

Abstract
Objective: To test the hypothesis that activation of peroxisome proliferator activated receptor γ (PPAR-γ) reduces experimental autoimmune myocarditis (EAM) associated with inhibitor κB (IκB) α induction, blockade of nuclear factor κB (NF-κB), and inhibition of inflammatory cytokine expression.
Methods: EAM was induced in Lewis rats by immunisation with porcine cardiac myosin. PPAR-γ activators 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) and pioglitazone (PIO) were administered to rats with EAM.
Results: Enhanced PPAR-γ expression was prominently stained in the nuclear and perinuclear regions of infiltrating inflammatory cells. Administration of 15d-PGJ2 and PIO greatly reduced the severity of myocarditis and suppressed myocardial mRNA and protein expression of inflammatory cytokines in rats with EAM. In addition, treatment with PPAR-γ activators enhanced IκB concentrations in the cytoplasmic fractions and nuclear fractions from inflammatory myocardium. Concurrently, NF-κB was greatly activated in myocarditis; this activation was blocked in the 15d-PGJ2 treated and PIO treated groups.
Conclusions: PPAR-γ may have a role in the pathophysiology of EAM. Because an increase in IκB expression and inhibition of translocation of the NF-κB subunit p65 to the nucleus in inflammatory cells correlated with the protective effects of PPAR-γ activators, these results suggest that PPAR-γ activators act sequentially through PPAR-γ activation, IκB induction, blockade of NF-κB activation, and inhibition of inflammatory cytokine expression. These results suggest that PPAR-γ activators such as 15d-PGJ2 and PIO may have the potential to modulate human inflammatory heart diseases such as myocarditis.
Keywords: myocarditis, immunity, PPAR-γ, NF-κB, cytokine, inflammation
Proinflammatory cytokines have been implicated in the pathogenesis of cardiovascular diseases, including acute myocarditis, acute myocardial infarction, dilated cardiomyopathy, and congestive heart failure.1,2 Suppression of the inflammatory cytokines has beneficial effects on these clinical conditions.3,4 Despite the large body of data on the contribution of inflammatory cytokines to myocardial injury, pathways that may limit inflammatory cytokine production in myocardium remain largely unexplored in the context of inflammatory heart diseases.
Peroxisome proliferator activated receptor γ (PPAR-γ) is expressed in macrophages, T cells, endothelial cells, vascular smooth muscle cells, and cardiac myocytes.5–8 Recent data have shown that 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) and synthetic antidiabetic thiazolidinedione, which are PPAR-γ activators, suppress the T cell proliferative response in vitro7 and inhibit inflammatory cytokine production by cells of the monocyte–macrophage lineage.5,9,10 These activators inhibit gene expression in part by antagonising the activities of transcription factors such as activator protein 1 and nuclear factor κB (NF-κB).7,11 Importantly, PPAR-γ activators have been shown to ameliorate a variety of inflammatory conditions, including atherosclerosis,11 arthritis,12 inflammatory bowel disease,13 and experimental autoimmune encephalomyelitis.14,15
To explore the role of PPARγ activators during the pathogenesis of experimental autoimmune myocarditis (EAM), we examined myocardial PPAR-γ expression in rats with EAM. We also examined the effect of the PPAR-γ activators 15d-PGJ2 and pioglitazone (PIO) on expression of inflammatory cytokines, as well as on the activation of NF-κB from rats with EAM. Our results showed that administration of PPAR-γ activators reduced myocardial inflammation, inhibited the activation of NF-κB, and suppressed the myocardial proinflammatory cytokine expression in rats with EAM. These findings suggest that the anti-inflammatory properties of PPAR-γ activators may provide benefit in the treatment of myocarditis.
MATERIALS AND METHODS
Induction of EAM in rats and PPAR-γ activator treatment protocol
Acute EAM was induced by immunisation with porcine cardiac myosin in 6 week old Lewis rats as previously described.16 Rats with EAM were divided into three groups: 15d-PGJ2, PIO, and vehicles. 15d-PGJ2 (Cayman Chemical, Ann Arbor, Michigan, USA) was administered intraperitoneally at 200 μg/kg/day in 0.4 ml of sterile phosphate buffered saline (PBS).14,15 Vehicles were injected with PBS. PIO tablets were added to chow at 0.01% (wt/wt). Rats were provided with free access to diets and each rat ingested about 20 g of chow daily providing about 10 mg/kg/day of PIO.15 Normal controls were also prepared. Since myocardial inflammation usually begins around day 14 in this model,17 we conducted two sets of experiments: in experiment I, rats were treated from day 1 to day 21; in experiment II, they were treated from day 14 to day 21 after immunisation. To determine whether PPAR-γ activators also affect PPAR-γ expression in the healthy myocardium, a set of experiments was performed in normal rats: 6 week old Lewis rats were randomly divided into 15d-PGJ2 (Con-15d-PGJ2), PIO (Con-PIO), or vehicle (Con-Vehicle) groups. Drug administration was the same as for the rats in experiments I and II. All rats were killed on day 22. The protocol was approved by the Institutional Animal Research Committee of Xi’an Jiaotong University.
