

Abstract
Objective: To evaluate the clinical, exercise, and haemodynamic effects of chronic oral administration of the non-selective endothelin receptor antagonist bosentan on patients with pulmonary arterial hypertension (PAH) related to congenital heart disease (CHD).
Design: Prospective non-randomised open clinical study.
Setting: Cardiology tertiary referral centre.
Patients: 21 patients with a mean (SEM) age of 22 (3) years with chronic PAH related to CHD (15 with Eisenmenger’s syndrome). Patients were in World Health Organization (WHO) class II to IV with oxygen saturation 87 (2)%.
Intervention: Patients underwent clinical, exercise, and haemodynamic evaluations at baseline and after 16 weeks of treatment.
Results: Bosentan improved (p < 0.01) WHO class, peak oxygen consumption from 16.8 (1.4) to 18.3 (1.4) ml/kg/min, exercise duration from 9.0 (0.8) to 10.7 (0.6) minutes during the treadmill test, walking distance from 416 (23) to 459 (22) m, and Borg dyspnoea index from 2.8 (0.2) to 2.0 (0.1) during the six minute walk test. Bosentan treatment improved (p < 0.05) mean pulmonary artery pressure from 87 (4) to 81 (4) mm Hg, pulmonary blood flow index from 3.2 (0.4) to 3.7 (0.5) l/min/m2, pulmonary to systemic blood flow ratio from 1.2 (0.2) to 1.4 (0.2), and pulmonary vascular resistance index from 2232 (283) to 1768 (248) dyn·s·cm−5. Two patients died, presumably of arrhythmic causes, who were in WHO class IV at baseline and who had improved during treatment.
Conclusions: Bosentan induces short and mid term clinical, exercise, and haemodynamic improvements in patients with PAH related to CHD. Larger studies with long term endothelin receptor antagonism are needed to assess the safety and possible treatment role of bosentan in this population.
Keywords: congenital heart disease, endothelin antagonism, exercise, haemodynamic status, pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH), whether idiopathic1 or related to autoimmune diseases2 or congenital heart disease (CHD),3 is a rare and debilitating entity with poor prognosis. PAH related to CHD and especially Eisenmenger’s syndrome carries a high risk of morbidity and mortality in a relatively young patient population4 and has limited treatment options. PAH is characterised by remodelling of the vascular wall, plexiform lesions, and pulmonary vascular obstruction leading to progressive right heart failure and death. Despite differences in the pathogenesis of idiopathic or autoimmune PAH and PAH related to CHD, several clinical features and pathological characteristics5 have been found to be similar.
Endothelin is a promoter of cell proliferation, which also causes vasoconstriction through endothelin A and endothelin B receptors on smooth muscle cells and vasodilatation through endothelin B receptors on endothelium. Endothelin has been implicated in endothelial dysfunction and the pathogenesis of vascular remodelling,6 and increased endothelin concentrations have been reported in both adult7 and paediatric patients correlating with the severity of PAH.8 Bosentan is the first orally active, non-selective endothelin receptor antagonist shown to be effective in adult PAH placebo controlled trials.9,10 Endothelin A receptor antagonism was shown to improve haemodynamic function in infants with PAH after congenital heart surgery in a small study in the acute postoperative period, albeit with potentially adverse effects.11 Patients with PAH including PAH related to CHD also improved in the acute setting.12 Bosentan was shown to improve haemodynamic function in paediatric patients with PAH with good safety and tolerability in a recent study reporting its paediatric doses.13 Bosentan improved oxygenation and functional status of adult patients with Eisenmenger’s syndrome in a small study without exercise or haemodynamic evaluation.14
The purpose of this study was to assess the clinical, exercise capacity, and haemodynamic effects of oral bosentan treatment in patients with PAH related to CHD.
PATIENTS AND METHODS
Patient population
Patients in World Health Organization (WHO) functional class II to IV15 with severe PAH related to CHD with or without prior surgical repair and pulmonary vascular resistance index > 500 dyn·s·cm−5 were enrolled in this study. At the time of enrolment, all patients had fixed increased pulmonary vascular resistance that precluded further surgical intervention. Care was taken to exclude other causes of pulmonary hypertension such as pulmonary arterial hypoplasia, hypoventilation, and lung or liver disease. Patients’ treatment regimens were not changed within the last month before enrolment and during the study period aside from the addition of bosentan. None were taking epoprostenol, glibenclamide, or cyclosporine to avoid drug interactions. No patient was taking calcium channel blockers, as they were unresponsive to vasodilators, and warfarin was used only for clinical indications. Epoprostenol was not used in this population due to the long term stability of patients with Eisenmenger’s syndrome. Bosentan was elected based on the encouraging data on idiopathic and autoimmune PAH.
