

Abstract
Objective: To define the natural history of idiopathic restrictive cardiomyopathy in a paediatric population and to identify any factors predictive of outcome.
Design: Retrospective analysis of patients born between 1970 and 2002 were identified from the Children’s Hospital of Pittsburgh cardiology database. Demographic data, mode of presentation, echocardiographic and haemodynamic findings at diagnosis, survival time, and manner of death were evaluated.
Setting: Tertiary referral and transplant centre for paediatric patients with cardiac disease.
Patients: All local and referred patients with idiopathic restrictive cardiomyopathy born after 1970 and under 21 years of age at time of diagnosis.
Results: 21 patients were identified. Probability of survival at 1, 5, and 10 years was 80.5% (95% confidence interval (CI) 58 to 100), 39% (95% CI 17 to 61), and 20% (95% CI 0 to 42), respectively. Median age of presentation was 3.8 years (mean (SD) 5.7 (6.1) years). Median survival without transplantation was 2.2 years (mean (SD) 4.6 (5.4) years). Age at presentation, sex, and presence or absence of heart failure symptoms at presentation were not associated with clinical course. Right (p = 0.05) and left ventricular end diastolic pressures (p = 0.04) and ratio of left atrial to aortic root dimensions (LA:Ao) (p = 0.03) at presentation had a significantly negative correlation with survival time after diagnosis.
Conclusions: Without transplantation, most children with restrictive cardiomyopathy have a very poor prognosis. Longer survival from diagnosis was correlated with lower LA:Ao and cardiac filling pressures at diagnosis. Survival time was not influenced by the symptoms present at diagnosis.
Keywords: natural history, paediatrics, restrictive cardiomyopathy, survival, transplant
Restrictive cardiomyopathy is rare in children, accounting for 2–5% of all paediatric cardiomyopathy.1–3 Restrictive cardiomyopathy is characterised by diastolic dysfunction with preserved systolic function, dilated atria, and the absence of ventricular hypertrophy or dilatation.4 A small number of reports of restrictive cardiomyopathy in children have described mean survival from diagnosis ranging from a few months to 7.8 years.5–9 In all but rare cases in children, the underlying aetiology remains undetermined. A detailed natural history has been elusive because of small patient numbers in any given series. For the same reason, no clear prognostic indicators have been identified.
In each report, several patients with idiopathic restrictive cardiomyopathy have significantly longer survival from diagnosis than the remainder of the cohort.6,7,9 It is these patients who encourage the search for significant indicators of prognosis. Identifying factors for prolonged survival may allow the safe delay of cardiac transplantation. However, several studies describe significantly increased pulmonary vascular resistance at diagnosis,5–7 often as high as 10 Woods units/m2. In one study, the increased pulmonary vascular resistance worsened rapidly, effectively doubling in 1–4 years5 from diagnosis. For this reason, early consideration for heart transplantation has been recommended to avoid the need for combined heart–lung transplantation.
To date, however, no consistent risk factors predictive of survival have been identified. The objective of the current study was to evaluate findings at presentation to identify prognostic indicators for survival.
METHODS
This study was approved by the Children’s Hospital Human Rights Committee, Pittsburgh, Pennsylvania, USA. Patients were identified by retrospective search of over 68 000 patient records contained within the Children’s Hospital of Pittsburgh cardiology database. All patients born after 1 January 1970 were included. Patients with identifiable aetiologies (hypertrophic cardiomyopathy, primary muscle diseases, chemotherapy exposure) or with systolic dysfunction or ventricular dilatation at presentation were excluded. Patients referred to our paediatric heart transplantation programme from other institutions were included (n = 12). Patients who received a transplant at other institutions and followed up in our clinic (n = 2) were excluded.
