

Abstract
We have been developing FAMSBASE, a protein homology-modeling database of whole ORFs predicted from genome sequences. The latest update of FAMSBASE (http://daisy.nagahama-i-bio.ac.jp/Famsbase/), which is based on the protein three-dimensional (3D) structures released by November 2003, contains modeled 3D structures for 368,724 open reading frames (ORFs) derived from genomes of 276 species, namely 17 archaebacterial, 130 eubacterial, 18 eukaryotic and 111 phage genomes. Those 276 genomes are predicted to have 734,193 ORFs in total and the current FAMSBASE contains protein 3D structure of approximately 50% of the ORF products. However, cases that a modeled 3D structure covers the whole part of an ORF product are rare. When portion of an ORF with 3D structure is compared in three kingdoms of life, in archaebacteria and eubacteria, approximately 60% of the ORFs have modeled 3D structures covering almost the entire amino acid sequences, however, the percentage falls to about 30% in eukaryotes. When annual differences in the number of ORFs with modeled 3D structure are calculated, the fraction of modeled 3D structures of soluble protein for archaebacteria is increased by 5%, and that for eubacteria by 7% in the last 3 years. Assuming that this rate would be maintained and that determination of 3D structures for predicted disordered regions is unattainable, whole soluble protein model structures of prokaryotes without the putative disordered regions will be in hand within 15 years. For eukaryotic proteins, they will be in hand within 25 years. The 3D structures we will have at those times are not the 3D structure of the entire proteins encoded in single ORFs, but the 3D structures of separate structural domains. Measuring or predicting spatial arrangements of structural domains in an ORF will then be a coming issue of structural genomics.
Keywords: domain duplication, domain interactions, genome, homology modeling, P-loop, structural genomics
Introduction
Genome sequencing projects provided a huge number of amino acid sequences without functional information (Stein 2001). To discover biological functions of those proteins, both computational predictions and biochemical experiments are necessary (Tsoka and Ouzounis 2000). Most of the proteins perform functions after forming specific 3D structures, and therefore protein 3D structure is one of the most valuable sources of information to predict protein function (Domingues et al. 2000; Xie and Bourne 2005). Protein function prediction based on 3D structures, especially protein surface structures, with evolutionary and/or physicochemical characteristics have been extensively studied (Lichtarge and Sowa 2002; Campbell et al. 2003; Kinoshita and Nakamura 2003; Laskowski et al. 2003; Ota et al. 2003; Pieper et al. 2006). However, determining protein structures of all the function-unknown proteins for applying these types of study is not practical.
Proteins are classified into a large number of ‘families’ based on the amino acid sequence similarity (Dayhoff 1972), and proteins with similar amino acid sequences are known to have similar 3D structures (Chothia and Lesk 1986), all because the proteins in a family are evolutionary related (Doolittle 1995). Once we have 3D structure of at least one of the proteins in a family, then 3D structures of other proteins in the same family can be computationally deduced by ‘homology modeling’ (Burley 2000; Baker and Sali 2001). Based on this logic, structural genomics (SG) projects, which are to determine protein 3D structures of representatives for each family have been proposed and launched (Vitkup et al. 2001; Brenner 2000; Burley and Bonnano 2002). In homology modeling, corresponding residues between an amino acid sequence of structure unknown protein (target) and that of 3D structure known protein (template) in the same family are determined by sequence alignment and every residue in a template protein is replaced by that in a target protein (Marti-Renoma et al. 2000).
SG projects have been providing new protein structures (Todd et al. 2005; Xie and Bourne 2005; Chandonia and Brenner 2006). Protein Data Bank (PDB) (Berman et al. 2000) now contains more than 390 3D structures for function unknown or hypothetical proteins (Stark et al. 2004). Protein function predictions based on 3D structures determined by SG projects are also in progress (Goldsmith-Fischman and Honig 2003; Liu et al. 2005; Petrey and Honig 2005). There are some projects that focus on a specific species and try to determine the 3D structures of whole proteins encoded in the genome of the species (Kim 2000; Yokoyama et al. 2000; Kim et al. 2003). Those projects provide a considerable number of 3D structures in a single protein family. This results in providing multiple templates for a single protein family and it can improve quality of homology modeling (Contreras-Moreira et al. 2003).
We have developed FAMSBASE; a database for homology modeling 3D structures of whole proteins predicted on whole genome sequences, since 2001 (Yamaguchi et al. 2003; http://daisy.nagahama-i-bio.ac.jp/Famsbase/). FAMSBASE contains results of homology modeling by FAMS, a full automatic modeling software (Ogata and Umeyama 2000). Sequence alignments between whole ORFs and proteins in PDB are based on GTOP (Kawabata et al. 2002).
We report here the update of the database including differences in the amount of structural data from the previous version, estimation of the time that whole ORFs predicted out of genome sequences are covered by homology modeling 3D structures and upcoming issues for utilizing those modeled structures.
Methods
Data update of FAMSBASE
Correspondence between ORFs derived from whole genome sequences and protein amino acid sequences whose 3D structures are known is provided by GTOP database (Kawabata et al. 2002). The update in May 2005 of FAMSBASE is based on February 2004 version of GTOP. Protein 3D structures in PDB by November 2003 are used for homology modeling templates. FAMS (Ogata and Umeyama 2000) is applied by Umeyama et al. to pair-wise alignments between a predicted ORF sequence and an amino acid sequence with known 3D structure, and a 3D structure is modeled. All the results are stored in FAMSBASE.
Assessing annual difference of data in FASBASE
Based on the amount of data in FAMSBASE in 2001 and the amount of increase in the following years, a due year for whole proteome 3D structure models is estimated. Estimation is done residue-wise, not ORF-wise, since modeled structures in FAMSBASE are often limited to structural domains. In this report, structural domains refer to SCOP domains (Andreeva et al. 2004). All ORFs predicted out of genome sequences are divided into soluble and membrane proteins. The division is carried out by SOSUI (Hirokawa et al. 1998), and a protein with one or more transmembrane regions is classified into a membrane protein. The number of residues of whole soluble proteins encoded in the genome sequence (G) of species i is denoted as S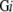 , and the number of residues of whole membrane proteins is denoted as M
, and the number of residues of whole membrane proteins is denoted as M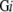 . The number of residues included in modeled 3D structures of soluble and membrane proteins are denoted as S3
. The number of residues included in modeled 3D structures of soluble and membrane proteins are denoted as S3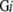 and M3
and M3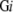 , respectively. For a certain genome Gi, the coverage of modeled 3D structures in whole soluble proteins is then S3
, respectively. For a certain genome Gi, the coverage of modeled 3D structures in whole soluble proteins is then S3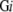 /S
/S
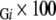 and the coverage for whole membrane proteins is M3
and the coverage for whole membrane proteins is M3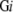 /M
/M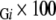 . The coverage is summarized in different kingdoms of life as in the following equations;
. The coverage is summarized in different kingdoms of life as in the following equations;
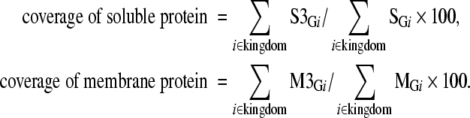 |
Both figures are calculated based on the data at the different times of FAMSBASE update, gradients in figures are then calculated, and the figures are extrapolated up to the year that coverage reaches to 100.
It is getting to be known that not all ORFs assume stable 3D structures. Some parts of ORFs are considered to be natively disordered (Oldfield et al. 2005; Dyson and Wright 2005). Hence it is unlikely that coverage by homology modeling reaches to 100. We, therefore, estimate disordered regions in whole ORFs by DisEMBL (Linding et al. 2003) and omit these disordered regions from the calculation.