Histopathology
At death, macroscopic findings were graded as described previously16: 0 (normal appearance), 1 (focal discoloured area), 2 (multiple or diffuse discoloured areas and not exceeding one third of the heart), 3 (diffuse discoloured areas not exceeding two thirds of the heart), and 4 (diffuse discoloured areas exceeding two thirds of the heart). Pericardial effusion was graded as 0 (none), 1 (mild), and 2 (massive). After macroscopic examination, the hearts were transversely sliced; a part of the ventricles was fixed in 10% formalin, embedded in paraffin, and stained with haematoxylin and eosin to examine the degree of myocardial damage and infiltration of inflammatory cells. Microscopic findings were graded as 0 (normal), 1 (lesion extent not exceeding 1% of a transverse section), 2 (not exceeding 10%), 3 (not exceeding 50%), and 4 (exceeding 50%). Three sections were obtained from each heart; the mean score of the three sections was recorded as the microscopic score. We measured the lesion area with a square lattice scale in an eye lens of a microscope. Two observers scored the histopathological scores blindly. After pathological examination, a part of the ventricles was embedded in cryoembedding compound (OCT) for immunohistochemistry and remnant ventricular tissues were kept at −80°C for western blotting and RNA extraction.
Ribonuclease protection assay
The total mRNA was extracted from the myocardium with TRIzol (GIBCO-BRL). Cytokine mRNA concentrations were measured by a ribonuclease protection assay as described previously18: the cytokine mRNA concentrations were measured with RiboQuant Multi-Probe template sets (BD Biosciences), in vitro transcription kits, and ribonuclease protection assay kits (Pharmingen, BD Biosciences) according to the Pharmingen/RiboQuant protocol. The NIH Image system (National Institutes of Health, Bethesda, Maryland, USA) was used to quantify the pixel intensity of cytokine bands, which were divided by the intensities in their L32 bands in the same lanes for normalisation.
Western blotting
Myocardial PPAR-γ, interleukin (IL) 1β, tumour necrosis factor α (TNF-α), and inhibitor IκB (IκB) α protein expressions were detected by western blotting as described previously.16 Equal amounts of protein (20–40 μg protein/lane) were electrophoresed by 10–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and sequentially electrophoretically transferred to a polyvinylidene difluoride microporous membrane (Millipore, Billerica, Massachusetts, USA). The membrane was incubated with antibodies to PPAR-γ (sc-7196; Santa Cruz Biotechnology Inc, Santa Cruz, California, USA), IL-1β (Secrotec), TNF-α (Secrotec), or IκBα (sc-37D, Santa Cruz Biotechnology Inc) and then with a peroxidase linked secondary antibody (Amersham, Wikstroms, Sweden). Chemiluminescence was detected with an ECL western blotting detection kit (Amersham) and analysed semiquantitatively with an NIH Image system.
Immunohistochemistry
We used an immunoperoxidase technique for immunohistochemical analysis of PPAR-γ.16,18 A primary antibody, anti-PPAR-γ (1:400, sc-7196; Santa Cruz Biotechnology Inc), was added and incubated overnight. Biotinylated and affinity purified IgG (Dako) was used as a secondary antibody and incubated for 30 minutes. An avidin-biotin complex was sequentially added for five minutes’ incubation with the substrate 0.1% 3′,3′-diaminobenzidine, followed by haematoxylin nuclear counterstaining.
Electrophoretic mobility shift assay for NF-κB
An electrophoretic mobility shift assay was run according to a previously described method.8,19 In brief, nuclear extracts from myocardium were prepared and gel shift assays with a double stranded oligonucleotide representing the consensus sequence for NF-κB(p65) binding (5′-AGTTGAGGGGACTTTCCCAGGC-3′, 32P labelled, Promega) were run. Protein-DNA complexes were separated by 5% polyacrylamide gel electrophoresis. Autoradiography was performed and densitometry in the region of NF-κB was semiquantitatively analysed with an NIH Image system. To test the specificity of NF-κB-DNA binding, antibodies (Santa Cruz Biotechnology Inc) to the p65 subunits of NF-κB were added to the proteins, resulting in further retardation of electrophoretic mobility, or a 160-fold molar excess of unlabelled oligonucleotide (cold probe) was added to the binding reaction, leading to a decrease in NF-κB bound radioactivity.
Statistical analysis
All values were expressed as mean (SD). Student’s t test or one way analysis of variance, followed by Fisher’s protected least significant difference test, were performed. A probability value of p < 0.05 was considered significant.
RESULTS
Myocardial PPAR-γ expression in rats with EAM
To examine the expression pattern of PPAR-γ in myocardium during the course of the acute stage of EAM, we ran an immunohistochemistry assay for PPAR-γ expression. Marginal or trivial immunoreactivity for PPAR-γ was detected in myocardium of control rats (fig 1A). In rats with EAM, PPAR-γ was strongly stained in infiltrating inflammatory cells, and the expression of PPAR-γ was prominently located in the nuclear and perinuclear regions of inflammatory cells (fig 1B). Immunostaining with normal rabbit serum was completely negative in all animals (data not shown). Administration of the PPAR-γ activators 15d-PGJ2 and PIO greatly suppressed PPAR-γ expression in experiment I (fig 1C, D) and in experiment II (data not shown).