The primary end point for analysis was time to death or cardiac transplantation (survival time). This was explored by Kaplan-Meier analysis. Associations between survival time and various continuous and categorical variables were explored by simple linear correlation and stratified Kaplan-Meier analyses, respectively. Variables analysed were sex, age at presentation, presence or absence of cardiovascular symptoms at presentation, echocardiographic left atrial to aortic root dimension ratio (LA:Ao), pulmonary capillary wedge pressure, right atrial mean pressure, indexed pulmonary vascular resistance (PVRI), right and left ventricular end diastolic pressures, mean pulmonary artery pressure, and cardiac index. The data set was considered too small to warrant multivariate analysis.
Echocardiographic and haemodynamic data at the time of diagnosis were obtained. In three cases, haemodynamic data were available only 2–3 years after diagnosis. These were included in our data set. Left ventricular systolic function (shortening fraction) at diagnosis was normal by echocardiography in all patients (data not shown).
RESULTS
Patient characteristics at presentation
Median age at presentation was 3.75 years (mean (SD) 5.7 (6.1) years). Twelve of the patients (57%) were boys and nine (43%) were girls. Seven (33%) patients presented without cardiac symptoms: one with murmur, four with extra heart sounds, one with middle cerebral artery infarction, and one with hypoglycaemia in the newborn period. Fourteen (66%) patients presented with symptoms of cardiovascular compromise: one with failure to thrive and fatigue, three with syncope, and 10 with congestive heart failure. The New York Heart Association functional class of these patients was not reported, as it is not applicable to the young paediatric population and was not often recorded in the medical record.
Several associated findings were identified but none were present in more than one patient: familial restrictive cardiomyopathy (one); middle cerebral artery stroke (one); hypothyroidism, retinal dysplasia, and heart block with hepatic lipid inclusions (one); arthrogryposis (one); calcification on dermal biopsy(one); chromosome 8 deletion with absent corpus callosum and high vanillylmandelic acid concentrations (one); and single right coronary artery and a seizure disorder (one).
Echocardiographic data were available for 18 patients (86%). The thickness of the left ventricular posterior wall and septum at presentation were compared with body surface area and z scores were generated. The posterior wall z scores ranged between −3.7 and 2.8 (mean −0.65). Only two z scores for the left ventricular posterior wall were above 2 with scores of 2.2 and 2.8. Septal thickness z scores ranged from −6.0 to 1.8 (mean −0.2). On the basis of absent or minimal hypertrophy and characteristic prominent biatrial dilatation of all our patients, none of this cohort was considered to have hypertrophic cardiomyopathy.
Haemodynamic data at presentation were as follows: mean pulmonary capillary wedge pressure 18 mm Hg, ranging from 9 to 26 mm Hg; cardiac index varying from 1.9 to 5.1 l/min/m2 with a mean of 2.9 l/min/m2; and PVRI at presentation varying widely between 1.8 index units and 15 index units (table 1).
Table 1.
Demographic and haemodynamic data at presentation and relation to survival time (to death or transplantation)
| No | Mean (SD) | Median | Range | Pearson correlation coefficients | ||
| r | p Value | |||||
| Survival time | ||||||
| Time to death or transplantation from diagnosis (years) | 21 | 4.6 (5.4) | 2.2 | 0.05–18.5 | NA | NA |
| Boys (years) | 12 | 7.4 | 6.2 | NA | NA | 0.20* |
| Girls (years) | 9 | 3.6 | 1.7 | NA | ||
| Asymptomatic patients (years) | 7 | 6.0 | 3.4 | NA | NA | 0.92* |
| Symptomatic patients (years) | 14 | 6.0 | 2.2 | NA | ||
| Data at presentation | ||||||
| Age (years) | 21 | 5.7 (6.1) | 3.8 | 0.01–18.5 | −0.19 | 0.41 |
| LA:Ao (mm) | 13 | 2.0 (0.4) | 2.1 | 1.4–2.5 | −0.61 | 0.03 |
| PCWP (mm Hg) | 16 | 18.8 (5.3) | 18 | 9–26 | −0.49 | 0.06 |
| Right atrial pressure (mm Hg) | 18 | 10.7 (4.3) | 9.5 | 5–21 | 0.27 | 0.29 |
| RVEDP (mm Hg) | 17 | 11.2 (4.7) | 11 | 2–20 | −0.48 | 0.05 |
| LVEDP (mm Hg) | 16 | 22.6 (7.3) | 25.5 | 10–34 | −0.51 | 0.04 |
| Pulmonary artery pressure (mm Hg) | 18 | 34.4 (14.1) | 28.5 | 21–60 | −0.05 | 0.84 |
| Cardiac index (l/min/m2) | 18 | 3.1 (1.0) | 2.9 | 1.9–5.1 | 0.07 | 0.78 |
| PVRI (index units) | 17 | 5.6 (3.8) | 4 | 1.8–15.0 | 0.39 | 0.12 |
*Log rank.