Study protocol
This was an open label non-controlled study approved by the Institutional Review Committee and conducted according to institutional guidelines after written informed consent was obtained from the patient or their parent or guardian. Patients received an oral bosentan regimen according to their body weight (table 1) following previously published guidelines.13
Table 1.
Bosentan dosing regimen according to body weight
| Body weight | Initial dose | Maintenance dose |
| >40 kg | 62.5 mg twice daily | 125 mg twice daily |
| 20–40 kg | 31.25 mg twice daily | 62.5 mg twice daily |
| 10–20 kg | 31.25 mg daily | 31.25 mg twice daily |
All patients were evaluated before initiation and at week 16 of treatment with a history and clinical examination to define WHO functional class, with maximum and submaximal exercise capacity and with a complete haemodynamic study. Treatment safety was evaluated bimonthly by monitoring adverse events such as flushing and nasal congestion, laboratory tests such as liver function tests and haemoglobin concentration, ECG, vital signs, and pulse oximetry recordings, and premature discontinuations.
Maximum exercise capacity was evaluated with the treadmill Dargie protocol with continuous ECG, blood pressure and pulse oximetry monitoring, and breath by breath gas analysis for oxygen consumption measurements. Peak oxygen consumption, defined as the average of data collected during the final 15 seconds of exercise, duration of exercise, and exercise capacity in metabolic equivalents were recorded. Submaximal exercise capacity was assessed by the six minute walk test16 (six minute walking distance was calculated as the mean distance covered during two performed tests) with continuous pulse oximetry monitoring and recording of the patients’ Borg dyspnoea index17 experienced at completion of the test.
Cardiac catheterisation was performed after an eight hour fast under local anaesthesia with additional intravenous midazolam 0.1–0.2 mg/kg for the six younger patients. Pulmonary arterial, right atrial, and pulmonary capillary wedge pressures and systemic pressures were recorded. Systemic and pulmonary arterial and venous saturations were obtained to calculate cardiac outputs with the Fick principle and table derived oxygen consumption values.18 Oxygen saturations obtained from pulmonary veins when possible or from several pulmonary capillary wedge positions were used to calculate pulmonary venous oxygen content. For great artery level shunts, oxygen saturations from both distal pulmonary arteries were used to calculate pulmonary arterial oxygen content. Calculation of systemic flow was adequate, as oxygen saturations were equal throughout the systemic circuit in all patients. The transpulmonary pressure gradient was defined as the difference between mean pulmonary artery pressure and mean pulmonary capillary wedge or left atrial pressure. Pulmonary and systemic vascular resistance indices were calculated with the standard formula.19 Patients with transposition of the great arteries physiology, where the small pulmonary arteriovenous oxygen content difference and the increased bronchial blood flow may render pulmonary blood flow and resistance calculations relatively unreliable,20 were excluded from the initial statistical analysis of blood flow and resistance parameters.
Statistical analysis
The descriptive data are presented as the mean (SEM) with median and range as appropriate. Changes from baseline are reported with 95% confidence interval (CI). Significance of the differences from baseline to week 16 for the exercise and haemodynamic parameters was calculated with Student’s paired t test. Change in the WHO functional class from baseline to week 16 was analysed with Wilcoxon’s rank sum test. All p values were two tailed with the required level of significance p < 0.05.
RESULTS
Patients
Twenty one patients, 11 (52%) male and 10 (48%) female, aged 22 (3) years (range 6–43 years, median 19 years) with severe chronic PAH related to CHD were enrolled in this study. Table 2 describes the patients’ baseline clinical characteristics including diagnosis, sex, pulse oximetry, medications, and supplemental oxygen.
Table 2.