Figure 1.

Immunohistochemical staining for peroxisome proliferator activated receptor γ (PPAR-γ). (A) In normal control, marginal or trivial immunoreactivity for PPAR-γ was detected in myocardium. (B) In rats with experimental autoimmune myocarditis, PPAR-γ was strongly stained in infiltrating inflammatory cells. The expression of PPAR-γ was prominently located in the nuclear and perinuclear regions of inflammatory cells (arrows). (C, D) Administration of PPAR-γ activators 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) and pioglitazone greatly suppressed PPAR-γ expression (arrows). Original magnification ×400.
Western blotting showed that control hearts had some PPAR-γ expression. PPAR-γ expression was upregulated 3.7-fold in rats with EAM and treated with PBS compared with that in controls. Treatment with 15d-PGJ2 and PIO reduced the upregulated PPAR-γ expression in experiment I (fig 2A, B). To determine whether PPAR-γ activators also affect PPAR-γ expression in the healthy myocardium, a set of experiments was performed in normal rats. Western blotting data showed that 15d-PGJ2 and PIO treatment did not significantly change PPAR-γ expression in the healthy myocardium (fig 2C, D). These results suggested that PPAR-γ may have a role in the pathophysiology of EAM.
Figure 2.
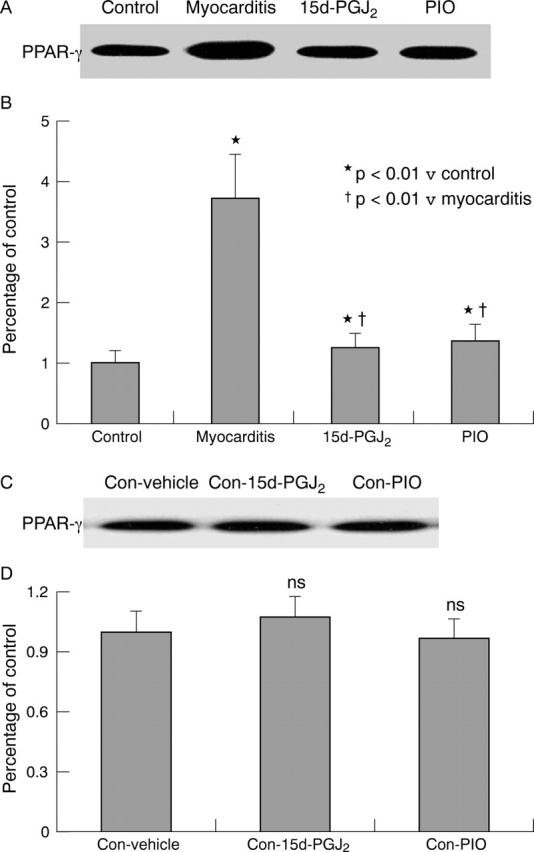
Myocardial PPAR-γ protein expression by western blotting. (A, C) Western blot analysis. (B, D) Densitometric analysis of relative protein concentrations. 15d-PGJ2, rats with myocarditis treated with 15d-PGJ2; Con-15d-PGJ2, normal rats treated with 15d-PGJ2; Control: normal rats; Con-PIO, normal rats treated with PIO; Con-Vehicle, normal rats treated with vehicle; Myocarditis: rats with myocarditis treated with phosphate buffered saline (PBS); PIO, rats with myocarditis treated with PIO. Values were derived from five animals and determined as a percentage of controls. *p < 0.01 v Control; †p < 0.01 v Myocarditis; NS, no significant difference v Con-Vehicle.
PPAR-γ activator attenuation of myocardial inflammation in rats with EAM
Because PPAR-γ activators had been shown to inhibit some inflammatory conditions, we determined whether administration of PPAR-γ activators would affect the pathogenesis of EAM. In experiment I, the hearts from immunised rats had severe and diffuse discoloured myocarditis with massive pericardial effusion. We observed extensive injuries to myocytes with inflammatory changes, such as fragmentation of necrotic myocardial fibres, mononuclear cells, polymorphonuclear neutrophils, eosinophils, and multinucleated giant cells (data not shown). Treatment with 15d-PGJ2 and PIO greatly reduced the severity of the disease, as assessed by measuring the ratio of heart weight to body weight, pericardial effusion, and macroscopic and microscopic scores (table 1). Having ascertained that PPAR-γ activators suppressed EAM, we then tested the effect of drug treatment on the later course of EAM. In experiment II, the severity of myocarditis was also significantly reduced (table 1). Thus, administration of 15d-PGJ2 and PIO before disease onset or at the time of onset of clinical disease had beneficial clinical effects in this EAM model.
Table 1.
