

Abstract
No single animal model for severe acute respiratory syndrome (SARS) reproduces all aspects of the human disease. Young inbred mice support SARS-coronavirus (SARS-CoV) replication in the respiratory tract and are available in sufficient numbers for statistical evaluation. They are relatively inexpensive and easily accessible, but their use in SARS research is limited because they do not develop illness following infection. Older (12- to 14-mo-old) BALB/c mice develop clinical illness and pneumonitis, but they can be hard to procure, and immune senescence complicates pathogenesis studies. We adapted the SARS-CoV (Urbani strain) by serial passage in the respiratory tract of young BALB/c mice. Fifteen passages resulted in a virus (MA15) that is lethal for mice following intranasal inoculation. Lethality is preceded by rapid and high titer viral replication in lungs, viremia, and dissemination of virus to extrapulmonary sites accompanied by lymphopenia, neutrophilia, and pathological changes in the lungs. Abundant viral antigen is extensively distributed in bronchial epithelial cells and alveolar pneumocytes, and necrotic cellular debris is present in airways and alveoli, with only mild and focal pneumonitis. These observations suggest that mice infected with MA15 die from an overwhelming viral infection with extensive, virally mediated destruction of pneumocytes and ciliated epithelial cells. The MA15 virus has six coding mutations associated with adaptation and increased virulence; when introduced into a recombinant SARS-CoV, these mutations result in a highly virulent and lethal virus (rMA15), duplicating the phenotype of the biologically derived MA15 virus. Intranasal inoculation with MA15 reproduces many aspects of disease seen in severe human cases of SARS. The availability of the MA15 virus will enhance the use of the mouse model for SARS because infection with MA15 causes morbidity, mortality, and pulmonary pathology. This virus will be of value as a stringent challenge in evaluation of the efficacy of vaccines and antivirals.
Author Summary
Severe acute respiratory syndrome (SARS) is a severe, sometimes fatal respiratory disease caused by a coronavirus (SARS-CoV). In order to study the disease and evaluate vaccines and antiviral drugs, animal models that mimic the disease are necessary. However, no single animal model for SARS reproduces all aspects of the disease as it affects humans. SARS-CoV replicates in the lungs of young mice, but they do not show signs of illness. Adaptation of SARS-CoV by serial passage in the lungs of mice resulted in a virus (MA15) that is lethal for young mice following intranasal inoculation. Lethality is preceded by rapid and high titer viral replication in lungs, viremia, and dissemination of virus to extrapulmonary sites accompanied by hematological changes and pathological changes in the lungs. Mice infected with MA15 virus die from an overwhelming viral infection with extensive, virally mediated destruction of pneumocytes, and ciliated epithelial cells. The MA15 virus has six coding mutations in its genome, which, when introduced into a recombinant SARS-CoV, confer lethality. The MA15 virus will enhance the use of the mouse model for SARS because infection with this virus in mice reproduces many aspects of severe human disease, including morbidity, mortality, and pulmonary pathology.
Introduction
The occurrence in late 2002 and early 2003 of cases of severe acute respiratory syndrome (SARS) in southeast China quickly drew international attention as the disease sickened more than 8,000 people and spread to more than 30 countries within six months. Since the identification of the etiological agent, the SARS-coronavirus (SARS-CoV), in 2003, development and characterization of animal models for evaluation of prophylaxis and treatment strategies have been of great interest. Although SARS-CoV has not been associated with a subsequent widespread outbreak since 2002–2003, the potential for such an outbreak remains. Identification of a SARS-like coronavirus in Chinese horseshoe bats (Rhinolophus species) that are indigenous across Southeast Asia suggests that they may represent a natural reservoir from which viruses may be introduced into the human population [1]. The course of infection in animal models is abbreviated compared with the course of SARS in humans; however, many aspects of SARS-CoV–associated disease are reproducible in animal models, including age-dependent susceptibility, recovery of SARS-CoV from respiratory tissues and secretions, infection of type I and type II pneumocytes and bronchial epithelial cells, detection of viral genome in blood and extrapulmonary tissues, and pulmonary pathology (including pneumonitis, edema, necrotic debris, and hyaline membrane formation) [2,3]. Clinical symptoms have been reported in some species, but the findings are not entirely reproducible in outbred species. Variability in clinical symptoms seen in outbred species may result from additional factors, including infection with co-pathogens, stress, existence of sub-species of test animals, and use of different virus strains. This variation can be problematic in studies of pathogenesis and vaccine efficacy unless a large enough number of animals are included in each treatment group [2]. The ideal animal model would demonstrate viral replication in respiratory tissues, histopathologic evidence of respiratory disease, and consistent clinical signs of disease, including mortality. A small animal model in which all of these aspects of virus-associated disease are seen would be desirable because reproducible data can be generated in inbred animals, and larger numbers of animals can be included for statistical analysis of biological outcomes.
To generate a model that satisfies these criteria, we have serially passaged SARS-CoV in the respiratory tract of young BALB/c mice, resulting in a lethal virus that causes dose-dependent weight loss and mortality associated with higher viral titers in the respiratory tract than are seen with the wild-type virus and with histopathologic findings of severe pulmonary disease. The characteristics of this lethal mouse-adapted SARS-CoV, (MA15), are reported here.
Results
Adaptation of SARS-CoV for Increased Virulence in a Young BALB/c Mouse Model
Adaptation of SARS-CoV (Urbani) was achieved by serial passage through lungs of BALB/c mice as previously described for influenza A and influenza B viruses [4,5]. Lightly anesthetized mice were inoculated intranasally with 105 50% tissue culture infectious dose (TCID50) per mouse of SARS-CoV (Urbani). Two to three days post-infection (d.p.i.), when peak viral titers are observed, lungs were harvested from infected mice and clarified homogenates were used as inocula for continued serial passage via intranasal inoculation in mice. To screen for virulence in mice, groups of young naïve mice (N = 5–8) were inoculated with lung homogenates collected at passage 2, 10, and 15 (P2, P10, and P15, respectively), weighed daily, and observed twice daily for signs of morbidity or mortality. Deaths were not observed following inoculation with P2 or P10; however, increased morbidity, as indicated by weight loss, was noted in P10-inoculated mice at day 3 post-infection (p.i.) (unpublished data). Mortality was observed in P15-inoculated mice; 60% of P15-inoculated mice died or displayed extreme morbidity and were euthanized by 5 d.p.i. Mortality was not observed in mice infected with intermediate passages (P11–P14), and accompanying morbidity was not measured (unpublished data). Supernatant from lung homogenates at P15 contained a heterogeneous virus pool as evident upon sequence analysis of cDNA fragments generated by reverse transcriptase PCR (RT-PCR) amplification from purified viral RNA (vRNA). Dual peaks were observed on sequence chromatograms at several nucleotide residues, indicating mixed populations in the virus pool. To obtain a clonal population, P15 virus was biologically cloned by three rounds of terminal dilution in Vero cells. Five clones were screened for lethality in 6- to 8-wk-old BALB/c mice; these clones caused mortality from 50% to 100%. One clone, designated MA15, resulted in 100% mortality within 6 d. BALB/c mice aged 6 to 8 wk, 4 mo, and 13 mo were all susceptible to MA15 infection, with severe morbidity or death occurring within 3 to 5 d following intranasal inoculation.
Mutations Identified in the Mouse-Adapted Virus
In order to identify the mutations in MA15 associated with the lethal phenotype, the sequence of MA15 was compared with that of SARS-CoV (Urbani). The sequence of the initial SARS-CoV (P0) virus was identical to the published SARS-CoV (Urbani) sequence, and six nucleotide substitutions were identified in the MA15 genome compared with that of SARS-CoV (Urbani). All six substitutions were predicted to cause amino acid substitutions. These six mutations were localized to open reading frame (ORF) 1ab (four mutations) and ORFs S and M (one each) of SARS-CoV (Figure 1). Independent analysis conducted by The Institute for Genomic Research (Rockville, Maryland, United States), under a National Institute of Allergy and Infectious Diseases (NIAID) contract, confirmed the same six mutations in the MA15 sequence compared with those in the SARS-CoV (Urbani) sequence.
Figure 1. Schematic Diagram of SARS-CoV Genome Indicating Mutations Found in MA15 Virus.

