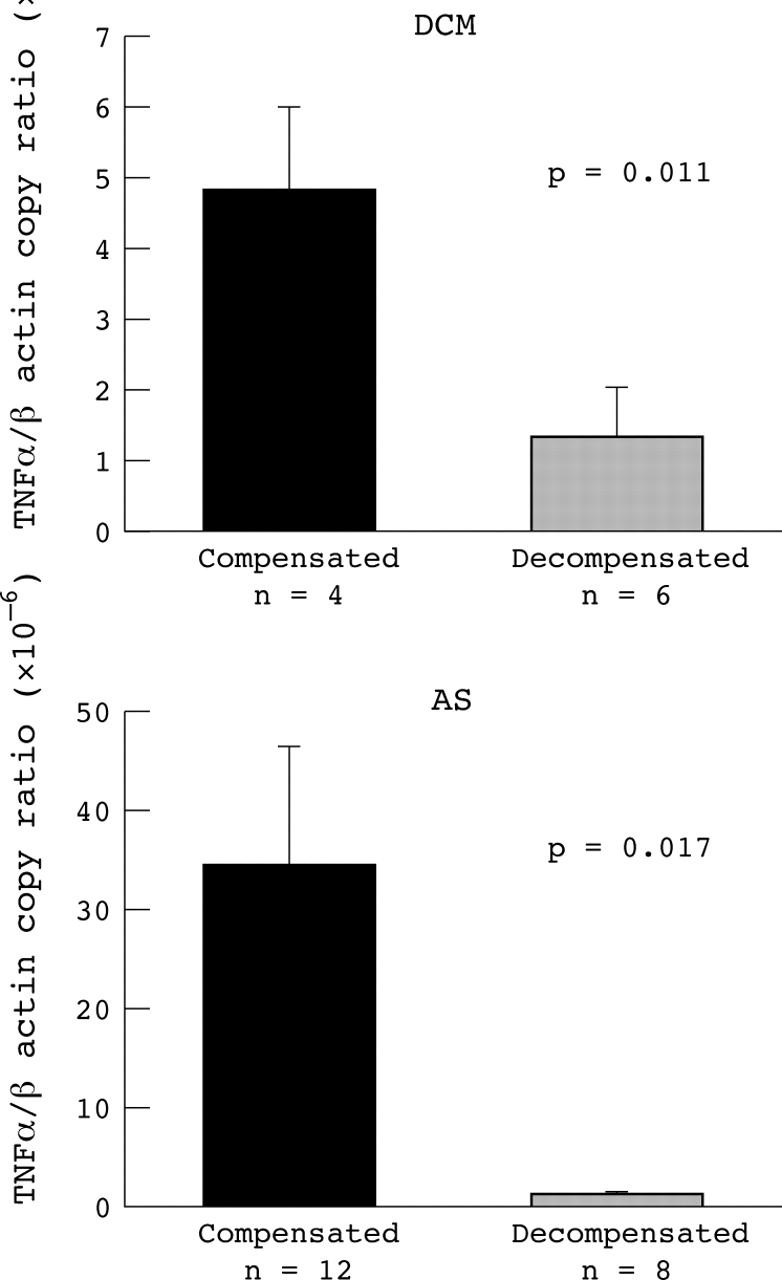
Level of gene expression of TNFα in patients with compensated and decompensated DCM (upper panel) and AS (lower panel). Number of copies of TNFα is indexed to a million copies of β actin.
Cytokine gene expression and circulating cytokine concentrations in patients with AS
Circulating TNFα protein concentrations ranged from 2.64–23.34 pg/ml (mean (SD) 5.64 (1.00) pg/ml), sTNFR1 ranged from 744–2414 pg/m l (mean (SD) 1147 (96) pg/ml), sTNFR2 ranged from 822–3965 pg/ml (mean (SD) 2132 (196) pg/ml), and IL-6 ranged from 0.58–34.20 pg/ml (mean (SD) 4.07 (1.66) pg/ml). As table 4 shows, TNFα, IL-6, sTNFR1, and sTNFR2 were closely linked to each other. A strong inverse relation was noted between TNFα protein and TNFα (r = −0.685, p = 0.014) and IL-1β gene expression (r = −0.664, p = 0.018) and between sTNFR2 and TNFα gene expression (r = −0.685, p = 0.020).