Haemodynamic data, histopathology, and weight ratios according to peroxisome proliferator activated receptor γ treatment in rats with and without experimental autoimmune myocarditis (EAM)
| Group | No | HW:BW (mg/g) | HR (beats/min) | SBP(mm Hg) | DBP(mm Hg) | Pericardial effusion score | Microscopic score | Macroscopic score |
| Healthy control rats | ||||||||
| Con-Vehicle | 7 | 2.98 (0.22) | 391 (13) | 130.3 (9.8) | 91.9 (8.7) | 0 | 0 | 0 |
| Con-15d-PGJ2 | 7 | 3.05 (0.14) | 411 (24) | 131.1 (11.1) | 92.7 (10.2) | 0 | 0 | 0 |
| Con-PIO | 7 | 3.09 (0.09) | 403 (21) | 132.5 (10.2) | 90.8 (9.8) | 0 | 0 | 0 |
| Experiment I | ||||||||
| Controls | 7 | 2.99 (0.11)** | 401 (20) | 132.5 (11.7) | 92.8 (9.1) | 0 | 0 | 0 |
| Myocarditis | 8 | 5.45 (0.57) | 432 (49) | 131.3 (10.7) | 91.5 (12.80 | 1.8 (0.4) | 3.4 (0.9) | 3.5 (1.1) |
| 15d-PGJ2 | 8 | 4.07 (0.69)** | 417 (31) | 131.8 (18.7) | 91.9 (16.9) | 0.9 (0.4)** | 1.5 (0.8)** | 1.8 (0.7)** |
| PIO | 8 | 4.19 (1.03)** | 412 (39) | 133.2 (13.8) | 92.8 (15.2) | 1.2 (0.8)* | 1.9 (0.8)** | 2.0 (0.3)** |
| Experiment II | ||||||||
| Controls | 7 | 3.07 (0.13)** | 396 (31) | 133.5 (10.7) | 93.1 (10.2) | 0 | 0 | 0 |
| Myocarditis | 8 | 5.56 (0.59) | 441 (59) | 129.3 (11.1) | 91.1 (13.2) | 1.9 (0.5) | 3.5 (1.0) | 3.6 (0.8) |
| 15d-PGJ2 | 8 | 4.41 (0.38)** | 427 (61) | 130.1 (12.7) | 92.0 (14.7) | 1.2 (0.7)* | 2.4 (0.7)* | 2.3 (0.6)** |
| PIO | 8 | 4.57 (0.63)* | 422 (49) | 131.2 (15.8) | 91.9 (16.1) | 1.3 (0.5)* | 2.7 (0.8) | 2.6 (0.4)** |
Data are mean (SD).
*p<0.05, **p<0.01 v Myocarditis rats.
15d-PGJ2, rats with EAM treated with 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2); Con-15d-PGJ2, normal rats treated with 15d-PGJ2; Con-PIO, normal rats treated with pioglitazone (PIO); Controls, normal rats treated with phosphate buffered saline (PBS); Con-Vehicle, normal rats treated with vehicle; DBP, diastolic blood pressure; HR, heart rate; HW:BW, ratio of heart weight to body weight; Myocarditis, rats with EAM treated with PBS; PIO, rats with EAM treated with PIO; SBP; systolic blood pressure.
PPAR-γ activator suppression of myocardial mRNA expression of inflammatory cytokines
In controls, myocardial mRNA expression of cytokines was detected only for macrophage inhibitory factor and interferon γ. In rats with EAM treated with PBS, mRNAs of type 1 T helper cell cytokines (such as IL-18), type 2 T helper cell cytokines (such as IL-6), and proinflammatory cytokines (such as macrophage inhibitory factor, interferon γ, IL-1β, and IL-1Ra) were greatly upregulated, and mRNA expression of IL-1α, IL-12p35, IL-12p40, and IL-10 were slightly upregulated. Administration of 15d-PGJ2 and PIO greatly reduced the expression of cytokine mRNAs in experiment I (fig 3). Results were similar in experiment II (data not shown), suggesting that PPAR-γ activator ameliorated EAM may be associated with inhibition of the expression of inflammatory cytokines.
Figure 3.
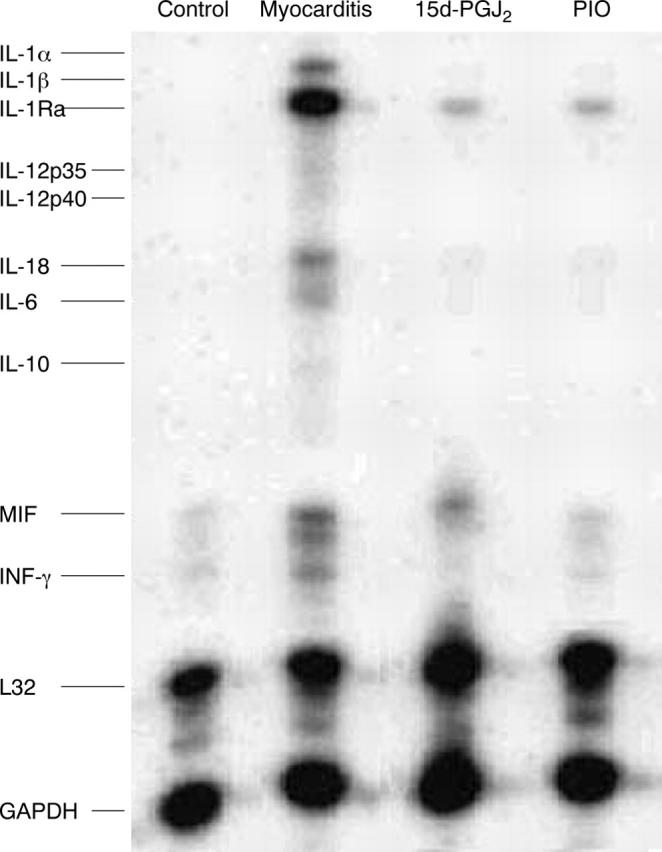
Ribonuclease protection assay for mRNAs of type 1 (Th1) and type 2 (Th2) T helper cells and of proinflammatory cytokines. In control, the myocardial mRNA expression of cytokines was detected only for macrophage inhibitory factor (MIF) and interferon γ (IFN-γ). In myocarditis rats treated with vehicle, mRNAs of Th1 cytokines (such as interleukin (IL) -18), Th2 cytokines (such as IL-6), and proinflammatory cytokines (such as MIF, IFN-γ, IL-1β, and IL-1Ra) were greatly upregulated, and mRNA expression of IL-1α, IL-12p35, IL-12p40, and IL-10 was slightly upregulated compared with controls. Treatment with 15d-PGJ2 and PIO greatly reduced the expression of cytokine mRNAs. L32 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) are housekeeping genes. A representative finding of three distinct experiments is shown.