LA:Ao, left atrial to aortic root dimension ratio; LVEDP, left ventricle end diastolic pressure, NA, not applicable; PCWP, pulmonary capillary wedge pressure; PVRI, indexed pulmonary vascular resistance; RVEDP, right ventricular end diastolic pressure.
Endomyocardial biopsy, explant, or necropsy reports were available for 11 patients (52%) and showed a variety of findings including varying degrees of endomyocardial and interstitial fibrosis ranging from mild to severe and mild myocyte hypertrophy. There was no evidence of myocarditis, amyloidosis, or sarcoidosis on any specimen. One specimen had myofibre disarray and increased mucin of unclear significance.
Outcomes
Probability of survival at one, five, and 10 years after diagnosis was 80.5% (95% confidence interval (CI) 58 to 102), 39% (95% CI 17 to 61), and 20% (95% CI 0 to 42) (fig 1). Median survival time to death or transplantation (any cause) was 2.2 years (mean (SD) 4.6 (5.4) years) (table 1). Eight patients (38%) died without undergoing transplantation, nine (43%) received a transplant, and four (19%) are alive without transplantation at 1–11 years from diagnosis. Three patients (14%) survived for greater than 10 years from diagnosis before transplantation or death.
Figure 1.

Freedom from death or transplantation for all patients with idiopathic restrictive cardiomyopathy.
Of the eight patients who died before transplantation, one died within a month of diagnosis from severe heart failure that could not be stabilised. One patient died during a catheterisation procedure at 21 months from diagnosis.
Three patients died suddenly at home; one died 20 months from diagnosis and had a preceding respiratory illness. One patient with atrial flutter diagnosed at initial presentation died suddenly at home four years later. The third patient to die at home survived 18.5 years from diagnosis and had progressive heart failure symptoms and a one year history of intermittent atrial flutter.
One patient, 16 years from diagnosis, died in the hospital after being admitted with a large pericardial effusion and subsequently decompensated with new onset refractory atrial flutter. There is insufficient information regarding the specific cause of death of two patients.
Predictors of outcome
The mean (SEM) survival of male patients was 7.4 (2) years with a median of 6.21 (2) years. For girls, mean survival was 3.6 (2.2) years with a median of 1.73 years (table 1). There was a trend to greater freedom from death or transplant for male patients. At one year and five years from diagnosis, male patient survival rates were 92% and 57%, respectively, compared with 65% and 13% for girls. This did not, however, reach significance (log rank p = 0.20) (fig 2A). There was also no statistical difference in long term survival based on mode of presentation (p = 0.92) (fig 2B).
Figure 2.