(A) The 29,727 nucleotide positive-sense RNA genome of SARS-CoV is depicted in this to-scale drawing with ORFs indicated by shaded boxes (dark gray, structural and non-structural proteins; light gray, accessory genes X1–X5 [37]; and straight lines, non-coding regions). Asterisks indicate the sites of the six nucleotide changes (compared with the published SARS-CoV (Urbani) sequence) resulting in six coding mutations found in the mouse-adapted SARS-CoV (MA15).
(B) The six mutations found in MA15. aORF, open reading frame. bCDS, coding sequence, sequence of nucleotides that corresponds with the sequence of amino acids in a protein (location includes start and stop codon). cnsp, non-structural protein, cleavage product of ORF 1ab; Mainpro, main 3C-like protease; Hel, helicase. dRBM, receptor binding motif (amino acids 424–494).
Generation of Recombinant Clones
To confirm that the six mutations identified in the MA15 virus were responsible for lethality in mice, the mutations were introduced into cDNA clones from which recombinant SARS-CoVs were recovered. Sequence analysis confirmed that recombinant viruses contained the appropriate mutation sets that were derived from the MA15 virus. In addition to generating a recombinant virus that included the six mutations (rMA15), two additional recombinants were generated containing either the two mutations in the structural protein genes or the four mutations in the ORF 1ab (rMA15SM and rMA15ORF1ab, respectively).
MA15 and Recombinant Viruses Replicate In Vitro with Similar Kinetics
The three recombinant viruses demonstrated similar kinetics and levels of viral replication compared with that of SARS-CoV infectious clone (icSARS-CoV), a wild-type recombinant SARS-CoV (Urbani) generated from cDNAs, the biologically derived MA15 virus, and SARS-CoV (Urbani) in in vitro single-cycle growth analyses in Vero E6 cells. At a multiplicity of infection (MOI) of 0.1, viruses reached peak replication (107.0–7.5 pfu/mL) at ∼24 h.p.i., with a slight delay in peak titers for rMA15ORF1ab (Figure S1). Northern blot analysis of RNA from infected Vero E6 cells indicated that genomic vRNA and viral mRNA and all eight sub-genomic mRNAs were present in similar ratios for the recombinant viruses and MA15 virus as for SARS-CoV (Urbani) (Figure 2A). The level of expression and mobility of the structural proteins (S and N) and an accessory protein (ORF 3a, also called X1) of the recombinant viruses and MA15 virus were comparable to those of SARS-CoV (Urbani), as determined by Western blot analysis (Figure 2B). Although S, N, and X1 are somewhat reduced in the lane (Figure 2B) from icSARS-CoV–infected cultures, this is associated with reduced amounts of total protein added to the well, rather than with any specific reduction in X1 expression. Thus, the recombinant viruses (rMA15SM, rMA15ORF1ab, rMA15, and icSARS-CoV) and the MA15 virus demonstrate no defects in replication or in RNA or protein expression compared with SARS-CoV (Urbani) in Vero E6 cells.
Figure 2. Recombinant SARS-CoVs Demonstrate Normal Processing of vRNAs and Proteins.