Table 4.
Correlations between serum cytokines in patients with AS
| TNFα | sTNFR1 | sTNFR2 | IL-6 | |||||
| r | p Value | r | p Value | r | p Value | r | p Value | |
| TNFα | NA | NA | 0.710 | <0.001 | 0.645 | 0.002 | 0.479 | 0.033 |
| sTNFR1 | 0.710 | <0.001 | NA | NA | 0.872 | <0.001 | 0.783 | <0.001 |
| sTNFR2 | 0.645 | 0.002 | 0.872 | <0.001 | NA | NA | 0.612 | 0.004 |
| IL-6 | 0.479 | 0.033 | 0.783 | <0.001 | 0.612 | 0.004 | NA | NA |
sTNFR, soluble tumour necrosis factor receptor.
DISCUSSION
The main findings in this study are the following. Firstly, myocardial cytokine gene expression is upregulated in the myocardium of both patients with AS and patients with DCM compared with normal hearts, with significantly higher upregulation of TNFα in AS than in DCM. Secondly, patients with decompensated AS or DCM are characterised by a lower TNFα gene expression than are patients with well compensated heart failure. Thirdly, in patients with AS an inverse correlation was noted between circulating TNFα concentrations and TNFα gene expression. These data suggest that, both in AS and in DCM, cytokine gene expression has an adaptive role in LV remodelling.
Myocardial cytokine gene expression in AS and DCM
Previous experimental and human studies have shown that cardiomyocytes themselves are an important producer of relevant cytokines.12,25–27 Of note, TNFα is present in immunodetectable concentrations in the cytosolic fraction of human explanted failing myocytes and the TNFα gene is upregulated in patients with chronic mitral regurgitation, whereas neither TNFα protein nor TNFα gene expression is elaborated in the heart under non-failing condition.4 Similarly we noticed a significant increase in TNFα, IL-1β, and IL-6 expression in AS and DCM as compared with healthy controls, suggesting that pressure overload and DCM and the resultant stretch stimulus result in TNFα gene expression.28 An interesting, albeit unexpected, finding was that TNFα gene expression was two- to threefold higher in patients with AS than in patients with DCM. The exact reason why pressure overload results in a greater stimulus for cytokine production in DCM is unknown. Nevertheless our data are in accordance with those of Kapadia and colleagues,28 who reported higher TNFα and TNFα receptor plasma concentrations in patients with pressure overload than in patients with volume overload caused by mitral regurgitation.
Myocardial cytokine gene expression and LV haemodynamic function
In contrast to previous studies that reported a relation between filling pressures15 and TNFα gene expression, as well as between wall stress and myocardial cytokine gene expression,13 no such relation was detectable in the present study. On the contrary, in patients with AS, we noticed high TNFα mRNA concentrations despite normal LV systolic function. The reasons for this discrepancy may be related to differences in aetiology and the temporal stage of the heart failure phenotype studied. Indeed, Torre-Amione and colleagues17 reported similar findings after orthotopic heart transplantation. In routine surveillance endomyocardial biopsies of transplant recipients with normal LV systolic function and with no histological evidence of rejection, they noticed upregulated TNFα gene expression.17 They speculated that in accordance with transgenic mice overexpressing TNFα the initial contractile function remains normal despite the high cytokine concentrations, but that cellular abnormalities are being created that will produce a cardiomyopathic state over time. Thus, it appears that in these early stages the expression of low concentrations of TNFα for relatively brief periods of time may provide the heart with a short term adaptive response to environmental stress, both by increasing sarcomeric protein synthesis and by expressing heat shock proteins in the cells.29 These observations may explain why, at least in patients with AS, LV systolic function can adapt despite high myocardial cytokine concentrations.