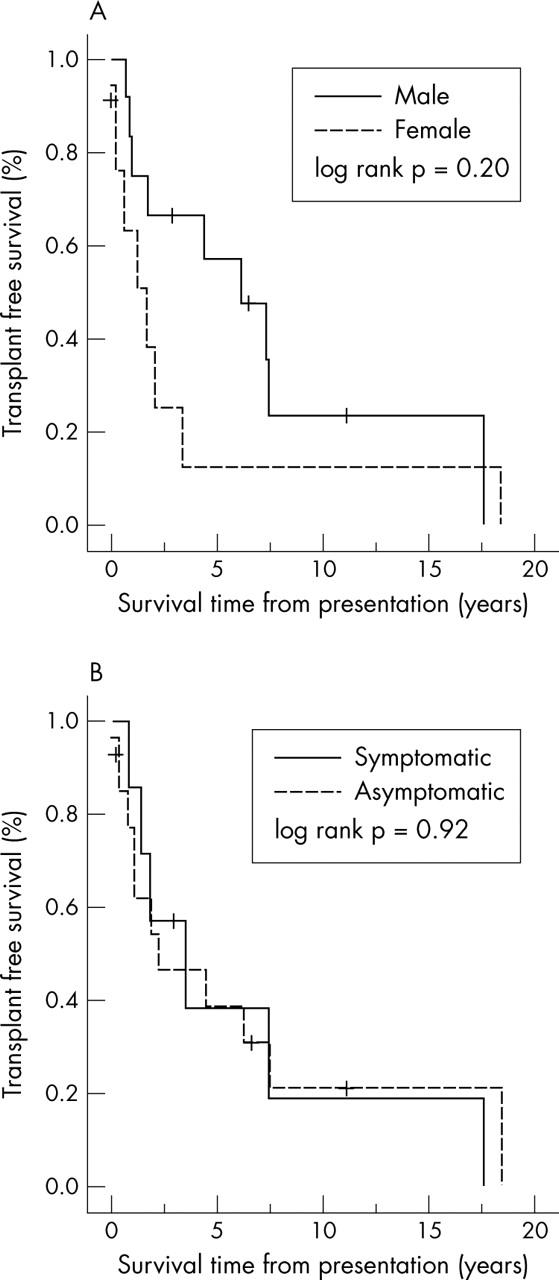
Influence of (A) sex and (B) presence or absence of cardiac symptoms at presentation on survival time.
By echocardiography, LA:Ao at presentation had a negative correlation with survival (r = −0.61, p = 0.03) (table 1). Pearson correlation coefficients showed a moderate negative correlation with survival for the following catheterisation data: pulmonary artery wedge pressure (r = −0.49, p = 0.06) and right and (r = −0.48, p = 0.05) left ventricular end diastolic pressures (r = −0.51, p = 0.04). Other parameters did not correlate with survival.
DISCUSSION
Idiopathic restrictive cardiomyopathy continues to be one of the least understood conditions in paediatric cardiology. It has been estimated at 2–5% of all paediatric cardiomyopathies.1–3 In our series it accounted for 2.3% of all cardiomyopathies, our series being one of the largest published to date. The small numbers of patients in all series has made it difficult to define the natural history of this condition. Most data, including ours, show that the vast majority of patients do not survive more than 2–5 years from diagnosis unless they receive a transplant. Because of the lack of effective medical treatment for these patients,8,10 it has been proposed that paediatric patients with a diagnosis of restrictive cardiomyopathy be listed for transplantation immediately.11 The difficulty with this approach is that there are rare patients who survive much longer, as long as 8–12 years.5–7,9 In our institution, three patients (14%) lived over 10 years after diagnosis. Only about half of all transplant recipients survive this long. Patients with restrictive cardiomyopathy also have a high risk of sudden death,10 although the mechanisms are not clear. These observations make the optimal planning of transplantation quite challenging.
Several institutions have reviewed their experience with this disease to glean some information that may help identify predictors of survival. In those studies, patients with cardiomegaly,5 age < 5 years,5 thromboembolism,5 increased PVRI,6 pulmonary venous congestion,7 progressive increase of PVRI,8 syncope, or chest pain at diagnosis10 all had a poor prognosis. These results were not consistent between studies. In our cohort, the medical regimen was not consistent across this population and changed over time for individual patients. It was not possible to assess the impact of differing medical regimens on survival.
The strongest predictor of poor survival in our study was the extent of increase of the LA:Ao measured by echocardiography at presentation. Ammash and colleagues12 report a similar finding in their study of adult patients that a left atrial size > 60 mm was correlated with poorer outcome. We also found that poor prognosis is associated with the degree of increase of end diastolic and pulmonary capillary wedge pressures.