Clinical data
| Patient | Diagnosis | Age (years) | Sex | pOx | Symptoms | Medications | WHO class |
| 1 | Aortopulmonary window | 31 | F | 93% | DOE, cyanosis | None | III |
| 2 | VSD | 28 | M | 86% | DOE, cyanosis | Warfarin | III |
| 3 | Aortopulmonary window | 14 | M | 87% | DOE, cyanosis | None | III |
| 4 | VSD | 15 | M | 90% | DOE, cyanosis | None | III |
| 5 | DORV, VSD | 43 | M | 75% | DOE, cyanosis | Warfarin | IV |
| 6 | Aortopulmonary window | 19 | M | 85% | DOE, cyanosis | None | III |
| 7 | VSD | 9 | M | 96% | DOE, exertion cyanosis | None | II |
| 8 | VSD, PDA, Down’s syndrome | 6 | F | 95% | DOE | None | II |
| 9 | PDA | 40 | F | 96% | DOE | Digoxin, warfarin, furosemide | III |
| 10 | Congenitally corrected TGA, VSD | 10 | M | 98% | DOE | Furosemide | II |
| 11 | VSD, small PDA | 22 | F | 92% | DOE, cyanosis | Warfarin | III |
| 12 | Atrioventricular canal, Down’s syndrome | 30 | F | 74% | DOE, cyanosis | None | III |
| 13 | VSD | 35 | F | 66% | DOE, cyanosis | Warfarin | III |
| 14 | DORV, VSD | 22 | M | 90% | DOE, cyanosis | None | III |
| 15 | DORV, VSD, TGA | 19 | M | 82% | DOE, cyanosis | Warfarin | III |
| 16 | DILV, TGA | 16 | F | 78% | DOE, cyanosis | Warfarin | III |
| 17 | DILV, TGA | 9 | F | 80% | DOE, cyanosis | Digoxin, furosemide | II |
| 18 | ASD | 36 | F | 73% | DOE, cyanosis | Warfarin, oxygen | IV |
| 19 | VSD, PDA, repaired at 20 years | 43 | M | 95% | DOE, atrial flutter | Warfarin, amiodarone | IV |
| 20 | TGA, VSD, repaired at 15 days | 12 | M | 98% | DOE | None | II |
| 21 | VSD repaired at 8 months, ASD | 8 | F | 72% | DOE, cyanosis | Warfarin, oxygen | IV |
ASD, atrial septal defect; DILV, double inlet left ventricle; DOE, dyspnoea on exertion; DORV, double outlet right ventricle; F, female; M, male; PDA, persistent ductus arteriosus; pOx, pulse oximetry; TGA, transposition of the great arteries; VSD, ventricular septal defect; WHO, World Health Organization.
Patients 1 to 17 had PAH associated with uncorrected unrestrictive communications at the ventricular or great artery level and no pulmonary stenosis allowing complete mixing and equal pressures in the pulmonary and systemic circulation. Of those, patients 15 to 17 additionally had transposition of the great arteries physiology and were thus excluded from the initial statistical analysis of the blood flow and resistance parameters. Patient 18 had unrestrictive atrial level shunt with fixed increased pulmonary vascular resistance due to a > 35 mm atrial septal defect. Patients 7 to 10 had only left to right shunt at baseline and the rest had baseline right to left shunt and cyanosis, as in Eisenmenger’s syndrome. All patients with unrestrictive communications had evidence of congestive heart failure during infancy, which disappeared gradually as pulmonary vascular resistance increased. Patients 19 to 21 were born with unrestrictive ventricular and great artery level communications allowing complete mixing and systemic pulmonary hypertension; all their defects, except for an atrial septal defect in patient 21, had been surgically repaired. All patients were clinically evaluated and performed the six minute walk test. All patients except the two youngest (patients 8 and 21) and the adult patient with Down’s syndrome (patient 12) underwent treadmill exercise testing with oxygen consumption measurements. Haemodynamic parameters were assessed invasively in all patients except patients 11 to 14 and 21, who declined the assessment.
Baseline characteristics
Most patients (76%) were significantly symptomatic (12 patients in WHO functional class III and four patients in class IV) except for five patients less than 12 years of age who were in WHO class II (table 2). Most patients (71%) were cyanotic with pulse oximetry < 95% at rest in room air. Four patients with unrestrictive mixing but absence of right to left shunt (patients 7 to 10) and the two patients with complete repair (patients 19 and 20) had normal baseline pulse oximetry (table 2). No patient had significant iron deficiency. All patients had severe right ventricular dilatation and hypertrophy on echocardiography with moderate to severe tricuspid regurgitation. Baseline maximum and submaximal exercise capacity and haemodynamic parameters were significantly impaired with median mean pulmonary artery pressure 92 mm Hg (range 57–105 mm Hg), median systemic blood flow index 2.5 l/min/m2 (range 1.6–3.8 l/min/m2), and median pulmonary vascular resistance index 2048 dyn·s·cm−5 (range 762–4387 dyn·s·cm−5).