Myocardial cytokine gene expression in decompensated heart failure
Although no correlation was noted between TNFα gene expression and LV haemodynamic function in the whole study population, patients with decompensated heart failure in the AS and DCM groups had lower TNFα gene expression than did patients with well compensated heart failure. It is important to recognise that the design of this study does not establish a clear cut cause and effect relation between altered TNFα gene expression and the development of cardiac failure. Indeed, it is likely that a variety of mechanisms contribute to the observed change in myocyte phenotype. This statement notwithstanding, it bears emphasising that our data corroborate previous studies that showed that altered myocardial expression parallels LV deterioration. Firstly, after transaortic constriction14 proinflammatory cytokine production rises transiently and declines over time when heart failure develops. Secondly, similar time dependent changes in expression of other genes occur during the transition of compensated to decompensated heart failure. Myocardial inducible nitric oxide synthase is higher in patients with New York Heart Association class II heart failure than in patients with more advanced heart failure30 and growth factors such as insulin-like growth factor and endothelin are known to be upregulated in the early phase of hypertrophy but are no longer detectable when wall stress increases.23 Although it is likely that a variety of mechanisms contribute to the observed change in myocyte phenotype, all these data indirectly raise the intriguing possibility that load, and wall stress in particular, is an important stimulus for cytokine gene expression with high gene expression if wall stress is normal but a blunted response whenever wall stress increases.
Circulating cytokines and gene expression
The relation between circulating cytokine concentrations and myocardial cytokine production remains controversial. Documented cardiac spillover as an explanation for high TNFα concentrations has been called upon by some investigators and refuted by others. Of note neither Munger et al31 nor Petretta et al32 found any significant difference between plasma cytokine concentrations of arterial and coronary sinus blood. On the other hand, Tsutamoto et al33 reported a significant transmyocardial TNFα gradient in patients with DCM.33 In patients with AS we observed an inverse correlation between circulating TNFα and sTNFR2 concentrations and myocardial gene expression. Although Kapadia et al28 never reported a similar correlation, Torre-Amione et al4 reported significantly lower TNFα concentrations in patients with high myocardial TNFα concentrations. In addition transgenic mice that overexpress TNFα have undetectable peripheral TNFα concentrations.34,35 Although the exact mechanism for this inverse relation is unknown, we speculate that the circulating TNFα concentrations may act as a negative feedback system that modulates myocardial gene expression. Higher circulating TNFα results in lower gene expression and vice versa.
Limitations
Firstly, because of the small sample size of endomyocardial biopsies, we could establish the presence of cytokines in the myocardium only by observing cytokine mRNA by reverse transcription PCR and not by concomitant direct evidence in the myocardium of the cytokine protein itself. This would have provided definite proof of the myocardial presence of the cytokine because of post-transcriptional modification of cytokine protein translation. However, in a previous study concordance between myocardial TNFα gene expression and protein expression was reported. In the failing human heart, TNFα immunostaining was detected in all biopsies in which TNFα mRNA expression was inferred by reverse transcription PCR.17 Secondly, it is well known that cytokine gene expression is altered during extracorporeal circulation in a time dependent matter. As it is impossible to obtain percutaneous transvalvar endomyocardial biopsies from patients with AS, we tried to avoid this bias by taking the biopsy immediately after starting the pump run through the excised aortic valve. By this approach the impact of extracorporeal circulation upon gene expression could be limited. Thirdly, mechanical forces may vary at different layers of the ventricular wall and may affect gene expression. To overcome this problem we did not take biopsies from the right ventricle but in both patients with AS and patients with DCM we took endomyocardial biopsies at the level of the distal left interventricular septum, which is subject to the sustained load. In addition in patients with DCM, mRNA expression of TNFα did not differ significantly between right ventricular and LV endomyocardial biopsies.36
Clinical implications and conclusion
Haemodynamic load (AS) modulates proinflammatory cytokine gene expression in AS and DCM. Normal LV systolic function, together with the high myocardial cytokine gene expression, in patients with well compensated heart failure suggests that cytokine gene expression may serve as a short term adaptive response and initiate cardiac remodelling. Therefore, wall stress induced TNFα production may be a novel autocrine or paracrine mechanism for regulating cardiac structure; treatments that selectively interfere with the cytokine pathway may be used to modulate myocyte structure in the failing heart. Further studies are needed to address this concept.