Non-overlap multiple model structures in single ORFs
Modeled 3D structures in FAMSBASE are often limited to structural domains. To find an ORF of which most of the entire 3D structure is modeled in pieces of structural domains, an ORF covered by non-overlapping three or more modeled 3D structures in eukaryotic genome is surveyed based on the following criteria; (1) 70% or more residues in the ORF are included in one of the modeled 3D structures, (2) the ORF contains three or more non-overlapping modeled structures, and (3) the sequence identity between a template protein and a target domain is no less than 25%. At the time of FAMSBASE building, five model structures are at most built for each ORF (Yamaguchi et al. 2003). Therefore, the expected number of modeled structures in the above criteria is between three and five.
Prediction of domain interfaces
The 3D structure in pieces for a single ORF needs to be assembled to model the entire 3D structure. For this procedure, a prediction of domain interfaces of each 3D structure is needed. A hydrophobicity index based on protein 3D structures is built for domain interface prediction. Hydrophobicity of amino acid residue is measured by buriedness of a residue inside the protein 3D structures. A representative 4,529 chains in PDB among which sequence identities are less than 30% were selected and solvent accessibility of each residue is calculated on a monomer state. For each amino acid residue type i (i = 1,...,20), the number of residue with accessibility no less than b (=0.0 − 1.0) is counted (Sb,i). Database derived hydrophobicity index (Ib,i) is obtained by;
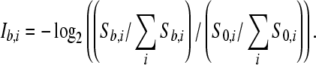 |
b is set to 0.15 to maximize the difference of Ib,i among different residues. The index has good correlation with Kyte and Doolittle hydrophobicity index (Kyte and Doolittle 1982). The index I0.15,i is assigned to every residue on the surface (accessibility no less than 0.15) of a modeled 3D structure. The hydrophobicity of each residue on a surface of a protein is then obtained by averaging the assigned values of residues within 7.0 Å from the residue in concern. A hydrophobic patch on the surface of the modeled structure is found as a cluster of surface residues with the hydrophobicity no less than 0.0.
Results and discussion
Coverage of whole protein space by homology modeling
The latest update of FAMSBASE at May 2005 uses protein 3D structures deposited to PDB by the end of Nov. 2003 and ORFs predicted from genome sequences deposited by February 2004 (http://daisy.nagahama-i-bio.ac.jp/ Famsbase/). The latest FAMSBASE contains 1,396,272 modeled 3D structures of 368,724 ORFs derived from 17 archaebacterial, 130 eubacterial, 18 eukaryotic and 111 phage genomes; in total 276 genomes. Five models at maximum are built for each ORF in FAMSBASE. Those five models are the structure for the same or different regions in the ORF. When multiple models are built for the same region of ORF, we can evaluate the reliability of the model. When the model based on different templates have the similar 3D structures, then the 3D structure would be reliable. When the structures are different, the modeled structure would be less reliable. We further test the quality of modeled 3D structure by ProsaII (Sippl 1993) and find that about 72% of the modeled 3D structures are energetically ranked as number one and comparable to experimentally determined 3D structures. Some of the structures that fail the test are structures of a part of a large protein, mostly structural domains of large proteins. It is difficult to assess the quality of this type of domain structures, because interfaces of the domain for other parts of the protein are exposed in the modeled structures. Tendency of amino acid residue appearance in the interface is supposed to be different from that at the surface as we discuss down below.
In the genome of 276 species, 734,193 ORFs are predicted. Therefore, in FAMSBASE, 3D structure of 50% (368,724/734,193) of ORFs have been built and stored (Table 1). These are about 47% of ORFs in archaebacterial genomes, about 52% in eubacterial genomes and about 49% of eukaryotic genomes.
Table 1.
Number of ORFs and those with modeled 3D structures in 276 genomes
| Species | ORF | Model | % |
|---|---|---|---|
| Archaea | |||
| Archaeoglobus fulgidus DSM4304 | 2,407 | 1,233 | 51.2 |
| Aeropyrum pernix K1 | 2,694 | 789 | 29.3 |
| Halobacterium sp. NRC-1 | 2,605 | 1,195 | 45.9 |
| Methanosarcina acetivorans C2A | 4,544 | 2,124 | 46.7 |
| Methanocaldococcus jannaschii DSM2661 | 1,770 | 875 | 49.4 |
| Methanopyrus kandleri AV19 | 1,687 | 784 | 46.5 |
| Methanosarcina mazei Goe1 | 3,371 | 1,634 | 48.5 |
| Methanothermobacter thermautotrophicus | 1,869 | 998 | 53.4 |
| Nanoarchaeum equitans Kin4-M | 536 | 264 | 49.3 |
| Pyrococcus abyssi Orsay | 1,784 | 942 | 52.8 |
| Pyrobaculum aerophilum IM2 | 2,605 | 1,047 | 40.2 |
| Pyrococcus furiosus DSM 3638 | 2,065 | 1,035 | 50.1 |
| Pyrococcus horikoshii OT3 | 2,061 | 879 | 42.6 |
| Sulfolobus solfataricus P2 | 2,994 | 1,365 | 45.6 |
| Sulfolobus tokodaii 7 | 2,826 | 1,228 | 43.5 |
| Thermoplasma acidophilum DSM1728 | 1,478 | 844 | 57.1 |
| Thermoplasma volcaniumGSS1 | 1,526 | 839 | 55.0 |
| sum | 38,822 | 18,075 | 46.6 |
| Eubacteria | |||
| Aquifex aeolicus VF5 | 1,553 | 929 | 59.8 |
| Nostoc sp. PCC 7120 | 6,132 | 2,765 | 45.1 |
| Agrobacterium tumefaciens C58 | 5,301 | 3,017 | 56.9 |
| A. tumefaciens C58 (Dupont) | 5,402 | 3,028 | 56.1 |