PPAR-γ activator suppression of myocardial IL-1β and TNF-α protein expression in rats with EAM
Western blotting showed that myocardial IL-1β and TNF-α expression was upregulated 3.3-fold and 4.3-fold, respectively, in rats with EAM and treated with PBS compared with controls. Treatment with 15d-PGJ2 and PIO decreased the upregulated IL-1β and TNF-α expression in experiment I (fig 4A, B) and in experiment II (data not shown). These results suggesting that PPAR-γ activator treatment suppressed myocardial IL-1β and TNF-α protein expression in myocarditis.
Figure 4.

Myocardial IL-1β and tumour necrosis factor α (TNF-α) protein expression by western blotting. (A) Western blot analysis. (B) Densitometric analysis of relative protein concentrations. In rats with myocarditis treated with PBS, IL-1β and TNF-α protein expression was greatly increased; 15d-PGJ2 and PIO treatment decreased IL-1β and TNF-α protein expression. Values are derived from five animals and determined as the percentage of control values. *p < 0.01 v control; †p < 0.01 v vehicle.
PPAR-γ activator inhibition of the activation of NF-κB in myocardium of rats with EAM
In acute myocarditis, NF-κB was greatly activated (enhanced DNA binding activity by 5.6-fold versus normal controls) in myocardium, and this activation was inhibited in the 15d-PGJ2 treated and PIO treated groups in experiment I (fig 5A, B) and in experiment II (data not shown). IκBα decreased remarkably in the cytoplasmic fractions from inflammatory myocardium, and treatment with 15d-PGJ2 and PIO enhanced the concentrations of IκBα by 2.2-fold and 2.1-fold, respectively, versus vehicle (fig 5C, D). Moreover, the concentration of IκBα in the nuclear fraction was enhanced by 1.7-fold and 1.5-fold (both p < 0.01) in the 15d-PGJ2 treated and PIO treated groups versus the vehicle group (fig 5C, D). These results suggesting that PPAR-γ activator treatment inhibited the activation of NF-κB.
Figure 5.
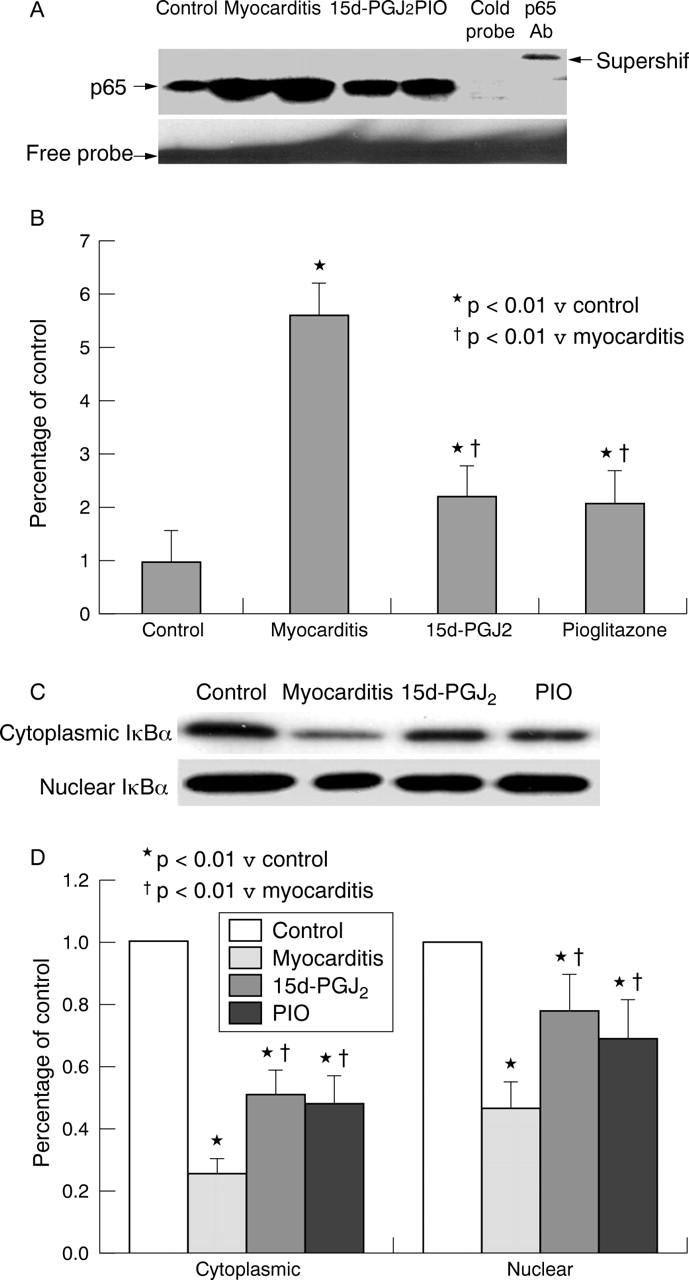
Electrophoretic mobility shift assay for nuclear factor κB (NF-κB) and western blot analysis of inhibitor IκB (IκB) α in cytoplasmic fraction of myocardium. (A) Representative electrophoretic mobility shift assay of NF-κB activity in myocardium. Specificity was determined by addition of p65 antibody (supershift) or unlabelled (cold) NF-κB oligonucleotide to the myocardium. Four separate experiments yielded similar results. (B) Densitometric analysis of DNA binding activity in the region of NF-κB (p65) was quantified. In acute myocarditis, NF-κB was greatly activated (enhanced DNA binding activity by 5.6-fold versus normal controls). This activation was inhibited in the 