Abbreviations
AS, aortic valve stenosis
DCM, dilated cardiomyopathy
ELISA, enzyme linked immunosorbent assay
IL, interleukin
LV, left ventricular
LVID, left ventricular internal dimension
PCR, polymerase chain reaction
PWT, posterior wall thickness
RT, reverse transcription
sTNFR, soluble tumour necrosis factor receptor
TNFα, tumour necrosis factor α
REFERENCES
- 1.Wang F, Seta Y, Baumgarten G, et al. Functional significance of hemodynamic overload-induced expression of leukemia-inhibitory factor in the adult mammalian heart. Circulation 2001;103:1296–302. [DOI] [PubMed] [Google Scholar]
- 2.Shioi T, Matsumori A, Furukawa Y, et al. Increased expression of interleukin-1 beta and monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 in the hypertrophied and failing heart with pressure overload. Circ Res 1997;81:664–71. [DOI] [PubMed] [Google Scholar]
- 3.Nagueh SF, Stetson SJ, Lakkis NM, et al. Decreased expression of tumor necrosis factor-alpha and regression of hypertrophy after nonsurgical septal reduction therapy for patients with hypertrophic obstructive cardiomyopathy. Circulation 2001;103:1844–50. [DOI] [PubMed] [Google Scholar]
- 4.Torre-Amione G, Kapadia S, Lee J, et al. Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human heart. Circulation 1996;93:704–11. [DOI] [PubMed] [Google Scholar]
- 5.Levine BKalman J, Mayer L, et al. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med 1990;323:236–41. [DOI] [PubMed] [Google Scholar]
- 6.Yokoyama T, Vaca L, Rossen RD, et al. Cellular basis for the negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian heart. J Clin Invest 1993;92:2303–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finkel MS, Oddis CV, Jacob TD, et al. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science 1992;257:387–9. [DOI] [PubMed] [Google Scholar]
- 8.Birks EJ, Latif N, Owen V, et al. Quantitative myocardial cytokine expression and activation of the apoptotic pathway in patients who require left ventricular assist devices. Circulation 2001;104 (12 suppl 1) :I233–40. [DOI] [PubMed] [Google Scholar]
- 9.Arstall MA, Sawyer DB, Fukazawa R, et al. Cytokine-mediated apoptosis in cardiac myocytes: the role of inducible nitric oxide synthase induction and peroxynitrite generation. Circ Res 1999;85:829–40. [DOI] [PubMed] [Google Scholar]
- 10.Niebauer J, Volk HD, Kemp M, et al. Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet 1999;353:1838–42. [DOI] [PubMed] [Google Scholar]
- 11.Anker SD, Egerer KR, Volk HD, et al. Elevated soluble CD14 receptors and altered cytokines in chronic heart failure. Am J Cardiol 1997;79:1426–30. [DOI] [PubMed] [Google Scholar]
- 12.Plenz G, Song ZF, Tjan TD, et al. Activation of the cardiac interleukin-6 system in advanced heart failure. Eur J Heart Fail 2001;3:415–21. [DOI] [PubMed] [Google Scholar]
- 13.Kapadia SR, Oral H, Lee J, et al. Hemodynamic regulation of tumor necrosis factor-alpha gene and protein expression in adult feline myocardium. Circ Res 1997;81:187–95. [DOI] [PubMed] [Google Scholar]
- 14.Baumgarten G, Knuefermann P, Lalra D, et al. Load-dependent and -independent regulation of proinflammatory cytokine and cytokine receptor gene expression in the adult mammalian heart. Circulation 2002;105:2192–7. [DOI] [PubMed] [Google Scholar]
- 15.Torre-Amione G, Stetson SJ, Youker KA, et al. Decreased expression of tumor necrosis factor-alpha in failing human myocardium after mechanical circulatory support: a potential mechanism for cardiac recovery. Circulation 1999;100:1189–93. [DOI] [PubMed] [Google Scholar]
- 16.Mann DL. Mechanisms and models in heart failure: a combinatorial approach. Circulation 1999;100:999–1008. [DOI] [PubMed] [Google Scholar]
- 17.Torre-Amione G, MacLellan W, Kapadia S, et al. Tumor necrosis factor-alpha is persistently expressed in cardiac allografts in the absence of histological or clinical evidence of rejection. Transplant Proc 1998;30:875–7. [DOI] [PubMed] [Google Scholar]