| Bacillus anthracis str. Ames | 5,311 | 2,463 | 46.4 |
| Buchnera aphidicola Sg | 552 | 410 | 74.3 |
| B. aphidicola | 507 | 385 | 75.9 |
| Bordetella bronchiseptica RB50 | 4,994 | 2,934 | 58.8 |
| Borrelia burgdorferi | 1,639 | 535 | 32.6 |
| Bacillus cereus ATCC 14579 | 5,255 | 2,534 | 48.2 |
| Candidatus Blochmannia floridanus | 583 | 447 | 76.7 |
| Bacillus halodurans C-125 | 4,066 | 2,127 | 52.3 |
| Bradyrhizobium japonicum | 8,317 | 4,449 | 53.5 |
| Bifidobacterium longum NCC2705 | 1,731 | 985 | 56.9 |
| Brucella melitensis 16M | 3,198 | 1,801 | 56.3 |
| Bordetella parapertussis | 4,185 | 2,525 | 60.3 |
| B. pertussis Tohama I | 3,447 | 2,179 | 63.2 |
| Bacillus subtilis 168 | 4,106 | 2,153 | 52.4 |
| Brucella suis 1330 | 3,264 | 1,677 | 51.4 |
| Bacteroides thetaiotaomicron VPI-5482 | 4,816 | 2,462 | 51.1 |
| Buchnera sp. APS | 574 | 436 | 76.0 |
| Clostridium acetobutylicum ATCC824 | 3,848 | 2,053 | 53.4 |
| Coxiella burnetii RSA 493 | 2,045 | 925 | 45.2 |
| Chlamydophila caviae GPIC | 1,005 | 505 | 50.2 |
| Caulobacter crescentus | 3,737 | 2,084 | 55.8 |
| Corynebacterium diphtheriae NCTC13129 | 2,272 | 1,165 | 51.3 |
| Corynebacterium efficiens YS-314 | 2,998 | 1,513 | 50.5 |
| Corynebacterium glutamicum ATCC 13032 | 3,099 | 1,554 | 50.1 |
| Campylobacter jejuni | 1,634 | 893 | 54.7 |
| Chlamydia muridarum Nigg | 911 | 483 | 53.0 |
| Clostridium perfringens 13 | 2,723 | 1,470 | 54.0 |
| Chlamydophila pneumoniae AR39 | 1,116 | 495 | 44.4 |
| Chlamydophila pneumoniae CWL029 | 1,052 | 496 | 47.1 |
| Chlamydophila pneumoniae J138 | 1,069 | 501 | 46.9 |
| Chlamydophila pneumoniae TW-183 | 1,113 | 501 | 45.0 |
| Chlorobium tepidum TLS | 2,252 | 1,166 | 51.8 |
| Clostridium tetani E88 | 2,432 | 1,306 | 53.7 |
| Chlamydia trachomatis D/UW-3/CX | 894 | 485 | 54.3 |
| Chromobacterium violaceum ATCC 12472 | 4,385 | 2,343 | 53.4 |
| Deinococcus radiodurans R1 | 3,102 | 1,579 | 50.9 |
| Escherichia coli K-12 MG1655 | 4,284 | 2,398 | 56.0 |
| E. coli O157:H7 | 5,447 | 2,607 | 47.9 |
| E. coli O157:H7 EDL933 | 5,449 | 2,629 | 48.2 |
| E. coli CFT073 | 5,379 | 2,558 | 47.6 |
| Enterococcus faecalis V583 | 3,265 | 1,568 | 48.0 |
| Fusobacterium nucleatum ATCC 25586 | 2,067 | 1,011 | 48.9 |
| Geobacter sulfurreducens PCA | 3,445 | 1,902 | 55.2 |
| Gloeobacter violaceus PCC 7421 | 4,430 | 2,208 | 49.8 |
| Haemophilus ducreyi 35000HP | 1,717 | 865 | 50.4 |
| Helicobacter hepaticus ATCC 51449 | 1,875 | 902 | 48.1 |
| Haemophilus influenzae Rd | 1,709 | 1,038 | 60.7 |
| Helicobacter pylori 26695 | 1,566 | 741 | 47.3 |
| Helicobacter pylori J99 | 1,491 | 747 | 50.1 |
| Listeria innocua Clip11262 | 3,043 | 1,641 | 53.9 |
| Leptospira interrogans serovar | 4,725 | 1,719 | 36.4 |
| Lactococcus lactis IL1403 | 2,266 | 1,254 | 55.3 |
| Listeria monocytogenes EGD-e | 2,846 | 1,653 | 58.1 |
| Lactobacillus plantarum WCFS1 | 3,009 | 1,647 | 54.7 |
| Mycobacterium bovis subsp. | 3,920 | 2,018 | 51.5 |
| Mycoplasma gallisepticum R | 726 | 371 | 51.1 |
| Mycoplasma genitalium G37 | 480 | 305 | 63.5 |
| Mycobacterium leprae TN | 1,605 | 918 | 57.2 |
| Mesorhizobium loti MAFF303099 | 7,281 | 3,829 | 52.6 |
| Mycoplasma penetrans | 1,037 | 472 | 45.5 |
| Mycoplasma pneumoniae M129 | 688 | 333 | 48.4 |
| Mycoplasma pulmonis UAB CTIP | 782 | 398 | 50.9 |
| Mycobacterium tuberculosis H37Rv | 3,918 | 2,036 | 52.0 |
| Mycobacterium tuberculosis CDC1551 | 4,187 | 1,990 | 47.5 |
| Nitrosomonas europaea ATCC 19718 | 2,461 | 1,366 | 55.5 |
| Neisseria meningitidis MC58 | 2,025 | 1,016 | 50.2 |
| Neisseria meningitidis Z2491 | 2,065 | 1,025 | 49.6 |
| Oceanobacillus iheyensis HTE831 | 3,496 | 1,892 | 54.1 |
| Phytoplasma asteris, OY strain | 754 | 423 | 56.1 |
| Pseudomonas aeruginosa PAO1 | 5,566 | 3,206 | 57.6 |
| Porphyromonas gingivalis W83 | 1,909 | 944 | 49.4 |
| Photorhabdus luminescens laumondii | 4,683 | 2,286 | 48.8 |
| Prochlorococcus marinus MED4 | 1,712 | 933 | 54.5 |
| Prochlorococcus marinus MIT9313 | 2,265 | 1,122 | 49.5 |
| Prochlorococcus marinus marinus | 1,882 | 939 | 49.9 |
| Pasteurella multocida PM70 | 2,014 | 1,237 | 61.4 |
| Pseudomonas putida KT2440 | 5,350 | 2,968 | 55.5 |
| Pseudomonas syringae pv. tomato str. | 5,608 | 2,938 | 52.4 |
| Pirellula sp. 1 | 7,325 | 2,588 | 35.3 |
| Rickettsia conorii Malish 7 | 1,374 | 572 | 41.6 |
| Rhodopseudomonas palustris | 4,814 | 2,739 | 56.9 |
| Rickettsia prowazekii Madrid E | 834 | 498 | 59.7 |
| Ralstonia solanacearum GMI1000 | 5,116 | 2,698 | 52.7 |
| Streptococcus agalactiae | 2,124 | 1,159 | 54.6 |
| Streptococcus agalactiae NEM316 | 2,094 | 1,174 | 56.1 |
| Staphylococcus aureus Mu50 | 2,748 | 1,451 | 52.8 |
| Staphylococcus aureus N315 | 2,624 | 1,447 | 55.1 |
| Staphylococcus aureus MW2 | 2,659 | 1,410 | 53.0 |
| Streptomyces avermitilis | 7,671 | 4,001 | 52.2 |
| Streptomyces coelicolor A3(2) | 8,154 | 4,195 | 51.4 |
| Staphylococcus epidermidis ATCC 12228 | 2,485 | 1,303 | 52.4 |
| Shigella flexneri 2a 301 | 4,452 | 2,306 | 51.8 |
| Shigella flexneri 2a str. 2457T | 4,068 | 2,159 | 53.1 |
| Sinorhizobium meliloti 1021 | 6,205 | 3,499 | 56.4 |
| Streptococcus mutans UA159 | 1,960 | 1,136 | 58.0 |
| Shewanella oneidensis MR-1 | 4,778 | 2,291 | 47.9 |
| Streptococcus pneumoniae R6 | 2,094 | 1,101 | 52.6 |
| Streptococcus pneumoniae TIGR4 | 2,043 | 1,135 | 55.6 |
| Streptococcus pyogenes SF370 | 1,696 | 956 | 56.4 |
| Streptococcus pyogenes MGAS8232 | 1,845 | 996 | 54.0 |
| Streptococcus pyogenes MGAS315 | 1,865 | 986 | 52.9 |
| Streptococcus pyogenes SSI-1 | 1,861 | 976 | 52.4 |