15d-PGJ2 and PIO treated groups. (C) Representative western blots. (D) Densitometric analysis (n = 5). IκBα concentration decreased greatly in the cytoplasmic and nuclear fractions in rats with EAM treated with PBS. Treatment with 15d-PGJ2 and PIO enhanced the cytoplasmic concentrations of IκBα by 2.2-fold and 2.1-fold, and increased the nuclear concentrations of IκBα by 1.7-fold and 1.5-fold, respectively, versus myocarditis. * p < 0.01 v control; †p < 0.01 v myocarditis.
DISCUSSION
The present findings clearly show that PPAR-γ activators greatly ameliorated the severity of EAM in rats and were associated with suppression of myocardial proinflammatory cytokine expression.
In the present study, immunohistochemical results showed that PPAR-γ expression was strongly stained in infiltrating inflammatory cells and that the expression of PPAR-γ was prominently located in the nuclear and perinuclear regions of inflammatory cells. Although Takano et al8 reported that PPAR-γ was expressed in neonatal normal cardiac myocytes, in the present study we detected only marginal or trivial immunoreactivity for PPAR-γ in myocardium of normal rats. Administration of PPAR-γ activators ameliorated EAM in rats; concurrently, PPAR-γ expression of inflammatory cells in myocardial tissue was reduced. These results suggest that PPAR-γ may have a role in the pathophysiology of EAM and that PPAR-γ activators have the potential to suppress myocardial inflammation in autoimmune myocarditis.
It has been shown that inflammatory cytokines have an important role in the development of inflammatory myocardial diseases.18,20 IL-1β and TNF-α may promote coxsackievirus B3 induced myocarditis in resistant B10.A mice.21 Transgenic mice with cardiac overexpression of TNF-α develop cardiac hypertrophy, fibrosis, and dilated cardiomyopathy.22 The proinflammatory cytokine TNF-α is known to depress myocardial contractility by uncoupling β adrenergic receptor signalling,23 increasing cardiac nitric oxide and peroxynitrite.24 Suppression of inflammatory cytokines has beneficial effects on ameliorating acute myocarditis.3,20 In the present study, administration of 15d-PGJ2 and PIO suppressed the myocardial mRNA expression of inflammatory cytokines and the expression of IL-1β and TNF-α protein in myocarditis. These results are consistent with reports that PPAR-γ activators inhibited the expression of proinflammatory cytokines interferon γ, IL-1β, and TNF-α in isolated human CD4+ T cells.5,25 Accordingly, in the present study the finding that 15d-PGJ2 and PIO attenuated myocardial inflammation in EAM may be, at least partly, due to the suppression of inflammatory cytokines.
