

Abstract
Purpose
Mutations in RPGRIP1 cause Leber congenital amaurosis. The human and bovine RPGRIP1 undergo alternative splicing. A single murine rpgrip1 transcript has been reported, but distinct expression profiles of RPGRIP1 isoforms exist between species in the retina. To elucidate the heterogeneity of RPGRIP1 isoforms and the degree of functional redundancy among these, we extended the analysis of RPGRIP1 to the region between exons 12 and 14, which undergoes significant alternative splicing.
Methods
Identification of alternative splice transcripts of murine and human RPGRIP1 was performed by reverse transcription–polymerase chain reaction (RT-PCR). The murine rpgrip1 isoforms were analyzed by immunoblot and immunocytochemistry analysis of murine retinas and transient transfected cultured cells.
Results
A novel murine-specific transcript, rpgrip1b was identified. It arises from the extension of exon 13, leading to the premature truncation of rpgrip1 and deletion of its C2 and RID domains. It is predominantly expressed in the retina, where it is more abundant than the transcript(s) encompassing the constitutive exons 12 to 14. Conversely, the human retina lacks rpgrip1b, and the constitutive transcript is the major isoform. The subcellular distribution of rpgrip1b is distinct from its larger isoform, rpgrip1. In the photoreceptor inner segments and cells expressing enhanced green fluorescent protein (EGFP)-rpgrip1b, rpgrip1b is dispersed as punctate foci throughout the perikarya, where it colocalizes with a subpopulation of lysosomes.
Conclusions
These data support the RPGR-independent function of the isotype- and species-specific rpgrip1b in lysosome-related processes. The results further strengthen the model of the selective participation of distinct RPGRIP1 isoforms in different subcellular processes and molecular pathogenesis of RPGRIP1-allied diseases.
Leber congenital amaurosis (LCA, Mendelian Inheritance in Man [MIM] 204000) is the most severe and the leading cause of blindness in children.1 Affected children exhibit strongly reduced or extinguished electroretinograms (ERGs), typically by 12 months of life. Several genes with diverse retinal function such as the aryl hydrocarbon receptor-interacting protein-like 1 (AILP1),2 Crumbs homolog 1 (CRB1),3,4 cone-rod homeobox (CRX),5 guanylate cyclase 2D (GUCYD2),6 RPE65,7 retinal dehydrogenase 12 (RDH12),8 and RPGRIP1,9,10 have been implicated in LCA and account for approximately 50% of cases. All mutations in RPGRIP1 lead only to LCA, whereas genetic lesions in the remaining genes also lead to other clinically heterogeneous retinal dystrophies with early postnatal and adult onsets.1
RPGRIP1 was originally found to be an interacting substrate of retinitis pigmentosa GTPase regulator (RPGR),11–14 thus implicating RPGRIP1 in the molecular pathogenesis of X-linked retinitis pigmentosa type 3 (XlRP3). All missense mutations in RPGR are clustered in its RCC1-homologous domain,15 and some of these have been shown to uncouple the interaction of RPGR with RPGRIP1.11 Human mutations in RPGR lead to retinitis pigmentosa (RP)16–19 and several other retinal degenerative diseases such as cone–rod,20 cone,21 and recessive atrophic macular degeneration.22 In addition, two distinct mutations (845-846delTG and G173R) in exon 8 of RPGR segregate with systemic disorders associated with hearing loss, sinusitis, and chronic recurrent respiratory tract and ear infections.23–26 Immunocytochemical analysis of human retina, bronchi, sinuses, and cochlea localized RPGR to the outer segments of photoreceptors and to nonocular tissues, such as the epithelial cells lining the lumen of the sinuses and bronchi cavities and the nonciliated cochlear tissues, stria vascularis, suprastrial cells, and spiral limbus.24 This finding is consistent with the manifestations of the ocular and systemic diseases described.23–26 Likewise, we have found that RPGR and RPGRIP1 isoforms localize to the outer segment of human and bovine photoreceptors,11,27 whereas in mouse photoreceptors, they localize to the connecting cilium.27,28 However, RPGRIP1 was also strongly expressed in a subset of inner retinal neurons, the amacrine cells.27,28 Hence, the differential expression of RPGR and RPGRIP1 among retinal neurons may provide a rationale for the distinct phenotypes caused by genetic lesions in RPGR and RPGRIP1 in the human.28 RPGRIP1 is subjected to significant alternative splicing in the human and bovine,11,13 and products thereof have been shown to be susceptible to various degrees of limited proteolysis, depending on the subcellular localization of RPGRIP1.28 This led to the proposal that the repertoire of RPGRIP1 products generated may mediate distinct functions and subcellular processes with pathologic outcomes still to be determined.28
To investigate further the implications of the heterogeneity of RPGRIP1 isoforms among and within species and the function of these in subcellular processes, we report the identification of novel murine- and human-specific RPGRIP1 isoforms with distinct expression profiles and subcellular properties.
Materials and Methods
All experiments described in the present study were performed in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and guidelines for the welfare of experimental animals issued by the National Institutes of Health and the Medical College of Wisconsin.
Tissue Sources, Primary Antibodies, and Reagents
Mice retinas were from 3- to 6-month-old C57Bl/6. All tissue manipulation procedures complied with institutional and federal guidelines. Antibodies against the murine rpgrip1b were raised in two hens (Aves Laboratories, Inc., Tigard, OR) against the keyhole limpet hemocyanin (KLH)-conjugated peptide, CZLPTSGKS (where Z is a molecular spacer). Anti-peptide ELISAs were performed before the affinity purification of the purified IgY from two hens. There was a > 1000-fold difference in the concentration of antibody recognizing the peptide sequence in the immune IgY fraction compared with the preimmune IgY fraction, thus indicating that the epitope sequence was very immunogenic. Half-maximum antibody binding occurred at 5 and 40 μg/mL for the purified IgY collected from hens 4227 and 4228. Purified and pooled IgY fractions were affinity-purified against the epitope peptide. Approximately 0.3% (~7 mg) of the original IgY represented the affinity-purified antibody. This antibody seems to require a free C-terminal end in rpgrip1b for proper antigen recognition. The affinity-purified antibodies, MCW3 and MCW4, Abs 22, and 38, respectively, against the C2 domain peptide sequence of RPGRIP1 and the coiled-coil N- and RID C-terminal domains of bovine RPGRIP1 have been described.11,27,28 Other primary antibodies were monoclonal anti-lysosome-associated membrane protein (Lamp-1; Stressgen Biotechnologies, Victoria, British Columbia, Canada), anti-Lamp2 (H4B4 and ABL-93; Developmental Hybridoma Bank, Department of Biological Sciences, University of Iowa, Iowa City, IA), rabbit anti-CI-MPR serum (gift of Nancy Dahms, Department of Biochemistry, Medical College of Wisconsin), rabbit anti-COP1 and -EEA1 (Affinity Bioreagents, Golden, CO). Anti-rabbit, -mouse, and -chicken secondary antibodies conjugated to Alexa 594 and Alexa 488 were from Molecular Probes (Eugene, OR). Prestained SDS-PAGE markers were from New England Biolabs (Beverly, MA).