Treatment effect
Figures 1 and 2 depict the changes from baseline observed after 16 weeks of bosentan treatment in the WHO classification, exercise tolerance, pulse oximetry, and haemodynamic status. At week 16, 13 patients had improved WHO functional class by one (the four patients in class IV improved to III and nine in class III improved to II), and two patients in class III and the five in class II remained stable (fig 1). All patients reported “feeling significantly better” during treatment and seven patients reported less cyanosis, especially during exercise.
Figure 1.
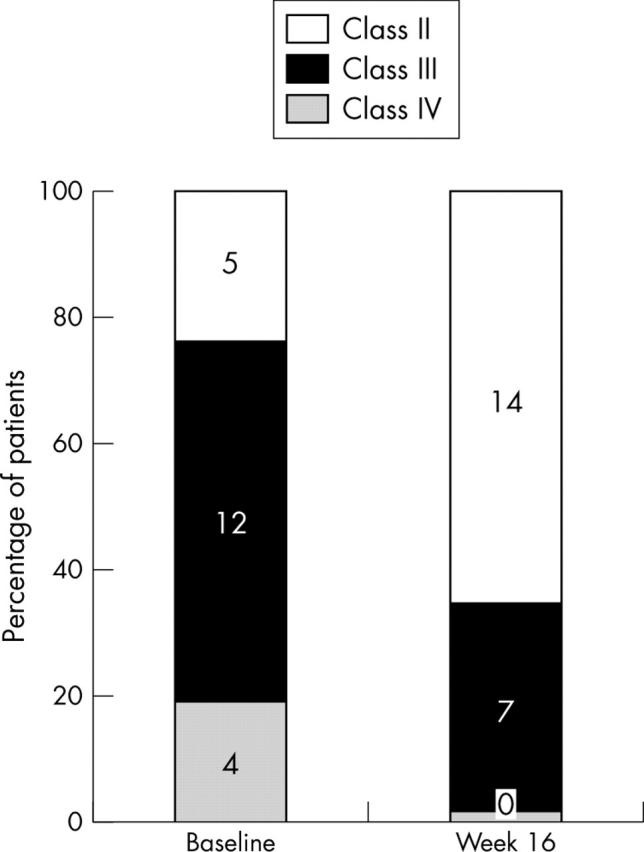
Change in World Health Organization functional class from baseline to week 16 of bosentan treatment (higher classes indicate greater severity of disease). Number of patients in each class are indicated inside the bars.
Figure 2.
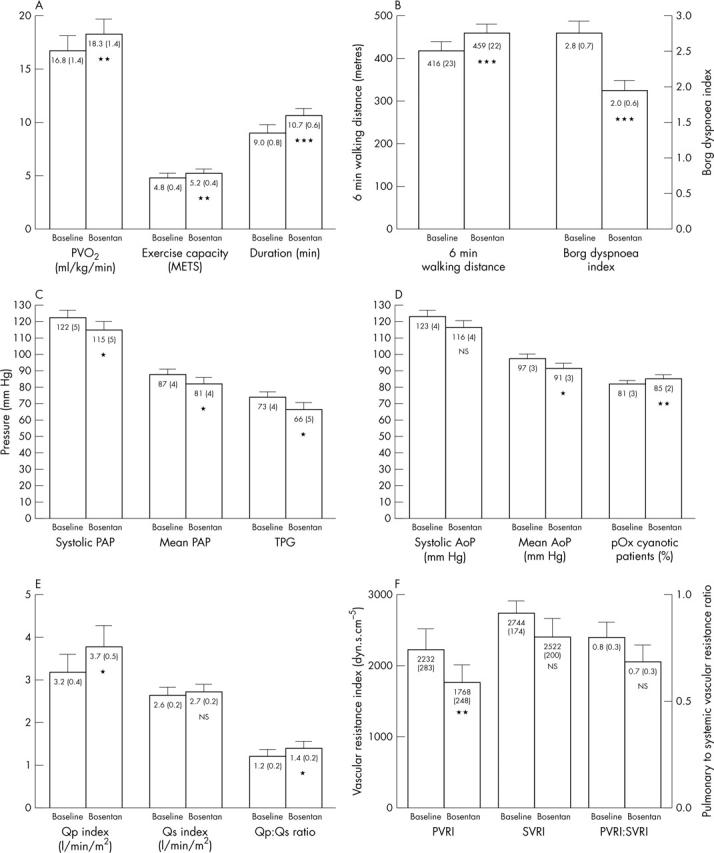
Change in exercise (A, B) and haemodynamic parameters (C, D, E, F) from baseline to week 16 of bosentan treatment. Numbers inside bars are mean (SEM). AoP, aortic pressure; METS, metabolic equivalents; NS, not significant; PAP, pulmonary artery pressure; pOx, pulse oximetry; PVo2, peak oxygen consumption; PVRI, pulmonary vascular resistance index; Qp, pulmonary blood flow; Qs, systemic blood flow; SVRI, systemic vascular resistance index; TPG, transpulmonary pressure gradient. *p < 0.05 v baseline; **p < 0.01 v baseline; ***p < 0.0001 v baseline.