The mRNAs of types 1 and 2 T helper cell cytokines and of proinflammatory cytokines were all significantly attenuated by 15d-PGJ2 and PIO treatment in our present study. The results suggest that activation of PPAR-γ may inhibit a common regulatory factor in the control of transcription of these proinflammatory mediators. Among the inducible transcription factors involved in myocardial inflammation, NF-κB has a central role.26 NF-κB binding sites in the promoter region have been found for a variety of proinflammatory cytokines. The data shown in fig 5A and B indicate that treatment with 15d-PGJ2 and PIO inhibited the activation of NF-κB induced by myocardial inflammation. In the present study, we have also found that 15d-PGJ2 and PIO enhanced cytoplasmic and nuclear concentrations of IκBα, an NF-κB inhibitor. The increase in IκBα protein would halt p65 mediated gene activation and result in acceleration of NF-κB nuclear deactivation. Moreover, treatment with 15d-PGJ2 and PIO enhanced the accumulation of IκBα in the nucleus, consistent with data from an in vitro study that observed enhanced nuclear IκBα, accompanied by a decrease in NF-κB DNA binding activity.27 Studies have suggested that IκBα, accumulated in the nucleus, is resistant to signal induced degradation. The presence of IκB in the nucleus contributed to the inhibition of binding of active NF-κB complexes to the κB sites located in the regulatory sequences of various genes.28 Therefore, the enhanced accumulation of IκBα in the cytoplasm and the nucleus by the PPAR-γ agonists would exert an inhibitory effect on NF-κB activity.
PPAR-γ activators have previously been shown to effectively inhibit inflammatory disorders.11–15 More recent studies have shown that PPAR-γ activators may reduce experimental myocardial infarction size29 and attenuate left ventricular remodelling and failure after myocardial infarction.30 Various mechanisms have been proposed to explain these treatment efficacies. Mechanistically, PPAR-γ activators act at least in part by inhibiting the activity of transcription factor activator protein 1, signal transducers and activators of transcription 1, and NF-κB.9,31,32 In the case of NF-κB, PPAR-γ activators inhibit the activity of IκB kinase, which normally phosphorylates the NF-κB inhibitor IκB, resulting in trans-activation of NF-κB target genes.31,32 Inhibition of the activation of NF-κB may attenuate EAM in rats.33
Overall, our results have shown that PPAR-γ activators suppress EAM. Because an increase in IκB expression and inhibition of translocation of the NF-κB subunit p65 to the nucleus in inflammatory cells correlated with the protective effects of PPAR-γ activators, our results suggest that PPAR-γ activators act sequentially through PPAR-γ activation, IκB induction, blockade of NF-κB activation, and inhibition of inflammatory cytokine expression. These results suggest that PPAR-γ activators such as 15d-PGJ2 and PIO may have the potential to modulate human inflammatory heart diseases such as myocarditis.
Abbreviations
15d-PGJ2, 15-deoxy-Δ12,14-prostaglandin J2
EAM, experimental autoimmune myocarditis
IL, interleukin
NF-κB, nuclear factor κB
PBS, phosphate buffered saline
PIO, pioglitazone
PPAR-γ, peroxisome proliferator activated receptor γ
TNF-α, tumour necrosis factor α
Published Online First 17 March 2005
Supported in part by the Natural Science Foundation of China (30170371).
REFERENCES
- 1.Matsumori A, Yamada T, Suzuki H, et al. Increased circulating cytokines in patients with myocarditis and cardiomyopathy. Br Heart J 1994;72:561–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neumann FJ, Ott I, Gawaz M, et al. Cardiac release of cytokines and inflammatory responses in acute myocardial infarction. Circulation 1995;92:748–55. [DOI] [PubMed] [Google Scholar]
- 3.Henriksen PA, Newby DE. Therapeutic inhibition of tumour necrosis factor with heart failure: cooling the inflamed heart. Heart 2003;89:14–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuan Z, Shioji K, Kihara Y, et al. Cardioprotective effects of carvedilol on acute autoimmune myocarditis: anti-inflammatory effects associated with antioxidant property. Am J Physiol Heart Circ Physiol 2004;286:83–90. [DOI] [PubMed] [Google Scholar]
- 5.Sidhu JS, Kaski JC. Peroxisome proliferator activated receptor γ: a potential therapeutic target in the management of ischaemic heart disease. Heart 2001;86:255–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark RB, Bishop-Bailey D, Estrada-Hernandez T, et al. The nuclear receptor PPAR gamma and immunoregulation: PPAR gamma mediates inhibition of helper T cell responses. J Immunol 2000;164:1364–71. [DOI] [PubMed] [Google Scholar]
- 7.Marx N, Schonbeck U, Lazar MA, et al. Peroxisome proliferator-activated receptor gamma activators inhibit gene expression and migration in human vascular smooth muscle cells. Circ Res 1998;83:1097–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takano H, Nagai T, Asakawa M, et al. Peroxisome proliferator-activated receptor activators inhibit lipopolysaccharide-induced tumor necrosis factor-alpha expression in neonatal rat cardiac myocytes. Circ Res 2000;87:596–602. [DOI] [PubMed] [Google Scholar]