| Salmonella typhi CT18 | 4,767 | 2,347 | 49.2 |
| Salmonella typhimurium LT2 | 4,554 | 2,457 | 54.0 |
| Salmonella enterica subsp. enterica | 4,323 | 2,263 | 52.3 |
| Synechocystis sp. PCC 6803 | 3,167 | 1,679 | 53.0 |
| Synechococcus sp. WH 8102 | 2,517 | 1,243 | 49.4 |
| Thermosynechococcus elongatus BP-1 | 2,475 | 1,303 | 52.6 |
| Thermotoga maritima MSB8 | 1,846 | 1,051 | 56.9 |
| Treponema pallidum subsp. | 1,031 | 517 | 50.1 |
| Thermoanaerobacter tengcongensis MB4T | 2,588 | 1,403 | 54.2 |
| Tropheryma whipplei TW08/27 | 783 | 494 | 63.1 |
| Tropheryma whipplei str. Twist | 808 | 499 | 61.8 |
| Ureaplasma urealyticum | 611 | 303 | 49.6 |
| Vibrio cholerae N16961 | 3,828 | 1,971 | 51.5 |
| Vibrio parahaemolyticus RIMD 2210633 | 4,832 | 2,461 | 50.9 |
| Vibrio vulnificus CMCP6 | 4,537 | 2,461 | 54.2 |
| Vibrio vulnificus YJ016 | 5,028 | 2,499 | 49.7 |
| Wigglesworthia brevipalpis | 611 | 441 | 72.2 |
| Wolinella succinogenes DSMZ 1740 | 2,044 | 1,208 | 59.1 |
| Xanthomonas axonopodis pv. citri 306 | 4,427 | 2,374 | 53.6 |
| Xanthomonas campestris pv. campestris | 4,181 | 2,287 | 54.7 |
| Xylella fastidiosa 9a5c | 2,832 | 1,158 | 40.9 |
| Xylella fastidiosa Temecula1 | 2,036 | 1,066 | 52.4 |
| Yersinia pestis CO92 | 4,083 | 2,116 | 51.8 |
| Yersinia pestis KIM | 4,281 | 2,123 | 49.6 |
| sum | 396,126 | 206,311 | 52.1 |
| Eukaryotes | |||
| Arabidopsis thaliana | 28,723 | 14,394 | 50.1 |
| Caenorhabditis briggsae | 14,713 | 7,063 | 48.0 |
| Caenorhabditis elegans | 22,220 | 8,841 | 39.8 |
| Ciona intestinalis | 15,865 | 7,994 | 50.4 |
| Drosophila melanogaster | 18,302 | 9,541 | 52.1 |
| Danio rerio | 26,587 | 16,443 | 61.8 |
| Encephalitozoon cuniculi | 1,996 | 887 | 44.4 |
| Guillardia theta Nucleomorph | 632 | 307 | 48.6 |
| Homo sapiens (ENSEMBLE) | 28,063 | 15,467 | 55.1 |
| Leishmania major Friedlin | 173 | 62 | 35.8 |
| Mus musculus | 24,928 | 14,382 | 57.7 |
| Neurospora crassa | 10,088 | 3,800 | 37.7 |
| Oryza sativa | 16,724 | 4,517 | 27.0 |
| Plasmodium falciparum 3D7 | 5,268 | 1,905 | 36.2 |
| Rattus norvegicus | 28,682 | 16,740 | 58.4 |
| Saccharomyces cerevisiae | 5,869 | 2,913 | 49.6 |
| Schizosaccharomyces pombe | 5,261 | 2,807 | 53.4 |
| Takifugu rubripes rubripes | 37,452 | 15,202 | 40.6 |
| sum | 291,546 | 143,265 | 49.1 |
| Phages/Viruses | |||
| 186 | 46 | 8 | 17.4 |
| 44AHJD | 21 | 1 | 4.8 |
| 44RR2.8t | 252 | 51 | 20.2 |
| 933W | 80 | 9 | 11.3 |
| A118 | 72 | 9 | 12.5 |
| A511 | 11 | 0 | 0.0 |
| Aeh1 | 331 | 51 | 15.4 |
| APSE-1 | 54 | 6 | 11.1 |
| B1 | 11 | 1 | 9.1 |
| B103 | 17 | 4 | 23.5 |
| Bcep781 | 61 | 5 | 8.2 |
| BF23 | 8 | 1 | 12.5 |
| bIL170 | 64 | 2 | 3.1 |
| bIL285 | 62 | 5 | 8.1 |
| bIL286 | 61 | 7 | 11.5 |
| bIL309 | 56 | 6 | 10.7 |
| bIL310 | 29 | 4 | 13.8 |
| bIL311 | 22 | 6 | 27.3 |
| bIL312 | 27 | 3 | 11.1 |
| BK5-T | 63 | 6 | 9.5 |
| Bxb1 | 86 | 12 | 14.0 |
| C2 | 39 | 2 | 5.1 |
| Cp-1 | 28 | 2 | 7.1 |
| ϕCTX | 47 | 4 | 8.5 |
| D29 | 79 | 15 | 19.0 |
| D3 | 94 | 11 | 11.7 |
| Rb15 | 49 | 6 | 12.2 |
| ϕg1e | 49 | 6 | 12.2 |
| GA-1 | 35 | 3 | 8.6 |
| Gh-1 | 42 | 12 | 28.6 |
| H-19B | 22 | 4 | 18.2 |
| HF2 | 114 | 11 | 9.6 |
| HK022 | 57 | 8 | 14.0 |
| HK620 | 58 | 6 | 10.3 |
| HK97 | 61 | 10 | 16.4 |
| HP1 | 41 | 3 | 7.3 |
| HP2 | 36 | 3 | 8.3 |
| K139 | 44 | 4 | 9.1 |
| KVP40 | 381 | 57 | 15.0 |
| 2,389 | 57 | 7 | 12.3 |
| L-413C | 40 | 4 | 10.0 |
| L5 | 85 | 12 | 14.1 |
| λ | 66 | 18 | 27.3 |
| A2 | 61 | 8 | 13.1 |
| Mu | 53 | 6 | 11.3 |
| N15 | 60 | 13 | 21.7 |
| Mycoplasma virus P1 | 11 | 0 | 0.0 |
| Enterobacteria phage P1 | 11 | 0 | 0.0 |
| P2 | 42 | 5 | 11.9 |
| P22 | 36 | 9 | 25.0 |
| P27 | 58 | 9 | 15.5 |
| P335 | 49 | 6 | 12.2 |
| P4 | 12 | 2 | 16.7 |
| P60 | 80 | 13 | 16.3 |
| PA01 | 34 | 5 | 14.7 |
| PaP3 | 69 | 8 | 11.6 |
| ϕKZ | 306 | 25 | 8.2 |
| ϕCh1 | 98 | 9 | 9.2 |
| ϕYeO3-12 | 59 | 13 | 22.0 |
| ϕ105 | 51 | 8 | 15.7 |
| ϕC31 | 55 | 8 | 14.5 |
| ϕ3626 | 50 | 10 | 20.0 |
| ϕE125 | 71 | 12 | 16.9 |
| ϕETA | 66 | 8 | 12.1 |
| ϕNIH1.1 | 55 | 6 | 10.9 |
| ϕPV83 | 65 | 9 | 13.8 |
| ϕSLT | 62 | 12 | 19.4 |
| ϕadh | 63 | 8 | 12.7 |
| ϕBT1 | 55 | 9 | 16.4 |
| ϕA1122 | 50 | 10 | 20.0 |
| P68 | 22 | 2 | 9.1 |
| ϕKMV | 48 | 11 | 22.9 |
| PM2 | 22 | 1 | 4.5 |
| PRD1 | 22 | 4 | 18.2 |
| ΨM2 | 31 | 1 | 3.2 |
| ΨM100 | 37 | 4 | 10.8 |
| PY54 | 67 | 10 | 14.9 |
| PZA | 27 | 4 | 14.8 |
| R1t | 50 | 6 | 12.0 |
| RB69 | 256 | 56 | 21.9 |
| RB49 | 272 | 49 | 18.0 |
| Rd | 47 | 6 | 12.8 |
| RM378 | 146 | 17 | 11.6 |
| PVL | 62 | 8 | 12.9 |
| Sfi11 | 25 | 1 | 4.0 |
| V | 53 | 7 | 13.2 |
| SIO1 | 34 | 6 | 17.6 |
| Sk1 | 54 | 1 | 1.9 |
| SP6 | 20 | 6 | 30.0 |
| SP βc2 | 185 | 33 | 17.8 |
| SPP1 | 106 | 7 | 6.6 |
| MM1 | 53 | 6 | 11.3 |
| ST64B | 56 | 8 | 14.3 |
| ST64T | 65 | 9 | 13.8 |
| 7201 | 46 | 8 | 17.4 |
| DT1 | 47 | 7 | 14.9 |
| O1205 | 57 | 4 | 7.0 |
| Sfi19 | 45 | 6 | 13.3 |
| Sfi21 | 50 | 9 | 18.0 |
| Stx2 | 165 | 11 | 6.7 |
| T3 | 44 | 10 | 22.7 |
| T4 | 278 | 58 | 20.9 |
| T7 | 58 | 10 | 17.2 |
| TM4 | 89 | 5 | 5.6 |
| TP901-1 | 56 | 7 | 12.5 |
| Tuc2009 | 56 | 7 | 12.5 |
| Ul36 | 58 | 5 | 8.6 |
| VHML | 57 | 8 | 14.0 |
| VpV262 | 67 | 4 | 6.0 |
| VT2-Sa | 82 | 11 | 13.4 |
| Wϕ | 44 | 4 | 9.1 |
| Sum | 7,699 | 1073 | 13.9 |
| Total | 734,193 | 368,724 | 50.2 |