Expression Profile Analysis by RT-PCR and PCR
RT-PCR analysis of mouse retina RNA and human retina mRNA was performed according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA). Singleplex RT-PCR of mouse retina RNA was performed with the primers P12 (5′-CTGCAGCGCAAAATCAAC), P13c (5′-CTGCCTCTGCCTACGTCC), and P12 and P14a/c (5′-CTGGGAGGTGAAGTCATAG). Multiplex RT-PCR was performed with equimolar amounts of primers P12, P13c, and P14a/c. PCR reactions with the same primers were used with samples of a mouse-normalized cDNA tissue panel (BD Biosciences, Franklin Lakes, NJ). The P12 primer sequence is the same for the PCR reactions with murine and human samples. The primers P14b (5′-CATCTCCATGGGCTGGCAGTG) and P13d14b (5′-GTGCCATAAGCAACATCTTTGAGCTGTTCT) were used in PCR reactions of human samples. For the RT-PCR reactions, 20 ng of mouse retinal RNA and 5 ng of human retinal mRNA were used in the following reaction condition: 48°C for 30 minutes, 94°C for 2 minutes followed by 38 cycles at 94°C for 30 seconds, 60°C for 45 seconds, and 68°C for 45 seconds, and a final elongation step at 68°C for 3 minutes. PCR reaction with cDNA panels were performed with 10 ng of cDNA, and cycling conditions were 94°C, 2 minutes; 94°C, 30 seconds; 60°C, 45 seconds; and 68°C, 45 seconds for 38 cycles, with a final elongation step at 68°C for 3 minutes.
Immunohistochemistry of Retinal Sections and Cultured Cells
Immunolocalization of the murine rpgrip1 and rpgrip1b were performed by similar procedures, as described previously.27,28,29 Murine dissected eyes were fixed with 2% paraformaldehyde in 1× phosphate-buffered saline (PBS) and by rotating these overnight at room temperature. Fixed eyes were washed three times in 1× PBS for 10 minutes each time and then they were infiltrated with 10% sucrose in 1× PBS at 4°C for 8 hours, followed by 30% sucrose in 1× PBS at 4°C overnight. Eyecups were then embedded in tissue-freezing medium (Tissue-Tek; Triangle Biomedical Sciences, Durham, NC), and sections (5–7 μm) were cut on a cryostat (Leica, Nussloch, Germany), mounted on gelatin-coated slides (Fisher Scientific, Pittsburgh, PA), and rinsed with 1× PBS for 10 minutes, three times at room temperature. Triton X-100 (0.1% vol/vol) was added to all incubations and washing buffers, and all incubations were done at room temperature. The affinity-purified antibodies MCW3 and MCW4 against the C2 domain of RP-GRIP1 have been described.27,28 The sections were first incubated with affinity-purified rabbit polyclonal antibodies MCW3 (5 μg/mL) or MCW4 (3 μg/mL) for 2 hours, followed by three washes and a 1-hour incubation with the secondary antibody (2.5 μg/mL). Sections were washed three times and blocked with a blocking agent (10% Blokhen II; Aves Laboratories) in 0.1% Triton X-100/1× PBS for 30 minutes, followed by a washing step with 0.1% Triton X-100/1× PBS and three additional washes with 1× PBS for 10 minutes each. The sections were incubated with 10 μg/mL affinity-purified chicken mRPGRIP1b antibody in 0.1% Triton X-100/1× PBS/10% blocking agent (Blockhen II; Aves Laboratories) for 2 hours, followed by a wash with 0.1% Triton X-100/1× PBS and three washes with 1× PBS for 10 minutes each. Then, sections were incubated with the secondary antibody (2.5 μg/ mL) for 1 hour, washed three times, and mounted with antifade reagent (Prolong Gold; Molecular Probes). Visualization of retinal sections and localization of proteins were performed by wide-field epifluorescence microscopy on an upright research microscope (model E600; Nikon, Melville, NY), equipped with appropriate excitation and emission filter wheels, a 100-W mercury light source, and special optics (Nomarski/differential interference contrast [DIC] and Plan Apochromat; Nikon) and an encoded motorized Z-stage. Images were obtained with a charge-coupled device (CCD) camera (SPOT-RT; Diagnostic Instruments, Sterling Heights, MI) and under identical acquisition parameters, and were analyzed on computer (Metamorph, ver. 6.2r6; Universal Imaging Corp., West Chester, PA).
Cultured cells were processed for immunocytochemistry by fixing them in 2% paraformaldehyde/1× PBS and three washes in PBS. Incubation procedures with primary and secondary antibodies were similar to those described for the retinal sections. Concentrations of COP1, EEA1, Lamp1, and Lamp2 antibodies were 10 μg/mL each and of CI-MPR anti-serum was 1:150. Visualization and protein colocalization in cultured cells were determined by microscope (model TE2000U; Nikon), equipped as described elsewhere,28 and images were analyzed on computer (Metamorph; Universal Imaging Corp.).
Molecular Cloning of EGFP-rpgrip1b
The full open reading frame of the murine rpgrip1b was subcloned into the Kpn1 and BamHI sites of the pExpress-EGFP vector,30 by introducing the Kpn1 site upstream of the in-frame translation start site, and the FLAG tag sequence followed by a BamHI site at the end of the coding sequence of rpgrip1b. The engineered vector was used in the transfection assays.
Tissue Culture and Cell Transfection
The COS7, 661W, and RGC-5 cells were cultured under the same conditions in DMEM (Invitrogen-Gibco, Grand Island, NY) and collagen-coated 35-mm glass-bottomed culture dishes (MatTek Corp., Ashland, MA) at 5% CO2/37°C for ~16 to 36 hours after transfection and processed for immunocytochemistry. Transfection of cultured cells with EGFP-rpgrip1b was performed (FuGENE 6 transfection reagent; Roche Diagnostics, Indianapolis, IN) as per the manufacturer’s instructions, with the exception that 1.2 μg of vector DNA was used per 4.5 μL of transfection reagent on a 35-mm plate of semiconfluent cultured cells.
Western Blot Analysis
Mouse tissue homogenates were prepared by homogenizing tissues in 3× SDS sample buffer (5% [wt/vol] SDS, 0.15 M Tris HCl [pH 6.7])and 30% glycerol) followed by several passes through an 18-gauge, 1 ½-in. needle and then a 25-gauge, ⅝5-in. needle. Homogenates were then diluted 1:3 in RIPA buffer (25 mM Tris [pH 8.2], 50 mM NaCl, 0.5% Nonidet P40, 0.5% deoxycholate, 0.1% SDS, and 0.1% azide) containing 10 mM iodoacetamide and complete protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). Protein concentration of tissue homogenates was measured by the bicinchoninic acid (BCA; Pierce, Rockford, IL) protein assay method. All protein samples were boiled, 200 μg of tissue homogenates was loaded and resolved on 7.5% SDS-polyacrylamide gels, and Western blot analysis was performed as previously described.28 Affinity-purified antibodies against rpgrip1b and RPGRIP1 were used at 250 ng/mL. The HRP-conjugated secondary antibodies, goat anti-chicken (Promega, Madison, WI), and anti-rabbit IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) were used at 25 ng/mL, and the blots were developed with the chemiluminescence substrate (SuperSignal; Pierce).
The murine and human nucleotide sequences reported in this manuscript were deposited in GenBank with the accession numbers AY914560 and AY914561 (http://www.ncbi.nlm.nih.gov/Genbank; provided in the public domain by the National Center for Biotechnology Information, Bethesda, MD).