Exercise evaluation showed, compared with baseline, an increase in peak oxygen consumption by 1.5 ml/kg/min (95% CI 0.4 to 2.5 ml/kg/min), in exercise capacity by 0.4 metabolic equivalents (95% CI 0.1 to 0.7), and in duration by 1.7 minutes (95% CI 1.1 to 2.3 minutes) during the Dargie treadmill protocol (fig 2). The ventilatory equivalent for carbon dioxide (minute ventilation/carbon dioxide production) at anaerobic threshold also decreased significantly (p < 0.05) from 59 (4) to 53 (4) (95% CI 1.4 to 10.3). During the six minute walk test, patients increased their walk distance by 42 m (95% CI 27 to 58 m) and reduced their Borg dyspnoea index by 0.6 (95% CI 0.5 to 1.1) (fig 2). The cyanotic patients also had increased (p < 0.05) pulse oximetry from 64 (3) to 69 (3)% (95% CI 0.4% to 10%) at test completion.
Pulse oximetry in room air improved in all 15 cyanotic patients by 4% (95% CI 1.1% to 6%) (fig 2). Haemodynamic assessment showed significant (p < 0.05) improvement, compared with baseline, in systolic pulmonary artery pressure by 7 mm Hg (95% CI 1 to 14 mm Hg), in mean pulmonary artery pressure by 6 mm Hg (95% CI 1 to 11 mm Hg), in transpulmonary pressure gradient by 8 mm Hg (95% CI 2 to 13 mm Hg), in mean aortic pressure by 6 mm Hg (95% CI 1 to 11 mm Hg), in pulmonary blood flow index by 0.6 l/min/m2 (95% CI 0.1 to 1.0 l/min/m2), in pulmonary to systemic blood flow ratio by 0.2 (95% CI 0.02 to 0.35), and in pulmonary vascular resistance index by 464 dyn·s·cm−5 (95% CI 189 to 738 dyn·s·cm−5) (fig 2). Right atrial pressure, systemic and effective blood flow, systemic oxygen delivery, systemic vascular resistance index, and pulmonary to systemic vascular resistance ratio did not change significantly. Haemodynamic improvement was more pronounced in the four patients without baseline cyanosis (patients 7 to 10), whose pulmonary vascular resistance index decreased by 20–53% to 472–1120 dyn·s·cm−5 and pulmonary to systemic vascular resistance ratio decreased to 0.3 to 0.5.
Analysis of the haemodynamic data including patients with transposition of the great arteries showed the same results as above except for an absence of change in the pulmonary blood flow and pulmonary to systemic flow ratio, probably due to the inherent problems in flow calculations in this population. The effect observed did not differ significantly between the three patients who had undergone surgical intervention and the rest of the population.
Safety and tolerability
Flushing and dizziness were reported once each but resolved within two weeks without regimen changes. Abnormal hepatic function, interference with the international normalised ratio, and other adverse events were not observed in this study. Haemoglobin concentration decreased significantly (p < 0.001) from 173 (6) to 160 (6) g/l. No patient required phlebotomy or red cell depletion during the study.
Two patients (patients 5 and 21) in this cohort died suddenly nine and five months, respectively, after starting bosentan treatment. Both deaths were observed, occurred after complains of palpitations, and were presumably due to arrhythmia. Both patients had no previously documented arrhythmia and had moved from WHO class IV to class III with treatment reporting subjective improvement and return to daily activities (work and school, respectively) that they were unable to do before. Both had mildly increased their six minute walking distance from 349 and 187 m to 366 and 207 m, respectively, at 16 weeks of treatment. Patient 21 had declined haemodynamic assessment; haemodynamic evaluation of patient 5 at 16 weeks of treatment showed stable pulmonary and aortic pressures with improved resting pulmonary blood flow index from 2.7 to 4.2 l min/m2, pulmonary to systemic blood flow ratio from 1.2 to 1.8, pulmonary vascular resistance index from 2248 to 1512 dyn·s·cm−5, and pulmonary to systemic vascular resistance ratio from 0.61 to 0.47. Postmortem examination showed generalised myocardial ischaemia as a recent terminal event consistent with possible arrhythmic death in the case of patient 5. Permission was not given to conduct a postmortem examination of patient 21.