When a modeled 3D structure is counted based on the number of amino acid residues, not on the number of ORFs, a different aspect emerges. Figure 1 shows the percentage of amino acid residues per ORF included in the modeled structures. ORFs without a modeled structure are omitted. Of archaebacterial and eubacterial genomes, in 60% of ORFs, more than 80% of the residues are included in modeled 3D structures, however, of eukaryotic genomes, only in 30% of ORFs, more than 80% of the residues are included (red and blue sections in Fig. 1). The proportion of residues in modeled 3D structure can be measured by the number of residues in a typical structural domain as shown in SCOP (Andreeva et al. 2004). The average size of protein domain is around 100–150 residues (Copley et al. 2002). In ORFs with modeled structures, a continuous region of residues with one domain or more remains as structure unknown in only about 18% of ORFs of archaebacterial and eubacterial genomes, whereas in about 60% of ORFs of eukaryotic genomes, the regions with one domain or more remain as structure unknown.
Fig. 1.
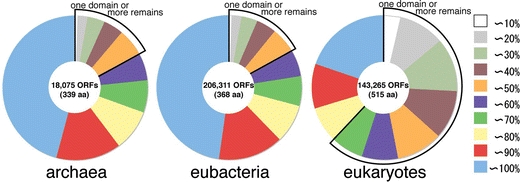
Percentage of amino acid residues included in modeled 3D structures in each ORF is classified by 10% bins and shown in pie charts. ORFs without a modeled structure are not included. A number of ORFs with modeled structures and an average length of the ORFs are shown at the center of each pie chart. Sections bordered by thick black lines indicate that the unmodeled region in the ORF is no less than the size of a domain (about 150 residues)
Annual difference of model structures
In FAMSBASE of 2001, 38% of amino acid residues in all ORFs of archaebacterial and 40% of eubacterial genomes were included in modeled 3D structures (Yamaguchi et al. 2003). In the current update of FAMSBASE based on data by around 2004, 42% of amino acid residues in all ORFs in archaebacterial and 46% of eubacterial genomes are included in modeled structures. In eukaryotic genomes, 24% of amino acid residues in 2003, and 26% in 2004 are included in modeled 3D structures. Those figures can be used to estimate the time when modeled 3D structures of whole proteins predicted from genomes are obtained. The estimation for the time obtaining the whole soluble and membrane proteins are treated separately, because the speed of structure determination for soluble and membrane proteins seems to differ. The assumption for the estimation is that the speed for structure determination would stay the same and no new protein family would appear.
For eubacterial genomes, 72.6% of residues in whole ORFs are predicted by SOSUI (Hirokawa et al. 1998) to encode soluble proteins and 27.4% to encode membrane proteins. This ratio is not so different from the previous prediction by Krogh et al. (2001). Of about 40% of whole eubacterial ORF that were with modeled 3D structures in 2001, approximately 90% were soluble proteins and 10% were membrane proteins. Therefore, about 50% (=0.40 × 0.90/0.726) of the whole soluble proteins were modeled. Of the whole membrane proteins in eubacterial genome, about 15% (=0.40 × 0.10/0.274) were modeled. In 2004, those figures are grown to 57% and 19%, respectively. In eubacterial whole ORFs, about 19.9% of amino acid residues are predicted to be included in disordered region by DisEMBL (Linding et al. 2003). Some of these regions are included in the modeled structures. These regions are either incorrectly predicted regions or incorrectly modeled regions. Assuming that the disordered regions without modeled 3D structures are correctly predicted, 10.8% of amino acid residues in soluble proteins were disordered and we would never obtain 3D structures of those regions. Then, by extrapolating the coverage of soluble proteins up to 89.2% (100–10.8) with the current growth rate, we can estimate that, by the year 2017, whole soluble proteins encoded in eubacterial genomes can be modeled (Fig. 2). Whole soluble proteins of archaebacterial genome can be modeled by 2021 and those of eukaryotic genomes, by 2031.
Fig. 2.
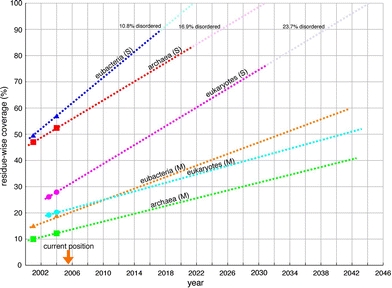
Annual differences of modeled structures classified by kingdoms of life. The percentage is the number of amino acid residues included in modeled structures over the whole number of residues in predicted sequences for soluble and membrane proteins in each kingdom. (S) stands for soluble proteins and (M) stands for membrane proteins. Some of the residues are predicted to be in a disordered region. The percentage of residues in disordered regions is shown at the top
Orengo et al. (1999) showed percentage of ORFs with protein 3D structures as between 30 and 46% in 1999. The genome sequences known by 1999 were mostly derived from prokaryotic species and the known protein 3D structures were mostly soluble proteins. Therefore, the figures they presented in 1999 should correspond to the figures of archaebacterial and eubacterial soluble proteins. When we extrapolate the figures of archaebacterial and eubacterial soluble proteins to the past in Fig. 2, the figures are around 40% in 1999, indicating that their figures approximately lie on the extrapolated lines.
The current estimation indicates that we will obtain 3D structures of whole soluble proteins of eubacteria in 11 years and archaebacteria in 15 years. This estimation does not take into account the acceleration of structure determination speed by automation (McPherson 2004; DeLucas et al. 2005), which makes the due days closer to the present. For membrane proteins, speed of structure determination has been drastically accelerated by recent technical innovations (Kyogoku et al. 2003; Lundstrom 2004; Walian et al. 2004; Dobrovetsky et al. 2005), and therefore we will not linearly extrapolate the present status to estimate the due day for membrane proteins.