Results
Identification of Novel rpgrip1 Isoforms in the Murine and Human Retina
For the sake of consistency, we designate hereafter exon 1 of the human and murine RPGRIP1, the exon comprising the starting codon. Isolation and previous characterization of cDNAs from human and bovine retinal libraries have revealed that the genomic region between exons 12 and 14 undergoes significant alternative splicing derived from alternative, non-and coding exons and introns (Fig. 1A).11,28 Preliminary investigations in our laboratory indicate that intron 13 is transcribed in the murine retina (data not shown). To investigate the splicing heterogeneity and efficiency of this genomic region in the murine, we performed single and multiplex RT-PCR analysis of murine retinal RNA, with primers complementary to the conserved regions of exons 12 and 14 and intron 13, designated P12, P14a/c, and P13c, respectively (Fig. 1A). Two amplicons of distinct sizes, 579 and 462 bp, with significantly different expression levels, were identified (Fig. 1B). Sequence analysis of the 462-bp amplicon generated from the combination of P12 and P14a/c primers (Fig. 1B) showed it encoded the canonical exons 12 through 14 (Fig. 1C). The 579-bp amplicon produced with P12 and P13c primers (12–13c, Fig. 1B) comprised the canonical exon 12 and an extended exon 13 containing the first 398 nucleotides of intron 13 and the sequence complementary to P13c primer (exon 13c; Fig. 1C). Comparison analysis of single and multiplex RT-PCR reactions also supported a much higher expression of the transcript containing the extended exon 13 (exon 13c) than that with just the canonical exon 13 (Fig. 1B).
Figure 1.
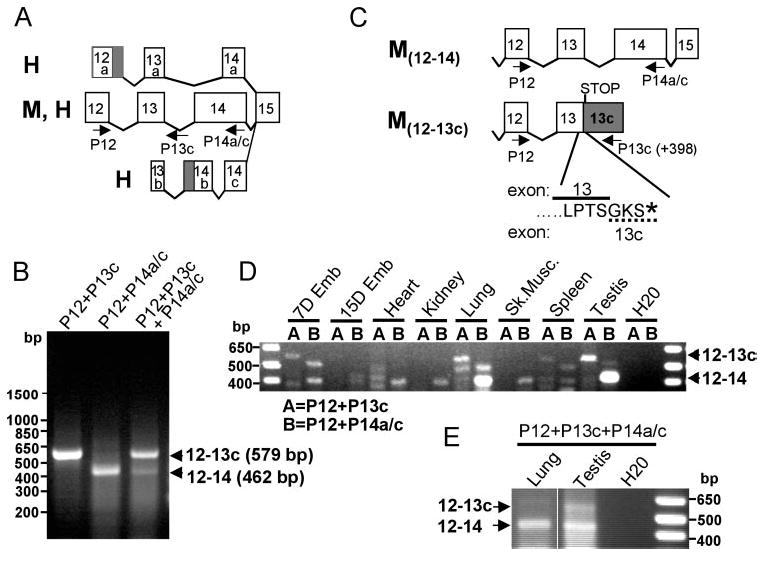
Identification of a novel and abundant rpgrip1 splice variant in the murine retina. (A) Schematic diagram of mRNA splice variants encompassing exons 12 and 14 of rpgrip1 identified to date in the human (H) and the murine (M) retinas. Only a single murine rpgrip1 isoform has been identified and molecularly defined. Primers used in this study are shown as arrows below the exons. Primers P12, P14a/c, and P13c are complementary to exons 12, 14a/c, and intron 13 (398 nucleotides downstream of the exon13–intron 13 boundary). (B) RT-PCR of retinal RNA by singleplex (lanes 1 and 2) and multiplex PCR (lane 3) with primers complementary to exon 12 (P12), exon 14 (P14a/c), and/or intron 13 (P13c). The combination of P12 with P13c and P12 with P14a/c amplified amplicons of 579 and 462 bp, respectively. The former was much more abundant than the latter in the retina. (C) Sequence analysis of the amplicons indicated the 462-bp product comprised the constitutive exons 12, 13, and 14 (M12–14), whereas the 579-bp product encompassed exons 12 and an extended exon 13 (exon 13c), which contained the constitutive exon 13 and 398 bp of intron 13 (M12–13c). This splice variant of rpgrip1 produced from the transcription of intron 13 leads to the premature termination of rpgrip1 and contains three unique residues (GKS) encoded by the extended exon 13c (dashed line). (D) Amplification of rpgrip1 isoforms comprising exons 12–14 and 12–13c from a normalized mouse tissue cDNA panel. In contrast to the retina, the truncated rpgrip1b isoform was detected in low abundance only in 7-day-old embryos, the testis, and the lung among all tissues tested. Conversely, the transcript containing exons 12–14 was very abundant in the lung and testis. (E) Multiplex PCR of normalized lung and testis cDNA confirmed the results obtained with the singleplex reactions in (D). Shaded blocks in (A) and (C) represent alternative extended exons previously reported.11,28
To corroborate further the existence and the identity of full-length transcripts encoding exon 1 through the extended exon 13c, we searched the murine GenBank and dbEST databases for rpgrip1 clones derived from murine retinal cDNA libraries. One clone (accession no. BC016092) contained the open reading frame encoded by exons 1 to 13c and several expressed sequence tag (EST) clones overlapped with the extended exon 13c. We sequenced fully two of the retinal EST clones (accession no. BU503646 and BI872172) and again, obtained similar results, albeit the 5′ untranslated region (UTR) of the EST clones was ~20 nucleotides longer than that reported for the GenBank clone entry (accession no. BC016092). These clones were generated by internal priming of the oligo dT primer to a poly(A) stretch present in the alternative exon 13c (intron 13). The clones contained a full open reading frame from residues 1 to 550 encoded by the canonical exons 1 to 13 and, three additional residues, a stop codon, and a 3′UTR, encoded by the extended and alternatively spliced exon 13 (exon 13c). Likewise, this matched exactly the sequence analysis of the amplicons isolated (Fig. 1C, M12–13c). In addition, the 3′-UTR of these clones was collinear with the intron 13/extended exon 13. Another signature feature of the novel murine transcript identified is that all three full-length and independent clones (accession nos. BC016092, BU503646, and BI872172) contain a microdeletion of three nucleotides caused by the alternative use of an adjacent and downstream 3′-splicing acceptor site (AG) in exon 10 (AGCAGCT). This leads to the deletion of the residue A399. Thus, the murine retina expresses an abundant rpgrip1 transcript in the retina that retains intron 13, leading to the production of a C-terminal truncated RPGRIP1 isoform with a predicted molecular mass of 63 kDa and lacking the C2 and RPGR-interacting (RID) domains. This novel isoform is hereafter designated as the murine rpgrip1b. The truncation and addition of three distinct amino acids at the C terminus generates three novel protein motifs comprising ATP/GTP-binding (P-loop), N-myristoylation, and PKC phosphorylation sites. The analysis of the tissue expression profile of a normalized mouse cDNA panel of transcripts containing the canonical and exon 13c showed that the former was mostly restricted to the lung and testis (Figs. 1D, 1E), and the latter was predominantly abundant in the retina (Fig. 1B).