- 18.Clark AL, Loebe M, Potapov EV, et al. Ventricular assist device in severe heart failure: effects on cytokines, complement and body weight. Eur Heart J 2001;22:2275–83. [DOI] [PubMed] [Google Scholar]
- 19.Remme WJ, Swedberg K. Guidelines for the diagnosis and treatment of chronic heart failure. Eur Heart J 2001;22:1527–60. [DOI] [PubMed] [Google Scholar]
- 20.Heymes C, Vanderheyden M, Bronzwaer JG, et al. Endomyocardial nitric oxide synthase and left ventricular preload reserve in dilated cardiomyopathy. Circulation 1999;99:3009–16. [DOI] [PubMed] [Google Scholar]
- 21.Hatle L, Angelsen BA, Tromsdal A. Non-invasive assessment of aortic stenosis by Doppler ultrasound. Br Heart J 1980;43:284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Douglas PS, Reichek N. Estimation of wall stress and left ventricular mass by noninvasive techniques and clinical implications. Cardiovasc Clin 1986;17:103–28. [PubMed] [Google Scholar]
- 23.Serneri GGN, Modesti PA, Boddi M, et al. Cardiac growth factors in human hypertrophy: relations with myocardial contractility and wall stress. Circ Res 1999;85:57–67. [DOI] [PubMed] [Google Scholar]
- 24.Vanderheyden M, Meerbeeck Van, Van Bouwel E. et al A rare case of inflammatory pseudotumour of the bronchus, occurring in an achondroplastic woman. Eur Respir J 1994;7:826–8. [DOI] [PubMed] [Google Scholar]
- 25.Francis SE, Holden H, Holt CM, et al. Interleukin-1 in myocardium and coronary arteries of patients with dilated cardiomyopathy. J Mol Cell Cardiol 1998;30:215–23. [DOI] [PubMed] [Google Scholar]
- 26.Habib FM, Springall DR, Davies GJ, et al. Tumour necrosis factor and inducible nitric oxide synthase in dilated cardiomyopathy. Lancet 1996;347:1151–5. [DOI] [PubMed] [Google Scholar]
- 27.Birks EJ, Burton PB, Owen V, et al. Elevated tumor necrosis factor-alpha and interleukin-6 in myocardium and serum of malfunctioning donor hearts. Circulation 2000;102 (19 suppl III) :III352–8. [DOI] [PubMed] [Google Scholar]
- 28.Kapadia SR, Yakoob K, Nader S, et al. Elevated circulating levels of serum tumor necrosis factor-alpha in patients with hemodynamically significant pressure and volume overload. J Am Coll Cardiol 2000;36:208–12. [DOI] [PubMed] [Google Scholar]
- 29.Blaxall BC, Tschannen-Moran BM, Milano CA, et al. Differential gene expression and genomic patient stratification following left ventricular assist device support. J Am Coll Cardiol 2003;41:1096–106. [DOI] [PubMed] [Google Scholar]
- 30.Haywood GA, Tsao PS, von der Leyen HE, et al. Expression of inducible nitric oxide synthase in human heart failure. Circulation 1996;93:1087–94. [DOI] [PubMed] [Google Scholar]
- 31.Munger MA, Johnson B, Amber IJ, et al. Circulating concentrations of proinflammatory cytokines in mild or moderate heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol 1996;77:723–7. [DOI] [PubMed] [Google Scholar]
- 32.Petretta M, Condorelli GL, Spinelli L, et al. Circulating levels of cytokines and their site of production in patients with mild to severe chronic heart failure. Am Heart J 2000;140:E28. [DOI] [PubMed] [Google Scholar]
- 33.Tsutamoto T, Hisanaga T, Wada A, et al. Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J Am Coll Cardiol 1998;31:391–8. [DOI] [PubMed] [Google Scholar]
- 34.Sivasubramanian N, Coker ML, Kurrelmeyer KM, et al. Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation 2001;104:826–31. [DOI] [PubMed] [Google Scholar]
- 35.Bryant D, Becker L, Richardson J, et al. Cardiac failure in transgenic mice with myocardial expression of tumor necrosis factor-alpha. Circulation 1998;97:1375–81. [DOI] [PubMed] [Google Scholar]
- 36.Satoh M, Nakamura M, Saitoh H, et al. Tumor necrosis factor-alpha-converting enzyme and tumor necrosis factor-alpha in human dilated cardiomyopathy. Circulation 1999;99:3260–5. [DOI] [PubMed] [Google Scholar]