Frequency of template structure in use
When the template 3D structures used in FAMSBASE are classified by SCOP superfamily, which is a group of proteins that have low sequence identities but whose structural and functional features suggest that a common evolutionary origin is probable (Lo Conte et al. 2002), and frequencies of superfamilies in use are counted, ‘P-loop containing nucleoside triphosphate hydrolases’ superfamily is found to be the most frequent one; 7,532 times (about 12%) in whole archaebacterial model structures, 77,806 (about 10%) in eubacterial structures and 35,468 times (about 6%) in eukaryotic structures. The templates that follow in frequency in archaebacterial and eubacterial protein structures are ‘NAD(P)-binding Rossmann fold domains’, ‘4Fe–4S ferredxin’, and ‘PLP-dependent transferases’ superfamilies. In eukaryotic protein structures, ‘protein-kinase’, ‘immunoglobulin’ and ‘C2H2 and C2HC zinc fingers’ superfamilies, which appear specifically in eukaryotic genomes, follow the top.
Differences in distribution of frequency of templates in different kingdoms of life are evident, when frequencies in use of template are plotted in descending order (Fig. 3). In any kingdoms of life, the frequencies of the most and the second most used templates exceed those of the remaining templates. The frequencies of templates in use drops first in archaebacterial protein structures and then in eubacterial protein structures. The descending curve of eukaryotic template frequency is less steep compared with the others, indicating that one template can produce a large number of domain 3D structures in eukaryotic ORFs. In other words, a significant number of proteins encoded in eukaryotic genomes are originated by domain duplication, as Koonin et al. (2000) demonstrated. Superfamilies with the 3D structures and with many copies in eukaryotic genomes, but seldom in prokaryotic genomes are ‘protein kinase-like’, ‘immunoglobin’, ‘RNA-binding domain’, ‘C2H2 and C2HC zinc fingers’, ‘WD40-repeat’, ‘glucocorticoid receptor-like’, ‘homeodomain-like’, ‘PH domain-like’, ‘RING-box’, ‘L domain’, ‘ankyrin repeat’, ‘ARM repeat’, ‘cytochrome P-450’ and ‘EF-hand’ superfamilies. These superfamilies are transcription factors, protein–protein interaction mediators and response factor for toxic substances, mostly known to be unique to eukaryotes.
Fig. 3.
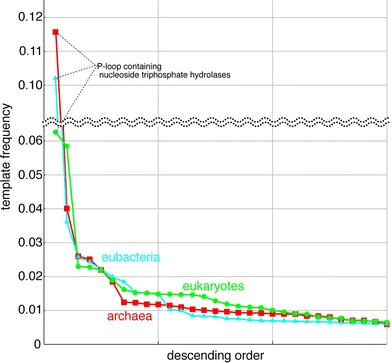
Frequency of template usage in descending order. Horizontal axis is a template and the vertical axis is a frequency of templates in use. Red line is a template usage in archaebacteria, blue line is eubacteria and green line is eukaryotes
The ‘P-loop containing nucleoside triphosphate hydrolases’ superfamily outnumbering other superfamily in template frequency corresponds to the previous finding that the enzyme is highly frequently used in every kingdom of life (Leipe et al. 2003). When biological functions of these ORFs with the 3D structure of ‘P-loop containing nucleoside triphosphate hydrolases’ superfamily are classified, about half of the proteins are ABC transporters in archaebacterial and eubacterial proteomes, but numbers of G-proteins and motor proteins in eukaryotic proteomes are noticeable (Fig. 4).
Fig. 4.
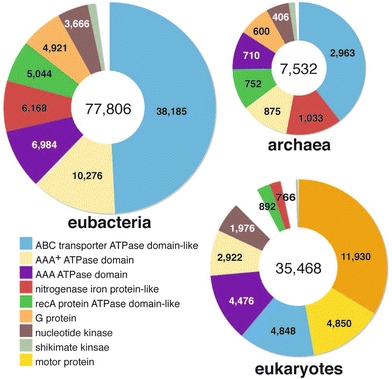
Protein family distribution of ‘P-loop containing nucleoside triphosphate hydrolases’ superfamily in each kingdom. In the three pie charts, the section with the same color is a category of the same family except for the white section
In the last two years, new protein structures were determined and contributed to an increase in the number of templates for homology modeling. A part of those template structures are listed in Table 2. A part of those top 15 templates contributed a lot for the growth of modeled 3D structure database. In Table 2, 3D structure derived from SG projects is rare. The ratio of SG products in Table 2 is the same as that in PDB (Editorial Board, Nature Structural & Molecular Biology 2004). As the SG projects in US and Europe have proceeded to phase 2 (Service 2005), SG products are expected to contribute to increase in the number of templates in the near future. The qualities of protein 3D structures, namely, size, resolution, R-factors and so forth, derived from SG projects were compared with those in PDB and no obvious compromise in quality of SG products were found (Todd et al. 2005). The quality of homology modeling based on products of SG projects in the future, therefore, will be expected to be no less than the current quality.
Table 2.
Top 15 modeling templates in the newly determined 3D structures between 2002 and 2003
| PDBID | Chain | Number of uses as a template | SGa | Protein name |
|---|---|---|---|---|
| 1q12 | A | 7,031 | N | Maltose/maltodextrin transport ATP-binding protein MalK |
| 1l2t | A | 6,529 | N | Hypothetical ABC transporter ATP-binding protein Mj0796 |
| 1oxx | K | 3,948 | N | ABC transporter ATP-binding protein GlcV |
| 1pf4 | A | 3,202 | N | Transport ATP-Binding Protein MsbA |
| 1nr0 | A | 2,640 | Y | Actin interacting protein 1 Aip1 |
| 1ixc | A | 2,495 | N | LysR-type regulatory protein CbnR |
| 1ld8 | A | 2,410 | N | Farnesyltransferase α subunit |
| 1ji0 | A | 2,331 | Y | ABC transporter |
| 1oyw | A | 2,251 | N | ATP-dependent DNA helicase; RecQ helicase |
| 1kt1 | A | 2,198 | N | Fk506-binding protein FKBP51 |
| 1mt0 | A | 1,961 | N | Haemolysin secretion ATP-binding protein; ATP-binding domain |
| 1mdb | A | 1,745 | N | 2,3-dihydroxybenzoate-AMP ligase DhbE |
| 1nnm | A | 1,730 | N | Acetyl-CoA synthetase |
| 1gxr | A | 1,715 | N | Transducin-like enhancer protein 1 Esg1 |
| 1uoh | A | 1,706 | N | 26S proteasome non-ATPase regulatory subunit 10 |
a PDB entry seemingly derived from the SG projects judged by description in PDB file is tagged Y, and the remaining entry is tagged N
Whole structure and function of proteins from homology modeling of domain structures
Protein function prediction, especially studies on enzyme specificity, based on homology modeling structures is intensively carried out in the field of drug design and related fields (Goldsmith-Fischman and Honig 2003; Kopp and Schwede 2004). Those studies are mostly based on homology modeling of domain structures. As mentioned above, most of the eukaryotic protein structures in FAMSBASE are 3D structures of structural domains, not the entire coding regions (Fig. 1). Protein functional sites are often located at a cleft of domains (Laskowski et al. 1996), and therefore understanding relative location of domains will be a critical issue. Xie and Bourne (2005) and O’Toole et al. (2003) also pointed out this problem and mentioned, “even if all the domains of a multiple-domain query sequence have determined structures, the individual structures will not enable accurate modeling of how they associate together in the structure of the entire proteins (O’Toole et al. 2003).”