Likewise, we performed similar expression analysis of the genomic region between exons 12 and 14 of RPGRIP1 in the human. RT-PCR reactions with primers complementary to exon 12 (P12) and intron 13 (hP13c) failed to produce any amplicons (data not shown). Conversely, RT-PCR reaction with primers complementary to exons 12 (P12) and 14 (P14b) (Fig. 2A) generated two amplicons of 223 and 256 bp (Fig. 2B, lane 2) with the latter being the most abundant. Sequence analysis of these showed that the latter encoded the canonical exons 12, 13, and 14, whereas the former had exon 13 truncated by 33 nucleotides (Fig. 2A). This led to an in-frame deletion of 11 amino acids from exon 13 (hereafter designated exon 13d). We also designed a hybrid primer comprising 2 and 28 nucleotides complementary to the truncated exon 13d and exon 14 (P13d14b), respectively. This primer when combined with P12 still produced two amplicons (209 and 176 bp) encompassing the segments of the alternatively spliced products described. This result was obtained because the temperature under which the reverse transcription (first) step of the RT-PCR had to be performed was not stringent enough for specific and sole recognition of the junction between exons 13d and 14 by the P13d14b primer (Fig. 2B, lane 3). The nonspecific amplification of the canonical exons 12 to 14 with P12/P13d14b by RT-PCR was overcome by the analysis of a normalized cDNA tissue panel in highly stringent temperature conditions. As shown in Figure 2C, exons 12 to 14 and exons 12 and 13d were weakly expressed in the liver, and the same was observed for the latter in the pancreas and placenta, where the expression of exons 12 to 14 were not detected.
Figure 2.
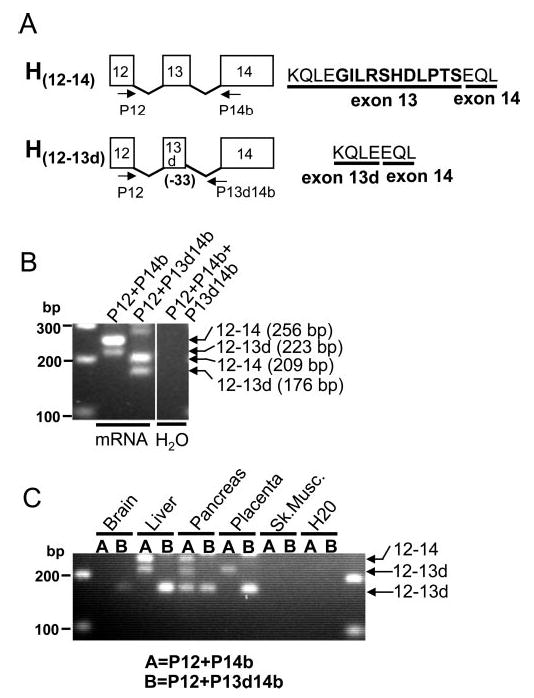
Identification of a novel RPGRIP1 splice variant in the human. (A) Schematic diagram of splice variants of transcripts encompassing exons 12 and 14 of RPGRIP1. The H12–14 isoform has been reported previously. H12–13d is a new isoform of RPGRIP1 identified and reported in this work. (B) RT-PCR of retinal mRNA by singleplex (lane 2) and multiplex PCR (lane 3) with primers (shown in A) complementary to exon 12 (P12), exon 14 (P14b), and the boundary sequence between the alternatively spliced exon 13 and the constitutive exon 14 (P13d14b). Sequence analysis of the amplicons indicated that the 256- and 209-bp products comprised the constitutive exons 12, 13, and 14 (H12–14), whereas the 223- and 176-bp products encompassed exons 12 and an exon 13 (exon 13d) truncated by 33 nucleotides (H12–13d). The expression of the RPGRIP1 transcript isoform containing the canonical exons 12–14 is much higher than that observed for the other RPGRIP1 splice variant in the retina. (C) Amplification of RPGRIP1 isoforms comprising exons 12–14 and exons 12–13d from a normalized human tissue cDNA panel. All isoforms are expressed at low levels in the tissues tested. With the exception of liver, the RPGRIP1 isoform comprising exons 12–14 is not expressed or it is transcribed at much lower levels than the other RPGRIP1 isoform.
Distinct Expression and Localization of rpgrip1 and rpgrip1b Isoforms in the Murine Retina
To probe the expression and localization of the novel and truncated rpgrip1b isoform in the murine retina, we raised and affinity purified antibodies (Ab rpgrip1b) against a peptide with a minimal epitope sequence comprising the last seven residues of exons 13–13c (Fig. 1C; see the Material and Methods section). As shown in Figure 3A, the antibody Ab rpgrip1b detected a single protein with the predicted and apparent molecular mass of 63 kDa, which was strongly expressed in the retina but not in other tissues tested (Fig. 3B). This protein was also detected by the antibody (Ab 22) against the coiled-coil domain of RPGRIP111,28 (Fig. 3B), but not with another (Ab 38) against the RID domain of RPGRIP1.11,28 Conversely, Ab rpgrip1b did not detect the counterpart 63-kDa murine protein in human retinal homogenates (data not shown). In addition, both Ab 22 (Fig. 3C) and Ab 38 (Fig. 3D) but not the Ab rpgrip1b (data not shown) detected a protein of ~110 kDa in the testis. This further supports the mRNA tissue expression profile analysis of the predominance of a murine transcript encoded by the canonical exons 12 and 14 in the testis (and lung). However, the testis rpgrip1 isoform must represent yet another and major product of an alternatively spliced transcript, because its molecular weight is lower than that predicted and consistently observed in the retina for the RID-containing RPGRIP isoform of ~150 kDa encoded by the constitutive exons 1–24.28
Figure 3.

Immunoblot analysis of rpgrip1b in murine tissues. Antibodies against the seven C-terminal residues of rpgrip1b (A, B) and the coiled-coil domain of RPGRIP1/rpgrip1b (C) detected a major protein with the predicated and apparent molecular mass of 63 kDa (A) and with the expression restricted to the murine retina (B). The 63-kDa protein was not immunoreactive to the rpgrip1b Ab preadsorbed with the cognate peptide epitope and an antibody (Ab38) against the C-terminal RID domain of RPGRIP1 (data not shown). The Ab22 detected also a testis-restricted and very abundant rpgrip1 isoform (rpgrip1T110) with an apparent molecular mass of 110 kDa (D), which was also immunoreactive to an antibody (Ab38) against the C-terminal RID domain of RPGRIP1 (E).
To localize the novel murine RPGRIP1 isoform in the mouse retina, we performed immunocytochemical analysis of murine eyecup cryosections with the Ab rpgrip1b against the C-terminal end of rpgrip1b and compared its cellular and subcellular distribution with that of two other previously characterized antibodies (MCW 3, MCW 4) against the C2 domain of RPGRIP1,27,28 at two distinct focal planes. As shown in Figure 4, different focal plane images of the retina immunostained with the antibodies MCW3, MCW4, and the Ab rpgrip1b, provided distinct localization patterns. The RPGRIP1 isoform was expressed strongly in amacrine neurons and neuronal processes thereof, branching toward the inner plexiform (synaptic) and ganglion cell layers as well as to the postsynaptic sublayer of the outer plexiform layer (Figs. 4A, 4D). The larger RPGRIP1 isoform localized also to the connecting cilium (Fig. 4D) and with a lower presence in the inner segments of photoreceptors (Fig. 4A), their nuclear rims (Fig. 4A), and the external limiting membrane (Fig. 4D). In contrast, the murine-specific rpgrip1b localized strongly to the neuronal cell bodies of retinal neurons across all the nuclear layers (Fig. 4B) and to the inner segment compartment of photoreceptors (Fig. 4E). Moreover, the RPGRIP1 and rpgrip1b isoforms did not colocalize in any retinal neurons or subcellular compartments thereof (Figs. 4C, 4F). Preabsorbed antibodies against the cognate epitopes did not immunostain the murine retina (Fig. 4E, inset).