The remaining 19 patients continued in stable clinical condition without changes in WHO functional class or exercise capacity on the same bosentan regimen during 1.3 (0.1) years (range 0.6 to 1.5 years) of follow up. Patient 1 reported disappearance of her bimonthly mild haemoptysis episodes and patient 18 was hospitalised once with her first moderate haemoptysis episode, which resolved with conservative treatment.
DISCUSSION
In this study, chronic oral administration of the non-selective endothelin receptor antagonist bosentan improved the clinical, exercise, and haemodynamic status of patients with PAH related to CHD. Factors affecting right ventricular performance, a key issue in PAH symptomatology, include increased afterload due to raised pulmonary pressure and vascular resistance, increased preload due to tricuspid regurgitation, and right ventricular ischaemia due to right ventricular hypertrophy and increased wall stress, which increase myocardial oxygen demand.21 Decreased right ventricular afterload may lead to improved right ventricular wall stress and performance, as manifested by the improved clinical and exercise status of our patients. This observation is in accordance with the improved right ventricular systolic function and size seen with bosentan in idiopathic PAH.22 Furthermore, the increased baseline pulse oximetry and blood oxygen content of our cyanotic patients may have contributed to the improved performance during exercise, a state of increased oxygen consumption.
The small number of studies showing benefit of endothelin antagonism in PAH enrolled only few patients with PAH due to CHD14 or administered endothelin A antagonists in the acute12 or chronic setting.23 The only previous study showing beneficial effect of bosentan in paediatric PAH included few patients with CHD, none with cyanosis and Eisenmenger’s syndrome.13 The present study is, to our knowledge, the first to examine in detail the clinical, exercise, and haemodynamic effects of chronic oral endothelin antagonism in patients with PAH associated with CHD, the majority of whom had Eisenmenger’s syndrome.
It is unclear whether treatment of pulmonary vascular disease is beneficial in PAH related to CHD; therefore, management should probably address the clinical condition, exercise capacity, and quality of life of this population. Conventional treatment including digitalis, diuretics, supplemental oxygen, warfarin, and calcium channel blockers has limited benefit, and lung transplantation presents issues with donor availability, correct timing for listing, and increased perioperative risk in this older patient population.24 Only continuous intravenous epoprostenol has been shown to improve haemodynamic status, exercise capacity, and quality of life in patients with PAH related to CHD but is associated with considerable inconvenience and morbidity from the indwelling catheter.25 The orally active bosentan benefited patients with PAH related to CHD in this study and its effects were sustained over one year in patients with idiopathic or autoimmune PAH.26 This treatment also prevented or reversed PAH and promoted pulmonary vascular remodelling in an animal model.27 PAH related to CHD possibly presents a novel indication for bosentan administration. Bosentan constitutes a promising treatment agent for a patient population with very limited treatment options. Further studies are warranted to assess whether bosentan will have long term effect and survival benefit in this population.
Two theoretical considerations arise with endothelin antagonism in PAH related to CHD. Firstly, in patients with left to right shunt, a decrease in pulmonary vascular resistance may lead to increased pulmonary blood flow and aggravation of congestive heart failure.13 This study included four such patients (patients 7 to 10), in whom clinical, exercise, and haemodynamic improvements were noted without need for additional anticongestive treatment. More important, pulmonary vascular resistance index and pulmonary to systemic vascular resistance ratio decreased significantly in these patients to values close to those permitting surgical repair in young infants and toddlers, considered not to have fixed pulmonary vascular resistance. Partial closure of CHD communications may be considered for such young patients if further long term endothelin antagonism studies prove sustained and possibly augmented improvement in pulmonary haemodynamic function.
The second theoretical consideration in patients with fixed increased pulmonary vascular resistance, as in Eisenmenger’s syndrome, is whether endothelin antagonism would act only on the systemic vascular bed without affecting the pulmonary vascular bed resulting in decreased systemic vascular resistance and increased right to left shunt and cyanosis. In this study, endothelin receptor antagonism was probably more selective to the pulmonary than the systemic vascular bed, as shown by the increased aortic saturation of our cyanotic patients and the improved pulmonary blood flow, pulmonary to systemic blood flow ratio, and pulmonary vascular resistance without change in the systemic vascular resistance. Pulmonary to systemic vascular resistance ratio did not change significantly, possibly due to the small values of this parameter.