- 9.Ricote M, Li AC, Willson TM, et al. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998;391:79–82. [DOI] [PubMed] [Google Scholar]
- 10.Cuzzocrea S, Pisano B, Dugo L, et al. Rosiglitazone, a ligand of the peroxisome proliferator-activated receptor-gamma, reduces acute inflammation. Eur J Pharmacol 2004;483:79–93. [DOI] [PubMed] [Google Scholar]
- 11.Ricote M, Huang J, Fajas L, et al. Expression of the peroxisome proliferator-activated receptor gamma in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc Natl Acad Sci USA 1998;95:7614–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawahito Y, Kondo M, Tsubouchi Y, et al. 15-deoxy-delta(12,14)-PGJ(2) induces synoviocyte apoptosis and suppresses adjuvant-induced arthritis in rats. J Clin Invest 2000;106:189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Su CG, Wen X, Bailey ST, et al. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J Clin Invest 1999;104:383–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daib A, Deng C, Smith JD, et al. Peroxisome proliferator-activated receptor-gamma agonist 15-deoxy-delta(12,14)-prostaglandin J(2) ameliorates experimental autoimmune encephalomyelitis. J Immunol 2002;168:2508–15. [DOI] [PubMed] [Google Scholar]
- 15.Feinstein DL, Galea E, Gavrilyuk V, et al. Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann Neurol 2002;51:694–702. [DOI] [PubMed] [Google Scholar]
- 16.Yuan Z, Liu Y, Liu Y, et al. Peroxisome proliferator-activated receptor-gamma ligands ameliorate experimental autoimmune myocarditis associated with inhibition of self-sensitive T cells. J Cardiovasc Pharmacol 2004;43:868–75. [DOI] [PubMed] [Google Scholar]
- 17.Kodama M, Matsumoto Y, Fujiwara M, et al. A novel experimental model of giant cell myocarditis induced in rats by immunization with cardiac myosin fraction. Clin Immunol Immunopathol 1990;57:250–62. [DOI] [PubMed] [Google Scholar]
- 18.Shioji K, Kishimoto C, Sasayama S. Fc receptor-mediated inhibitory effect of immunoglobulin therapy on autoimmune giant cell myocarditis: concomitant suppression of the expression of dendritic cells. Circ Res 2001;89:540–6. [DOI] [PubMed] [Google Scholar]
- 19.Yue T, Bao W, Jucker BM, et al. Activation of peroxisome proliferator-activated receptor-α protects the heart from ischemia/reperfusion injury. Circulation 2003;108:2393–9. [DOI] [PubMed] [Google Scholar]
- 20.Matsui Y, Inobe M, Okamoto H, et al. Blockade of T cell costimulatory signals using adenovirus vectors prevents both the induction and the progression of experimental autoimmune myocarditis. J Mol Cell Cardiol 2002;34:279–95. [DOI] [PubMed] [Google Scholar]
- 21.Lane JR, Neumann DA, Lafond-Walker A, et al. Interleukin 1 or tumor necrosis factor can promote Coxsackie B3-induced myocarditis in resistant B10.A mice. J Exp Med 1992;175:1123–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bryant D, Becker L, Richardson J, et al. Cardiac failure in transgenic mice with myocardial expression of tumor necrosis factor-alpha. Circulation 1998;97:1375–81. [DOI] [PubMed] [Google Scholar]
- 23.Chung MK, Gulick TS, Rotondo RE, et al. Mechanism of cytokine inhibition of β-adrenergic agonist stimulation of cyclic AMP in rat cardiac myocytes: impairment of signal transduction. Circ Res 1990;67:753–63. [DOI] [PubMed] [Google Scholar]
- 24.Finkel MS, Oddis CV, Jacob TD, et al. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science 1992;57:387–9. [DOI] [PubMed] [Google Scholar]
- 25.Marx N, Kehrle B, Kohlhammer K, et al. PPAR activators as antiinflammatory mediators in human T lymphocytes: implications for atherosclerosis and transplantation-associated arteriosclerosis. Circ Res 2002;90:703–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D’Acquisto F, May MJ, Ghosh S. Inhibition of nuclear factor κB (NF-κB): an emerging theme in anti-inflammatory therapies. Mol Intervent 2002;2:22–35. [DOI] [PubMed] [Google Scholar]
- 27.Delerive P, Gervois P, Fruchart JC, et al. Induction of I κBα expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-α activators. J Biol Chem 2000;275:36703–7. [DOI] [PubMed] [Google Scholar]
- 28.Vanden-Berghe W, Vermeulen L, Delerive P, et al. A paradigm for gene regulation: inflammation, NF-kappaB and PPAR. Adv Exp Med Biol 2003;544:181–96. [DOI] [PubMed] [Google Scholar]
- 29.Wayman NS, Hattori Y, McDonald MC, et al. Ligands of the peroxisome proliferator-activated receptors (PPAR-gamma and PPAR-alpha) reduce myocardial infarct size. FASEB J 2002;16:1027–40. [DOI] [PubMed] [Google Scholar]
- 30.Shiomi T, Tsutsui H, Hayashidani S, et al. Pioglitazone, a peroxisome proliferator-activated receptor-gamma agonist, attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation 2002;106:3126–32. [DOI] [PubMed] [Google Scholar]
- 31.Purcell NH, Tang G, Yu C, et al. Activation of NF-kappa B is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proc Natl Acad Sci USA 2001;98:6668–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rossi A, Kapahi P, Natoli G, et al. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature 2000;403:103–8. [DOI] [PubMed] [Google Scholar]
- 33.Yokoseki O, Suzuki J, Kitabayashi H, et al. cis Element decoy against nuclear factor-kappaB attenuates development of experimental autoimmune myocarditis in rats. Circ Res 2001;89:899–906. [DOI] [PubMed] [Google Scholar]