Figure 4.
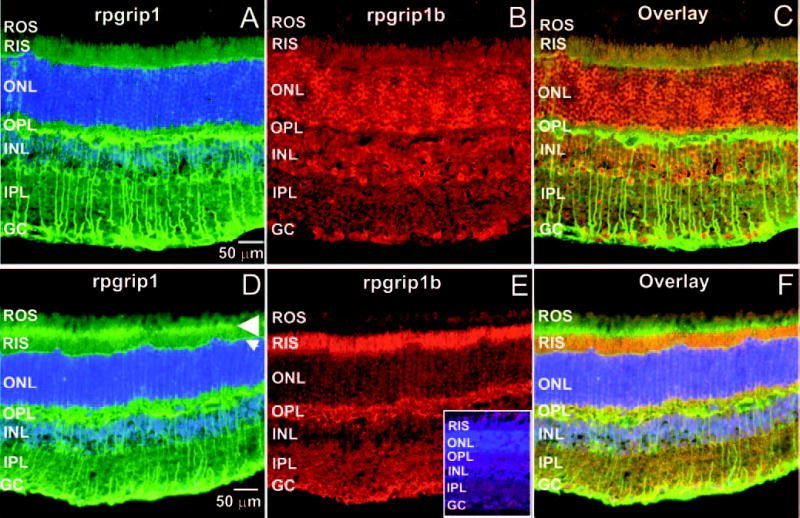
rpgrip1 and rpgrip1b have distinct subcellular localization in the murine retina. Radial cryosections of murine retinas immunostained with two antibodies, MCW 3 (A) and MCW 4 (D), against the C2 domain of rpgrip1 and with an antibody against seven C-terminal residues of rpgrip1b (B, E). (A–C) and (D–F) are images captured at different focal planes, with the former optimally focused on the retinal cell bodies (outer [ONL] and inner [INL] nuclear layers ONL and INL) and the latter on the connecting cilium region (arrowhead) of photoreceptor neurons. (C) and (F) are merged images of (A) and (B) and of (D) and (E), respectively. Rpgrip1b was localized throughout different retinal neurons and layers (B, E) with particular preponderance in the cell bodies (ONL and INL) of these (B) and in the inner segment compartment (RIS) of rod photoreceptor neurons (E). In contrast, rpgrip1 was strongly localized to the amacrine neurons and branching processes thereof, postsynaptic sublayer of the outer plexiform layer (OPL) (A). In addition, rpgrip1 was also localized to the connecting cilium (arrowhead) of photoreceptors, external limiting membrane (arrow), and the inner segment compartment of rod photoreceptors (D). rpgrip1 and rpgrip1b had a distinct subcellular distribution pattern, as they did not colocalize in the retinal neurons (C, F). Preabsorbed anti-rpgrip1b antibody with the cognate peptide epitope completely blocked the immunostaining of the retina (E, inset). ROS, rod outer segments; OPL, outer plexiform layer; IPL, inner plexiform layer; GC, ganglion cell layer. Scale bar, 50 μm.
Association of the Murine-Specific rpgrip1b Isoform with a Subpopulation of Lysosomes
To obtain clues about the role of the murine-specific rpgrip1b isoform in the retina and compare it with that of other isoforms, we double-tagged the murine rpgrip1b isoform with EGFP and a FLAG epitope at its N- and C-terminal ends, respectively, and ectopically expressed it in COS7 cells (Fig. 5), which are not immunoreactive against RPGRIP1, and retinal cells lines such as 661W and RGC5 (data not shown). First, we checked whether the EGFP signal colocalized with that obtained with an antibody against the N-terminal coiled-coil domain of rpgrip1b (Ab 22). To this effect, there was extensive and perfect colocalization between these two signals (Figs. 5A–C) and nontransfected cells lacked any staining (not shown), supporting further the antigenicity of Ab 22 and the lack of any difference in subcellular phenotype between the EGFP- and rpgrip1b-fused moieties. Similar results were obtained with the FLAG-tagged rpgrip1b at its C-terminal end (data not shown). Focal plane images of the EGFP and rpgrip1b signals showed that they were present throughout the cytoplasm as fine and coarse puncta, which were sometimes seen in fluorescent aggregates (Figs. 5A–C). To characterize the nature of these fluorescent signals, we used a battery of sub-cellular markers, many of which associated with various secretory pathways and organelles. Among these, we obtained partial colocalization of the murine rpgrip1b isoform with Lamp1 (Figs. 5D–F), a lysosomal marker,31,32 supporting that the rpgrip1b function is likely linked to the lysosome or a lysosome-related subcellular process. This colocalization was consistent among and within the cell culture lines used (data not shown), supporting its colocalization as independent of the transfection and/or expression efficiency of the fusion construct. Likewise, this observation was consistent for different time courses after transfection and colocalization could be observed as soon as 16 hours after transfection. Finally, we screened ~120 Lamp1 and rpgrip1b double-labeled foci and found that 18% exhibited 100% overlap, while in the remaining foci, Lamp1 and rpgrip1b signals interfaced with each other and partially overlapped (Fig. 5F, insets). In addition, we often observed multiple signals of rpgrip1b surrounding a Lamp1-immunopositive signal, which was always central to rpgrip1b puncta (Fig. 5F, inset pictures).
Figure 5.
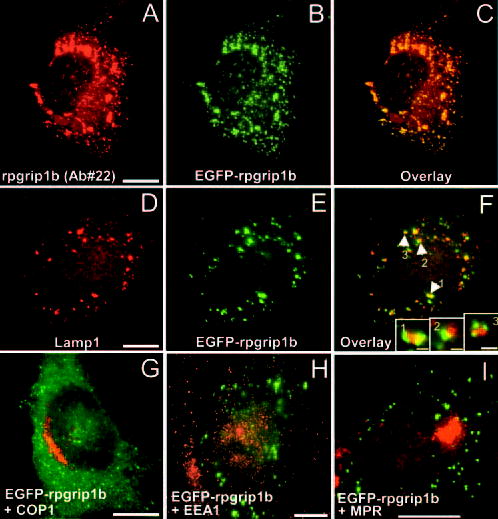
The EGFP-rpgrip1b fusion protein partially colocalized with a subpopulation of lysosomes in COS7 cells. Immunolabeling of EGFP-rpgrip1 fusion protein with Ab 22 against the counterpart coiled-coil domain of rpgrip1 labeled fine and aggregate foci (A) throughout the cytoplasm, which colocalized perfectly and extensively (C) with the EGFP signal of transfected cells (B). Nontransfected COS7 cells were immunonegative for rpgrip1 (not shown). The EGFP signal from the rpgrip1 fusion protein partially colocalized and interfaced closely with the lysosome-associated membrane protein Lamp1 (D–F), but not with another lysosomal marker, Lamp-2 (not shown). Screening of the double-labeled foci in transfected cells showed that the EGFP signal rarely overlapped completely with the Lamp-1-immunoreactive structures. Instead, the overlap of the signals was partial (F, inset 2). In addition, multiple and discrete EGFP signals (82% of double-labeled signals) often encircled and interfaced with a single Lamp-1-immunopositive signal (F, insets 1, 3). The EGFP-rpgrip1b fusion protein failed to colocalized with other subcellular and vesicular markers tested such as COP1 (G), EEA-1 (H), and CI-MPR (I). Scale bars: (A–I), 10 μm; (F, insets), 1 μm.