The two patients who died suddenly, probably of arrhythmic causes, were initially in WHO functional class IV with very poor prognosis. Interestingly, both patients had had subjective and objective clinical, exercise, haemodynamic, and quality of life improvements during treatment with bosentan. Arrhythmia has not been reported with bosentan treatment in the literature and endothelin antagonists may in fact block ischaemia induced arrhythmias in experimental models.28 The rest of our population did not report palpitations, nor did these two patients before their deaths. Although the role of increased activity of these two patients in their sudden death remains unclear, extreme caution should be exercised when applying novel treatments manipulating pulmonary and systemic haemodynamic function in this very sensitive population, especially in the case of patients in WHO functional class IV.
Study limitations
Limitations of this study include the lack of a placebo group, the relatively small sample size, and the age and diagnosis heterogeneity of the study population. Despite the lack of a placebo group, which may have influenced end points such as WHO functional classification, exercise performance, and Borg dyspnoea index, the observed effects were consistent and persisted over a significant follow up period, suggesting a true haemodynamic effect. Use of assumed oxygen consumption, especially for patients with PAH with possibly significantly increased measured values, may create problems in flow and resistance haemodynamic calculations but not their ratios. Against these limitations, this study reports in detail the clinical, exercise, and haemodynamic effects of the non-selective endothelin antagonist bosentan in the infrequently studied population of PAH related to CHD and especially Eisenmenger’s syndrome. It does not, however, definitely show reversibility of PAH leading to operability of patients with Eisenmenger’s syndrome.
In conclusion, this study shows that chronic oral administration of the non-selective endothelin antagonist bosentan induces clinical, exercise, and haemodynamic improvements in patients with chronic PAH related to CHD, including Eisenmenger’s syndrome. Additional larger detailed studies with long term endothelin antagonism in patients with PAH related to CHD are needed to assess safety and duration of the effect and whether there may be a treatment role of endothelin antagonism in this challenging patient population.
Abbreviations
CHD, congenital heart disease
CI, confidence interval
PAH, pulmonary arterial hypertension
WHO, World Health Organization
There are no competing interests concerning this manuscript.
This study was conducted with approval of the Onassis Cardiac Surgery Centre Ethics Committee and after informed consent was obtained from the patients and their parents.
REFERENCES
- 1.Rubin LJ. Primary pulmonary hypertension. N Engl J Med 1997;336:111–7. [DOI] [PubMed] [Google Scholar]
- 2.MacGregor AJ, Canavan R, Knight C, et al. Pulmonary hypertension in systemic sclerosis: risk factors for progression and consequences for survival. Rheumatology (Oxford) 2001;40:453–9. [DOI] [PubMed] [Google Scholar]
- 3.Ivy D. Diagnosis and treatment of severe pediatric pulmonary hypertension. Cardiol Rev 2001;9:227–37. [DOI] [PubMed] [Google Scholar]
- 4.Daliento L, Somerville J, Presbitero P, et al. Eisenmenger syndrome: factors relating to deterioration and death. Eur Heart J 1998;19:1845–55. [DOI] [PubMed] [Google Scholar]
- 5.Wagenvoort CA, Wagenvoort N. Pathology of the Eisenmenger syndrome and primary pulmonary hypertension. Adv Cardiol 1974;11:123–30. [DOI] [PubMed] [Google Scholar]
- 6.Wort SJ, Woods M, Warner TD, et al. Endogenously released endothelin-1 from human pulmonary artery smooth muscle promotes cellular proliferation: relevance to pathogenesis of pulmonary hypertension and vascular remodeling. Am J Respir Cell Mol Biol 2001;25:104–10. [DOI] [PubMed] [Google Scholar]
- 7.Dupuis J, Cernacek P, Tardif JC, et al. Reduced pulmonary clearance of endothelin-1 in pulmonary hypertension. Am Heart J 1998;135:614–20. [DOI] [PubMed] [Google Scholar]
- 8.Allen SW, Chatfield BA, Koppenhafer SA, et al. Circulating immunoreactive endothelin-1 in children with pulmonary hypertension: association with acute hypoxic pulmonary vasoreactivity. Am Rev Respir Dis 1993;148:519–22. [DOI] [PubMed] [Google Scholar]
- 9.Channick RN, Simonneau G, Sitbon O, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet 2001;358:1119–23. [DOI] [PubMed] [Google Scholar]
- 10.Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002;346:896–903. [DOI] [PubMed] [Google Scholar]
- 11.Prendergast B, Newby DE, Wilson LE, et al. Early therapeutic experience with the endothelin antagonist BQ-123 in pulmonary hypertension after congenital heart surgery. Heart 1999;82:505–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Apostolopoulou SC, Rammos S, Kyriakides ZS, et al. Acute endothelin A receptor antagonism improves pulmonary and systemic haemodynamics in patients with pulmonary arterial hypertension that is primary or autoimmune and related to congenital heart disease. Heart 2003;89:1221–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barst RJ, Ivy D, Dingemanse J, et al. Pharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertension. Clin Pharmacol Ther 2003;73:372–82. [DOI] [PubMed] [Google Scholar]
- 14.Christensen DD, McConnell ME, Book WM, et al. Initial experience with bosentan therapy in patients with the Eisenmenger syndrome. Am J Cardiol 2004;94:261–3. [DOI] [PubMed] [Google Scholar]
- 15.Rich S, ed. Primary pulmonary hypertension: executive summary from the World Symposium on Primary Pulmonary Hypertension, Evian, France, 6–10 September 1998.