Figure 5 shows all eukaryotic ORFs whose 3D structures are mostly modeled in pieces. There are three types of enzymes and four types of cell surface receptors. A protein structure of ENSP00000264705 which is an ORF found in human genome can be modeled based on Escherichia coli carbamoylphosphate synthetase (CPS) and Pyrococcus abyssi asparatate transcarbamoylase (ATC). E. coli CPS is composed of a large subunit and a small subunit. CPS and ATC are the first and the second enzymes, respectively, in pyrimidine biosynthesis pathway. In mammalian genomes, those proteins are coded by a single gene and active in a hexamer form (Serre et al. 2004). Interactions between the large subunit domain and the small subunit domain of human CPS are conjectured to be the same as those between the large and the small subunits of E. coli CPS. N-terminal residues of the large subunit and the C-terminal residues of the small subunit are spatially located close in E. coli CPS, which permits the two chains to be chemically connected without disrupting subunit interfaces. To be active, human CPS should form a hexamer supramolecule and the interfaces for the supramolecule formation should be predicted from the modeled 3D structures. At the moment, the interfaces are unknown.
Fig. 5.
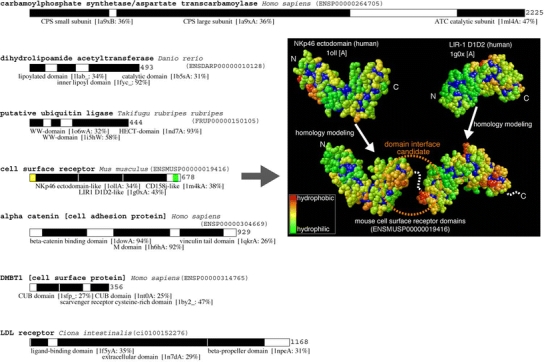
Eukaryotic ORFs with multiple model structures covering more than 70% of entire protein. In each of the bar representation of proteins, a black box is a region with 3D structure. A name and PDB ID of a template structure and amino acid sequence identity between template and target domains are given below the black box. A yellow box is a putative signal peptide and green box is a putative transmembrane region. Template and modeled structures of ENSMUSP00000019416 were shown on the right side of the figure. Each domain is colored by hydrophobicity. A hydrophilic residue is in green and a hydrophobic residue is in red. A buried residue is in deep blue
ENSMUSP00000019416 is an ORF found in mouse genome and encodes a putative cell surface receptor. The protein is predicted to consist of six consecutive Ig-fold domains. There is a putative transmembrane helix at the C-terminal region of the protein. Two consecutive Ig-fold domains are modeled without overlap, and no pieces of information for relative orientation of three modeled structures have been found. Information of interaction sites of those domains is required to build the entire structure of the protein and to predict a target molecule of this receptor. Computational analyses of domain interfaces and of protein–protein interfaces have been targets for extensive study for a long time, and some general characteristics have been found. One of them is the hydrophobicity of the interfaces (Wodak and Janin 2002). Hydrophobic clusters on the surface of modeled structures of ENSMUSP00000019416 are shown in right side of Fig. 5. One of the template structures, Nkp46 ectodomain, has hydrophilic surface (green) around the C-terminal residues of the domain, however the modeled structure has a hydrophobic surface (orange) at the corresponding area. The other template structure, LIR-1 D1D2, has a hydrophilic surface around the N-terminal residues of the domain, however the modeled structure has a hydrophobic surface at the corresponding area. The surfaces uniquely turned into hydrophobic in modeled structures are close to the residues that are chemically bonded in the target protein, and therefore both of the areas likely form interfaces of the two domains. The modeled structure based on LIR-1 D1D2 domain has another hydrophobic surface around the C-terminal residues, which may interact with CD158j-like domain located at the C-terminal side of the domain.
Accuracy of homology modeling
There are at least three major issues that affect accuracy in homology modeling; the best template selection, accuracy of an amino acid sequence alignment between template and target protein sequences and the accuracy of structure building procedure itself (Contreras-Moreira et al. 2005). Accuracy of the alignment is high, when sequence identity of template and target proteins is higher than 30%, and alignment of proteins with identity less than 30% is known to be less reliable, thereby accuracy of homology modeling deteriorates (Kopp and Schwede 2004). FAMS has been shown to construct relatively accurate model structures, even with low sequence identity between template and target sequences in CAFASP2, the homology modeling competition (Iwadate et al. 2001; Yamaguchi et al. 2003). A distribution of sequence identity between amino acid sequences of template and target proteins in FAMSBASE is shown in Fig. 6. Half of the model structures in FAMSBASE rely on alignments of sequence identity less than 20%. Figure 6 suggests that the current 3D structure database does not contain good enough structures for high quality homology modeling. SG projects will eventually provide better template structures, and improvement in target selection, alignment and modeling methods are also in pursuit to overcome the difficulties in homology modeling (John and Sali 2003; Wallace et al. 2005).
Fig. 6.
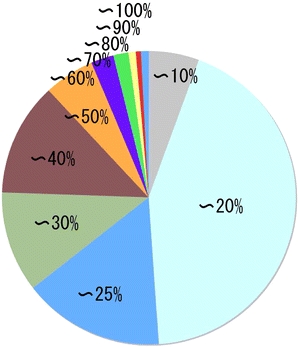
Distribution of sequence identity between template and target amino acid sequences in FAMSBASE
Conclusion
Construction of database of whole genome homology modeling clarified that protein 3D structures of about 50% of the protein coding regions in whole genome can now be modeled. Maintaining the current speed of 3D structure determination, it will take, at most, 11 years to have enough templates to cover whole soluble proteins of eubacterial genomes, and 25 years to cover those of eukaryotic genomes. The current advancement in technologies of protein structure determination is expected to make these due times closer to the present. What we obtain at those times are not the 3D structures of entire proteins, but domain structures in pieces. A homology modeled domain structure is now in use of predicting domain functions, but predicting spatial arrangement of domains in a protein will be an important issue for function prediction.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on Priority Area (C), Genome Information Science from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- Andreeva A, Howorth D, Brenner SE, Hubbard TJP, Chothia C, Murzin AG (2004) Nucleic Acids Res 32:D22 [DOI] [PMC free article] [PubMed]