We also probed whether rpgrip1b colocalizes with another lysosomal marker, Lamp2, which has been identified as a receptor for substrate proteins in chaperone-mediated autophagy,33 but we failed to observe any colocalization (data not shown). In light of the colocalization of rpgrip1b to the lysosomes, we investigated further whether the murine rpgrip1b was associated with other vesicular markers linked to the secretory pathway such as the Golgi and the endocytic pathways. To this effect, the murine rpgrip1b also failed to localize with COP1, the early endosome antigen 1 (EEA1), and the cation-independent mannose-6-phosphate receptor (Figs. 5G–I).
Then, the colocalization analysis of the rpgrip1b with Lamp1 was extended to the murine retina. As shown in Fig. 6, rpgrip1b, and Lamp1 partially colocalized throughout the inner segment subcellular compartment of photoreceptor neurons, but the extent of the colocalization of these appeared to be more limited in other retinal neurons.
Figure 6.
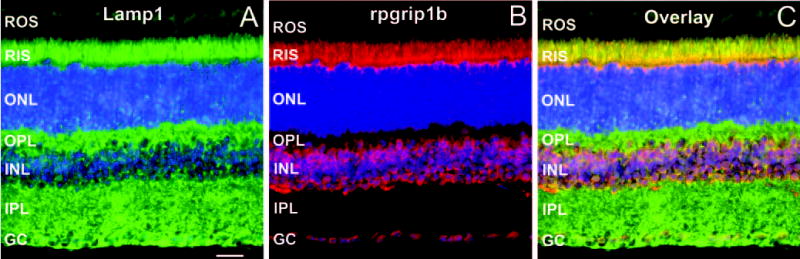
Lamp1 and rpgrip1b partially colocalized to the inner segment compartment of photoreceptor neurons. Radial cryosections of murine retinas immunostained with Lamp1 (A) and with an antibody against seven C-terminal residues of rpgrip1b (B). (C) is a merged image of (A) and (B). Lamp1 strongly localized to the inner segments (RIS) of rod photoreceptor neurons and throughout other subcellular compartments of other retinal neurons. Partial colocalization of Lamp1 with rpgrip1b was predominantly observed throughout the inner segment compartment of rod photoreceptor neurons. ROS, rod outer segments; RIS, rod inner segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GC, ganglion cell layer. Scale bar, 50 μm.
Discussion
We identified a novel and major murine-specific rpgrip1 isoform, rpgrip1b, that retains intron 13 and it is expressed predominantly in the retina. This generates a truncated rpgrip1 isoform with a truncated C terminus and thus lacks the C2 and RID domains. The murine-specific rpgrip1 transcript retaining intron 13 is much more abundant in the retina than the transcript(s) encompassing the canonical exons 12, 13, and 14 (Fig. 1B). The two EST clones sequenced and the other identified in the GenBank database comprised the complete coding region from exon 1 to 13, and the 3′ UTR encoded by the alternative exon 13c (intron 13). A careful search of the dbEST database also identified numerous clones derived from retina cDNA libraries and whose 5′- and/or 3′-sequences mapped to the alternative exon 13c (intron 13; e.g., accession nos. BM938450, BG295426, BU504218, BI736585, BF464064, and BU503095). In addition, the 5′-sequence of the EST clones with the accession nos. BI464064 and BU503095 covered the alternative exon 13c and the constitutive exon 14. Thus, the aforementioned full-length and overlapping clones strongly support the existence of a major alternatively spliced transcript containing an alternative exon 13c and downstream nonencoding exons encompassing a long 3′-UTR. The retention of intron 13 adds ~3.8 kb to the 4.7-kb transcript represented by the murine clone reported, which contains solely the spliced constitutive exons 1–24 of rpgrip1b and 3′-UTR.14 This closely approximates the size of 10 kb observed for the major transcript produced by rpgrip1 in the murine retina and cannot be accounted for by the ~4.8-kb size of the full-length rpgrip1 reported (accession no. AY008297).14
In contrast, the human retina lacks a transcript retaining intron 13 and, instead, it expresses a novel transcript generated by the use of an alternative donor splicing site that leads to the skipping (deletion) of 33 nucleotides from the canonical exon 13. This novel and alternatively spliced transcript is of very low abundance in the retina compared with the counterpart transcript isoform expressing the constitutive exons 13 and 14. Among other tissues analyzed, the expression of the novel human RPGRIP1 transcript was restricted to the liver, pancreas, and placenta, where its expression was rather low.
Altogether, these data support that the murine species express a truncated rpgrip1 isoform, rpgrip1b, that is much more abundant in the retina than its alternatively spliced counterpart containing all the constitutive and coding exons, whereas the human lacks the former and the latter is the predominant isoform among the alternatively spliced transcripts identified.11,13 These observations are supported further by immunoblot analysis of retinal homogenates of different species, where the high molecular mass and RID-containing RPGRIP1 isoform was much more abundant in the human and bovine than in rodent retinas.28 Moreover, there is another novel rpgrip1 isoform expressed strongly in the mouse testis that contains all or some of the coiled-coil and RID domains of rpgrip1. Finally, a word of caution regarding the murine rpgrip1 transcript reported14: Similar to the EST clones sequenced and the 3′-EST sequences in the dbEST, the rpgrip1 transcript lacked a canonical polyadenylation signal and a poly(A) tail. This finding is in contrast to that reported for the bovine and human RPGRIP1 transcripts characterized, albeit some of these had noncanonical polyadenylation signals (ATAAA).11 Hence, it remains unclear to what extent the 3′UTR may contribute to the regulation of the translational levels of rpgrip1/rpgrip1b observed in the murine retina,28 despite the very strong expression of another RID-containing RPGRIP1 isoform in the testis.