- 16.Guyatt GH, Sullivan MJ, Thompson PJ, et al. The 6-minute walk: a new measure of exercise capacity in patients with chronic heart failure. Can Med Assoc J 1985;132:919–23. [PMC free article] [PubMed] [Google Scholar]
- 17.Borg GA. Psychophysical bases of perceived exertion. Med Sci Sports Exerc 1982;14:377–81. [PubMed] [Google Scholar]
- 18.Bridges ND, Freed MD. Cardiac catheterization. In: Emmanouilides GC, Riemenscneider TA, Allen HD, et al, eds. Moss and Adams heart disease in infants, children, and adolescents. Baltimore: Williams & Wilkins, 1995:310–28.
- 19.Grossman W. Clinical measurement of vascular resistance and assessment of vasodilator drugs. In: Grossman W, ed. Cardiac catheterization and angiography. Philadelphia: Lea and Febiger, 1991:143–51.
- 20.Bush A, Busst CM, Knight WB, et al. Preoperative measurement of pulmonary vascular resistance in complete transposition of the great arteries. Br Heart J 1990;63:300–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gomez A, Bialostozky D, Zajarias A, et al. Right ventricular ischemia in patients with primary pulmonary hypertension. J Am Coll Cardiol 2001;38:1137–42. [DOI] [PubMed] [Google Scholar]
- 22.Galie N, Hinderliter AL, Torbicki A, et al. Effects of the oral endothelin-receptor antagonist bosentan on echocardiographic and Doppler measures in patients with pulmonary arterial hypertension. J Am Coll Cardiol 2003;41:1380–6. [DOI] [PubMed] [Google Scholar]
- 23.Barst RJ, Rich S, Widlitz A, et al. Clinical efficacy of sitaxsentan, an endothelin-A receptor antagonist, in patients with pulmonary arterial hypertension: open-label pilot study. Chest 2002;121:1860–8. [DOI] [PubMed] [Google Scholar]
- 24.Stoica SC, McNeil KD, Perreas K, et al. Heart-lung transplantation for Eisenmenger syndrome: early and long-term results. Ann Thorac Surg 2001;72:1887–91. [DOI] [PubMed] [Google Scholar]
- 25.Rosenzweig EB, Kerstein D, Barst RJ. Long-term prostacyclin for pulmonary hypertension with associated congenital heart defects. Circulation 1999;99:1858–65. [DOI] [PubMed] [Google Scholar]
- 26.Sitbon O, Badesch DB, Channick RN, et al. Effects of the dual endothelin receptor antagonist bosentan in patients with pulmonary arterial hypertension: a 1-year follow-up study. Chest 2003;124:247–54. [DOI] [PubMed] [Google Scholar]
- 27.Chen SJ, Chen YF, Meng QC, et al. Endothelin-receptor antagonist bosentan prevents and reverses hypoxic pulmonary hypertension in rats. J Appl Physiol 1995;79:2122–31. [DOI] [PubMed] [Google Scholar]
- 28.Burrell KM, Molenaar P, Dawson PJ, et al. Contractile and arrhythmic effects of endothelin receptor agonists in human heart in vitro: blockade with SB 209670. J Pharmacol Exp Ther 2000;292:449–59. [PubMed] [Google Scholar]