- Baker D, Sali A (2001) Science 294:93 [DOI] [PubMed]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) Nucleic Acids Res 28:235 [DOI] [PMC free article] [PubMed]
- Brenner SE (2000) Nat Struct Biol 7(Suppl):967 [DOI] [PubMed]
- Burley SK (2000) Nat Struct Biol 7(Suppl):932 [DOI] [PubMed]
- Burley SK, Bonanno JB (2002) Annu Rev Genomics Hum Genet 3:243 [DOI] [PubMed]
- Campbell SJ, Gold ND, Jackson RM, Westhead DR (2003) Curr Opin Struct Biol 13:389 [DOI] [PubMed]
- Chandonia J-M, Brenner SE (2006) Science 311:347 [DOI] [PubMed]
- Chothia C, Lesk AM (1986) EMBO J 5:823 [DOI] [PMC free article] [PubMed]
- Contreras-Moreira B, Ezkurdia I, Valencia TA (2005) FEBS lett 579:1203 [DOI] [PubMed]
- Contreras-Moreira B, Fitzjohn PW, Bates PA (2003) J Mol Biol 328:593 [DOI] [PubMed]
- Copley RR, Ponting CP, Schultz J, Bork P (2002) Adv Protein Chem 61:75 [DOI] [PubMed]
- Dayhoff MO (1972) Atlas of protein sequence and structure, vol 5. National Biomedical Research, Foundation Georgetown University, Washington, DC
- DeLucas LJ, Hamrick D, Cosenza L, Nagy L, McCombs D, Bray T, Chait A, Stoops B, Belgovskiy A, William Wilson W (2005) Prog Biophys Mol Biol 88:285 [DOI] [PubMed]
- Dobrovetsky E, Lu ML, Andorn-Broza R, Khutoreskaya G, Bray JE, Savchenko A, Arrowsmith CH, Edwards AM, Koth CM (2005) J Struct Funct Genomics 6:33 [DOI] [PubMed]
- Domingues FS, Koppensteiner WA, Sippl MJ (2000) FEBS Lett 476:98 [DOI] [PubMed]
- Doolittle RF (1995) Annu Rev Biochem 64:287 [DOI] [PubMed]
- Dyson HJ, Wright PE (2005) Nat Rev Mol Cell Biol 6:197 [DOI] [PubMed]
- Editorial Board (2004) Nat Struct Mol Biol 11:201 [DOI] [PubMed]
- Goldsmith-Fischman S, Honig B (2003) Protein Sci 12:1813 [DOI] [PMC free article] [PubMed]
- Hirokawa T, Boon-Chieng S, Mitaku S (1998) Bioinformatics 14:378 [DOI] [PubMed]
- Iwadate M, Ebisawa K, Umeyama H (2001) Chem-Bio Infor J 1:136 [DOI]
- John B, Sali A (2003) Nucleic Acids Res 31:3982 [DOI] [PMC free article] [PubMed]
- Kawabata T, Fukuchi S, Homma K, Ota M, Araki J, Ito T, Ichiyoshi N, Nishikawa K (2002) Nucleic Acids Res 30:294 [DOI] [PMC free article] [PubMed]
- Kim S-H, Shin DH, Choi I-G, Schulze-Gahmen U, Chen S, Kim R (2003) J Struct Funct Genomics 4:129 [DOI] [PubMed]
- Kim S-H (2000) Curr Opin Struct Biol 10:380 [DOI] [PubMed]
- Kinoshita K, Nakamura H (2003) Curr Opin Struct Biol 13:396 [DOI] [PubMed]
- Koonin EV, Aravind L, Kondrashov AS (2000) Cell 101:573 [DOI] [PubMed]
- Kopp J, Schwede T (2004) Nucleic Acids Res 32:D230 [DOI] [PMC free article] [PubMed]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) J Mol Biol 305:567 [DOI] [PubMed]
- Kyogoku Y, Fujiyoshi Y, Shimada I, Nakamura H, Tsukihara T, Akutsu H, Odahara T, Okada T, Nomura N (2003) Acc Chem Res 36:199 [DOI] [PubMed]
- Kyte J, Doolittle RF (1982) J Mol Biol 157:105 [DOI] [PubMed]
- Laskowski RA, Watson JD, Thornton JM (2003) J Struct Funct Genomics 4:167 [DOI] [PubMed]
- Laskowski RA, Luscombe NM, Swindells MB, Thornton JM (1996) Protein Sci 5:2438 [DOI] [PMC free article] [PubMed]
- Leipe DD, Koonin EV, Aravind L (2003) J Mol Biol 333:781 [DOI] [PubMed]
- Lichtarge O, Sowa ME (2002) Curr Opin Struct Biol 12:21 [DOI] [PubMed]
- Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB (2003) Structure 11:1453 [DOI] [PubMed]
- Liu G, Li Z, Chiang Y, Acton T, Montelione GT, Murray D, Szyperski T (2005) Protein Sci 14:1597 [DOI] [PMC free article] [PubMed]
- Lo Conte L, Brenner SE, Hubbard TJ, Chothia C, Murzin AG (2002) Nucleic Acids Res 30:264 [DOI] [PMC free article] [PubMed]
- Lundstrom K (2004) Curr Opin Drug Discov Devel 7:342 [PubMed]
- Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A (2000) Annu Rev Biophys Biomol Struct 29:291 [DOI] [PubMed]
- McPherson A (2004) J Struct Funct Genomics 5:3 [DOI] [PubMed]
- Ogata K, Umeyama H (2000) J Mol Graph Model 18:258 [DOI] [PubMed]
- Oldfield CJ, Ulrich EL, Cheng Y, Dunker AK, Markley JL (2005) Proteins 59:444 [DOI] [PubMed]
- Orengo CA, Pearl FM, Bray JE, Todd AE, Martin AC, Lo Conte L, Thornton JM (1999) Nucleic Acids Res 27:275 [DOI] [PMC free article] [PubMed]
- Ota M, Kinoshita K, Nishikawa K (2003) J Mol Biol 327:1053 [DOI] [PubMed]
- O’Toole N, Raymond S, Cygler M (2003) J Struct Funct Genomics 4:47 [DOI] [PubMed]
- Petrey D, Honig B (2005) Mol Cell 20:811 [DOI] [PubMed]
- Pieper U, Eswar N, Davis FP, Braberg H, Madhusudhan MS, Rossi A, Marti-Renom M, Karchin R, Webb BM, Eramian D, Shen M-Y, Kelly L, Melo F, Sali A (2006) Nucleic Acids Res 34:D291 [DOI] [PMC free article] [PubMed]
- Serre V, Penverne B, Souciet JL, Potier S, Guy H, Evans D, Vicart P, Herve G (2004) BMC Biochem 5:6 [DOI] [PMC free article] [PubMed]
- Service R (2005) Science 307:1554 [DOI] [PubMed]
- Sippl MJ (1993) Proteins Struct Funct Genet 17:355 [DOI]
- Stark A, Shkumatov A, Russell RB (2004) Structure (Camb) 12:1405 [DOI] [PubMed]
- Stein L (2001) Nat Rev Genet 2:493 [DOI] [PubMed]
- Todd AE, Marsden RL, Thornton JM, Orengo CA (2005) J Mol Biol 348:1235 [DOI] [PubMed]
- Tsoka S, Ouzounis CA (2000) FEBS Letters 480:42 [DOI] [PubMed]
- Vitkup D, Melamud E, Moult J, Sander C (2001) Nat Struct Biol 8:559 [DOI] [PubMed]
- Walian P, Cross T, Jap B (2004) Genome Biol 5:215 [DOI] [PMC free article] [PubMed]
- Wallace IM, Blackshields G, Higgins DG (2005) Curr Opin Struct Biol 15:261 [DOI] [PubMed]
- Wodak SJ, Janin J (2002) Adv Protein Chem 61:9 [DOI] [PubMed]
- Xie L, Bourne PE (2005) PLoS Comput Biol 1:e31 [DOI] [PMC free article] [PubMed]
- Yamaguchi A, Iwadate M, Suzuki E, Yura K, Kawakita S, Umeyama H, Go M (2003) Nucleic Acids Res 31:463 [DOI] [PMC free article] [PubMed]
- Yokoyama S, Hirota H, Kigawa T, Yabuki T, Shirouzu M, Terada T, Ito Y, Matsuo Y, Kuroda Y, Nishimura Y, Kyogoku Y, Miki K, Masui R, Kuramitsu S (2000) Nat Struct Biol 7(Suppl):943 [DOI] [PubMed]