There are several implications in the findings reported herein. First, it is very likely that the frequency of mutations in the human RPGRIP1 gene remain underreported, and more systematic screenings are therefore required to better assess the contribution of RPGRIP1 mutations in LCA and potentially, in other systemic and nonsystemic retinal dystrophies. The screening of patients with LCA thus far has taken into account only the constitutive exons of RPGRIP1.9,10 In light of the molecular diversity of the transcripts produced by the genomic region encompassing exons 12 and 14 (and others) in the human and across species,11,28 it is likely that translationally silent and other type of mutations, sometimes considered to be neutral polymorphisms, anywhere in this genomic region could lead to aberrant transcripts (e.g., exon skipping). To this effect, it is estimated that mutations on splice sites account for at least 15% of all human genetic mutations reported,34,35 a number that is also underestimated, because it does not account for cis-acting intronic, enhancer, and silencer elements regulating splicing.34 In genes that have been subjected to systematic genetic and mutational analysis, this number is significantly higher. For example, 13% to 20% and 50% of the mutations in CFTR, neurofibromatosis type 1 (NF1), and ataxia telangiectasia (ATM) are believed to affect splicing.36
Second, the production of several RPGRIP1 isoforms underlies the presence of isoform ratios, which may differ among and within neuronal cell types.28 Mutations in RPGRIP1 may change those ratios, which may be important for neuronal function. The change of transcript isoform ratios is known to have disease implications. For example, mutations (many of these affecting splicing) in the MAPT gene, which encodes six isoforms of tau in the brain, have been shown to lead to frontotemporal dementia with parkinsonism (FTDP-17).36–39 Among these, mutations that affect the inclusion of exon 10 lead to FTDP-17 by altering the ratio of isoforms containing a distinct number of microtubule-binding domains.40
Third, it will be interesting from an evolutionary perspective to understand the reason(s) behind the retention of intron 13 in the murine but not the human. The exon–intron boundaries of intron 13 are well conserved but do not follow the GT/AG rule,36,41 where GT is replaced by GC, which is a very rare splicing site.42,43 This finding probably does not account for the splicing efficiency of intron 13, because the GC is replaced by GT in the novel transcript isoform with a deletion of 33 nucleotides in the constitutive exon 13 found in the human and the alternative transcribed exon is much less abundant than its constitutive counterpart. Analysis of the intron 13 in the mouse and the human RPGRIP1 genes shows the existence of two and one, and three and two, poly-T- and poly-A-rich tracts, respectively, between 8 and 34 nucleotides long. In addition, the human intron 13 is significantly smaller (>1 kb) than the murine counterpart. It is tempting to speculate that this difference in tracts and/or intron size may affect the looping out of the pre-mRNA by differential sequestration of exonic splicing silencers (e.g., hn RNP I, hn RNP A/B, and hn RNP H).34 Finally, the species-specific alternative splicing of RPGRIP1 herein reported is reminiscent of that observed for RPGR, whose intron 14 is an exon (exon 14a) in the mouse, but not in the human.44
Fourth, the existence of an rpgrip1 isoform lacking the C2 and RID domains implies that its function is independent of RPGR. Moreover, the absence of those domains, in particular C2, may contribute significantly to its unique subcellular distribution. Similarly, the presence of three unique residues at the C terminus of rpgrip1b, which is otherwise identical to the N-terminal half-sequence of rpgrip1, may generate (in conjunction with neighboring residues) a novel targeting or localization signal to the lysosomes or subpopulation thereof. To this effect, it is intriguing to observe that rpgrip1b colocalized with the Lamp1, but not with the Lamp2, lysosomal marker—further supporting the participation of rpgrip1b in a subset of lysosomes or lysosomes-related structures.
Finally, genetic targeting of the murine rpgrip1 locus was recently reported by partial homologous recombination and insertion of exons 4 and 5 downstream of exon 13,45 leading to the dysmorphogenesis of the outer segments of photoreceptors. However, exons 1 to 13 and a portion of intron 13 were not targeted, and exon 13 may splice with an out-of-phase exon 4. This raises the strong possibility that the expression of the truncated and 63-kDa rpgrip1b isoform is not affected and/or the production of a chimeric rpgrip1 isoform containing at its C terminus 62 extra residues encoded by an out-of-frame exon 4. Any of these gene products may contribute to “off-target” phenotypes. To this end, it will be interesting to probe the degree of functional redundancy between rpgrip1b and other RPGRIP1 isoforms in the retina. Future genetic models will help to investigate the functional diversity of RPGRIP1 isoforms and their role in disease processes.
Acknowledgments
The authors thank Rolf Jakobi (Department of Biochemistry, University Health Sciences, Kansas City, MO) for the pExpress-EGFP vector.
Footnotes
Disclosure: X. Lu, None; P.A. Ferreira, None
Note Added in Proof
Following submission of this manuscript, a report by Lu et al. analyzes and contrasts phenotypically and quantitatively the subcellular distribution patterns of bRPGRIP1, bRPGRIP1b and mRPGRIP1b, ectopically expressed in COS7 cells (Lu X, Guruju M, Oswald J, Ferreira PA, 2005. Limited proteolysis differentially modulates the stability and subcellular localization of domains of RPGRIP1 that are distinctly affected by mutations in Leber’s congenital amaurosis. Hum Mol Genet. 2005;14: 1327–1340).
Supported by National Eye Institute Grants EY11993 and EY012665 (PAF).
References
- 1.Cremers FP, Van Den Hurk JA, Den Hollander AI. Molecular genetics of Leber congenital amaurosis. Hum Mol Genet. 2002;11:1169–1176. doi: 10.1093/hmg/11.10.1169. [DOI] [PubMed] [Google Scholar]
- 2.Sohocki MM, Bowne SJ, Sullivan LS, et al. Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nat Genet. 2000;24:79–83. doi: 10.1038/71732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.den Hollander AI, Heckenlively JR, van den Born LI, et al. Leber congenital amaurosis and retinitis pigmentosa with Coats-like exudative vasculopathy are associated with mutations in the crumbs homologue 1 (CRB1) gene. Am J Hum Genet. 2001;69:198–203. doi: 10.1086/321263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lotery AJ, Jacobson SG, Fishman GA, et al. Mutations in the CRB1 gene cause Leber congenital amaurosis. Arch Ophthalmol. 2001;119:415–420. doi: 10.1001/archopht.119.3.415. [DOI] [PubMed] [Google Scholar]
- 5.Freund CL, Wang QL, Chen S, et al. De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat Genet. 1998;18:311–312. doi: 10.1038/ng0498-311. [DOI] [PubMed] [Google Scholar]
- 6.Perrault I, Rozet JM, Calvas P, et al. Retinal-specific guanylate cyclase gene mutations in Leber’s congenital amaurosis. Nat Genet. 1996;14:461–464. doi: 10.1038/ng1296-461. [DOI] [PubMed] [Google Scholar]
- 7.Marlhens F, Bareil C, Griffoin JM, et al. Mutations in RPE65 cause Leber’s congenital amaurosis. Nat Genet. 1997;17:139–141. doi: 10.1038/ng1097-139. [DOI] [PubMed] [Google Scholar]
- 8.Perrault I, Hanein S, Gerber S, et al. Retinal dehydrogenase 12 (RDH12) mutations in leber congenital amaurosis. Am J Hum Genet. 2004;75:639–646. doi: 10.1086/424889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerber S, Perrault I, Hanein S, et al. Complete exon-intron structure of the RPGR-interacting protein (RPGRIP1) gene allows the identification of mutations underlying Leber congenital amaurosis. Eur J Hum Genet. 2001;9:561–571. doi: 10.1038/sj.ejhg.5200689. [DOI] [PubMed] [Google Scholar]