Patients with restrictive cardiomyopathy with advanced symptoms of heart failure warrant cardiac transplantation to improve the quality of life. The greatest dilemma in caring for this population is what to do with patients with relatively mild symptoms. One of the most intriguing findings in our review was that the survival from diagnosis was almost identical in patients who presented with symptoms of heart failure or syncope and in patients who were asymptomatic at presentation. This suggests that presence of cardiac symptoms should not be the key factor in determining timing of listing for transplantation as is generally the case in other types of paediatric cardiomyopathy.
PVRI in our study was often quite increased at presentation—as high as 15 index units. Despite this degree of increase, there was no statistical relation between PVRI and survival in our cohort, in contrast to other reports in the literature.5–7
These findings offer further insight into the natural history of paediatric restrictive cardiomyopathy. Although this is one of the largest cohorts at any one institution, the small number of patients limits the power of the findings. The number of patients is too small for a multivariate model to predict survival. Also, this study includes patients that were referred to our institution for transplantation, which may bias the data by presenting sicker patients. To limit this effect, data from time of presentation were obtained, rather than from time of referral.
The biases of this type of study can be overcome only by multicentre or national registries, such as the paediatric cardiomyopathy registry2 or the national Australian cardiomyopathy study.1 These larger studies can neutralise referral bias and low statistical power and allow for multivariate analysis of risk factors for survival.
Abbreviations
CI, confidence interval
LA:Ao, left atrial to aortic root dimension ratio
PVRI, indexed pulmonary vascular resistance
REFERENCES
- 1.Nuget AW, Daubeney P, Chondros P, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med 2003;248:1639–46. [DOI] [PubMed] [Google Scholar]
- 2.Lipschutz SE, Sleeper LA, Towbin JA, et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med 2003;348:1647–55. [DOI] [PubMed] [Google Scholar]
- 3.Malcic I, Jelusic M, Kniewald H, et al. Epidemiology of cardiomyopathies in children and adolescents: a retrospective study. Cardiol Young 2002;12:253–9. [DOI] [PubMed] [Google Scholar]
- 4.Richardson P, McKenna W, Bristow M, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology task force on the definition and classification of cardiomyopathies. Circulation 1996;93:841–2. [DOI] [PubMed] [Google Scholar]
- 5.Chen S, Balfour IC, Jureidini S. Clinical spectrum of restrictive cardiomyopathy in children. J Heart Lung Transplant 2001;20:90–2. [DOI] [PubMed] [Google Scholar]
- 6.Kimberling MT, Balzer DT, Hirsch R, et al. Cardiac transplantation for pediatric restrictive cardiomyopathy: presentation, evaluation and short-term outcome. J Heart Lung Transplant 2002;21:455–9. [DOI] [PubMed] [Google Scholar]
- 7.Cetta F, O’Leary PW, Seward JB, et al.Idiopathic restrictive cardiomyopathy in childhood: diagnostic features and clinical course. Mayo Clin Proc 1995;70:634–40. [DOI] [PubMed] [Google Scholar]
- 8.Denfield SW, Rosenthal G, Gajarski RJ, et al. Restrictive cardiomyopathies in childhood: etiologies and natural history. Tex Heart Inst J 1997;24:38–44. [PMC free article] [PubMed] [Google Scholar]
- 9.Weller RJ, Weintraub R, Addonizio LJ, et al. Outcome of idiopathic restrictive cardiomyopathy in children. Am J Cardiol 2002;90:501–6. [DOI] [PubMed] [Google Scholar]
- 10.Rivenes SM, Kearney DL, Smith EO, et al. Sudden death and cardiovascular collapse in children with restrictive cardiomyopathy. Circulation 2000;102:876–82. [DOI] [PubMed] [Google Scholar]
- 11.Towbin JA. Cardiomyopathy and heart transplantation in children. Curr Opin Cadiol 2002;17:274–9. [DOI] [PubMed] [Google Scholar]
- 12.Ammash NM, Seward JB, Bailey KB, et al. Clinical profile and outcome of idiopathic restrictive cardiomyopathy. Circulation 2000;101:2490–6. [DOI] [PubMed] [Google Scholar]