- 10.Dryja TP, Adams SM, Grimsby JL, et al. Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am J Hum Genet. 2001;68:1295–1298. doi: 10.1086/320113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roepman R, Bernoud-Hubac N, Schick DE, et al. The retinitis pigmentosa GTPase regulator (RPGR) interacts with novel transport-like proteins in the outer segments of rod photoreceptors. Hum Mol Genet. 2000;9:2095–2105. doi: 10.1093/hmg/9.14.2095. [DOI] [PubMed] [Google Scholar]
- 12.Roepman R, Schick D, Ferreira PA. Isolation of retinal proteins that interact with retinitis pigmentosa GTPase regulator by interaction trap screen in yeast. Methods Enzymol. 2000;316:688–704. doi: 10.1016/s0076-6879(00)16757-6. [DOI] [PubMed] [Google Scholar]
- 13.Boylan JP, Wright AF. Identification of a novel protein interacting with RPGR. Hum Mol Genet. 2000;9:2085–2093. doi: 10.1093/hmg/9.14.2085. [DOI] [PubMed] [Google Scholar]
- 14.Hong DH, Yue G, Adamian M, Li T. Retinitis pigmentosa GTPase regulator (RPGRr)-interacting protein is stably associated with the photoreceptor ciliary axoneme and anchors RPGR to the connecting cilium. J Biol Chem. 2001;276:12091–12099. doi: 10.1074/jbc.M009351200. [DOI] [PubMed] [Google Scholar]
- 15.Breuer DK, Yashar BM, Filippova E, et al. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 2002;70:1545–1554. doi: 10.1086/340848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meindl A, Dry K, Herrmann K, et al. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3) Nat Genet. 1996;13:35–42. doi: 10.1038/ng0596-35. [DOI] [PubMed] [Google Scholar]
- 17.Roepman R, Bauer D, Rosenberg T, et al. Identification of a gene disrupted by a microdeletion in a patient with X-linked retinitis pigmentosa (XLRP) Hum Mol Genet. 1996;5:827–833. doi: 10.1093/hmg/5.6.827. [DOI] [PubMed] [Google Scholar]
- 18.Vervoort R, Lennon A, Bird AC, et al. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet. 2000;25:462–466. doi: 10.1038/78182. [DOI] [PubMed] [Google Scholar]
- 19.Zito I, Thiselton DL, Gorin MB, et al. Identification of novel RPGR (retinitis pigmentosa GTPase regulator) mutations in a subset of X-linked retinitis pigmentosa families segregating with the RP3 locus. Hum Genet. 1999;105:57–62. doi: 10.1007/s004399900110. [DOI] [PubMed] [Google Scholar]
- 20.Demirci FY, Rigatti BW, Wen G, et al. X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am J Hum Genet. 2002;70:1049–1053. doi: 10.1086/339620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Z, Peachey NS, Moshfeghi DM, et al. Mutations in the RPGR gene cause X-linked cone dystrophy. Hum Mol Genet. 2002;11:605–611. doi: 10.1093/hmg/11.5.605. [DOI] [PubMed] [Google Scholar]
- 22.Ayyagari R, Demirci FY, Liu J, et al. X-linked recessive atrophic macular degeneration from RPGR mutation. Genomics. 2002;80:166–171. doi: 10.1006/geno.2002.6815. [DOI] [PubMed] [Google Scholar]
- 23.Iannaccone A, Wang X, Jablonski MM, et al. Increasing evidence for syndromic phenotypes associated with RPGR mutations (letter) Am J Ophthalmol. 2004;137:785–786. doi: 10.1016/j.ajo.2003.11.050. author reply 786. [DOI] [PubMed] [Google Scholar]
- 24.Iannaccone A, Breuer DK, Wang XF, et al. Clinical and immunohistochemical evidence for an X linked retinitis pigmentosa syndrome with recurrent infections and hearing loss in association with an RPGR mutation. J Med Genet. 2003;40:e118. doi: 10.1136/jmg.40.11.e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koenekoop RK, Loyer M, Hand CK, et al. Novel RPGR mutations with distinct retinitis pigmentosa phenotypes in French-Canadian families. Am J Ophthalmol. 2003;136:678–687. doi: 10.1016/s0002-9394(03)00331-3. [DOI] [PubMed] [Google Scholar]
- 26.Zito I, Downes SM, Patel RJ, et al. RPGR mutation associated with retinitis pigmentosa, impaired hearing, and sinorespiratory infections. J Med Genet. 2003;40:609–615. doi: 10.1136/jmg.40.8.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mavlyutov TA, Zhao H, Ferreira PA. Species-specific subcellular localization of RPGR and RPGRIP isoforms: implications for the phenotypic variability of congenital retinopathies among species. Hum Mol Genet. 2002;11:1899–1907. doi: 10.1093/hmg/11.16.1899. [DOI] [PubMed] [Google Scholar]
- 28.Castagnet P, Mavlyutov T, Cai Y, Zhong F, Ferreira P. RPGRIP1s with distinct neuronal localization and biochemical properties associate selectively with RanBP2 in amacrine neurons. Hum Mol Genet. 2003;12:1847–1863. doi: 10.1093/hmg/ddg202. [DOI] [PubMed] [Google Scholar]
- 29.Mavlyutov TA, Cai Y, Ferreira PA. Identification of RanBP2- and kinesin-mediated transport pathways with restricted neuronal and subcellular localization. Traffic. 2002;3:630–640. doi: 10.1034/j.1600-0854.2002.30905.x. [DOI] [PubMed] [Google Scholar]
- 30.Jakobi R, McCarthy CC, Koeppel MA. Mammalian expression vectors for epitope tag fusion proteins that are toxic in E. coli. BioTechniques. 2002;33:1218–20. doi: 10.2144/02336bm07. 22. [DOI] [PubMed] [Google Scholar]
- 31.Lewis V, Green SA, Marsh M, Vihko P, Helenius A, Mellman I. Glycoproteins of the lysosomal membrane. J Cell Biol. 1985;100:1839–1847. doi: 10.1083/jcb.100.6.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rohrer J, Schweizer A, Russell D, Kornfeld S. The targeting of Lamp1 to lysosomes is dependent on the spacing of its cytoplasmic tail tyrosine sorting motif relative to the membrane. J Cell Biol. 1996;132:565–576. doi: 10.1083/jcb.132.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cuervo AM, Dice JF. Unique properties of lamp2a compared to other lamp2 isoforms. J Cell Sci. 2000;113:4441–4450. doi: 10.1242/jcs.113.24.4441. [DOI] [PubMed] [Google Scholar]
- 34.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 35.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 36.Garcia-Blanco MA, Baraniak AP, Lasda EL. Alternative splicing in disease and therapy. Nat Biotechnol. 2004;22:535–546. doi: 10.1038/nbt964. [DOI] [PubMed] [Google Scholar]
- 37.Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9:4225–4230. doi: 10.1002/j.1460-2075.1990.tb07870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 39.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 40.D’Souza I, Poorkaj P, Hong M, et al. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA. 1999;96:5598–5603. doi: 10.1073/pnas.96.10.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pagani F, Baralle FE. Genomic variants in exons and introns: identifying the splicing spoilers. Nat Rev Genet. 2004;5:389–396. doi: 10.1038/nrg1327. [DOI] [PubMed] [Google Scholar]
- 42.Fischer HD, Dodgson JB, Hughes S, Engel JD. An unusual 5′ splice sequence is efficiently utilized in vivo. Proc Natl Acad Sci USA. 1984;81:2733–2737. doi: 10.1073/pnas.81.9.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aebi M, Hornig H, Padgett RA, Reiser J, Weissmann C. Sequence requirements for splicing of higher eukaryotic nuclear pre-mRNA. Cell. 1986;47:555–565. doi: 10.1016/0092-8674(86)90620-3. [DOI] [PubMed] [Google Scholar]
- 44.Kirschner R, Rosenberg T, Schultz-Heienbrok R, et al. RPGR transcription studies in mouse and human tissues reveal a retina-specific isoform that is disrupted in a patient with X-linked retinitis pigmentosa. Hum Mol Genet. 1999;8:1571–1578. doi: 10.1093/hmg/8.8.1571. [DOI] [PubMed] [Google Scholar]
- 45.Zhao Y, Hong DH, Pawlyk B, et al. The retinitis pigmentosa GTPase regulator (RPGR)-interacting protein: subserving RPGR function and participating in disk morphogenesis. Proc Natl Acad Sci USA. 2003;100:3965–3970. doi: 10.1073/pnas.0637349100. [DOI] [PMC free article] [PubMed] [Google Scholar]