(A) Northern analysis. Intracellular RNA was isolated 10.5 h.p.i. from Vero E6 cells infected with indicated viruses or from mock-infected cells. RNA (0.1 μg) was treated with glyoxal, separated on 1% agarose gel, transferred to a BrightStar-Plus membrane, and probed with an N gene–specific biotinylated oligomer as described in Materials and Methods.
(B) Western analysis. Cell lysates were separated on two 7.5% SDS-PAGE gels, transferred to polyvinylidene fluoride and probed with either mouse anti-S antisera (top panel) or probed first with a mouse anti-X1 antisera (sera raised to accessory protein X1 [37]; middle panel), and then stripped and probed again with a mouse anti-N antisera (bottom panel). Each primary antibody was followed by goat anti-mouse HRP-conjugated secondary antibody and visualized by enhanced chemiluminescence.
Replication of MA15 and rMA15 in Mice
Groups of naïve mice were inoculated with serial 10-fold dilutions of MA15 virus, and the dose-dependent weight loss and lethality observed following infection is summarized in Table 1. Mice receiving a dose ≥103.9 TCID50 of MA15 virus died or lost more than 20% of their initial body weight between days 3 and 5 p.i. Mice experiencing weight loss in excess of 20% initial body weight are euthanized in accordance with our animal study protocol. The 50% lethal dose (LD50) was 104.6 TCID50. At an intermediate, non-lethal dose (102.9/mouse), mice lost 8.4% ± 2.5% of initial body weight by day 4 p.i. At a lower dose, weight loss was not significant (4.3% ± 1.0% by day 4 p.i.)
Table 1.
Morbidity and Mortality in Mice following Intranasal Administration of MA15 and Recombinant MA15 Viruses

Similarly, groups of naïve mice were inoculated with serial 10-fold dilutions of rMA15, rMA15ORF1ab, or rMA15SM, and followed daily for signs of morbidity (indicated by weight loss) and mortality. Mortality was observed only in rMA15-inoculated mice at doses equal to or in excess of 104.4 TCID50/mouse (Table 1). The LD50 for rMA15 of 103.9 TCID50 is similar to the LD50 observed for the MA15 virus. Consistent with observations following infection with MA15 virus, mice inoculated with low concentrations of rMA15 (100.4–2.4 TCID50/mouse) had little to no significant weight loss, but an intermediate dose of rMA15 (103.4 TCID50/mouse) resulted in significant weight loss but no mortality. Thus rMA15 recapitulated the phenotype of the MA15 virus in mice. Inoculation with the highest doses of rMA15SM and rMA15ORF1ab (106.2 TCID50/mouse) did not result in mortality (Table 2), and only rMA15ORF1ab-infected mice demonstrated significant morbidity as indicated by weight loss.
Table 2.
Morbidity and Mortality in Mice following Intranasal Administration of MA15 and Recombinant Viruses

The rapid lethality observed following administration of MA15 virus (or its recombinant clone rMA15) to BALB/c mice could result from changes in tissue tropism with or without viremia, increased viral load and subsequent necrosis in pulmonary or extrapulmonary tissues, failure of innate or early adaptive immune responses, immunopathology in pulmonary or extrapulmonary tissues, or a combination of these or other factors. In order to evaluate whether changes in tissue tropism or levels of viral replication could contribute to the lethal phenotype of the MA15 virus, viral titers in lungs, spleen, liver, and brain of BALB/c mice were determined at various time points following intranasal inoculation with SARS-CoV (Urbani) or MA15.
Efficient Replication of MA15 Virus in Pulmonary Tissues
Mice were inoculated intranasally with SARS-CoV (Urbani) (105 TCID50/mouse, a dose that results in peak viral titers in lungs of mice 2 d.p.i.) or with lethal (105.6 TCID50/mouse) or sub-lethal (103.6 TCID50/mouse) doses of MA15 virus. At various time points p.i., mice were sacrificed and tissues harvested for subsequent processing. Data from replicate experiments were combined. High titers of virus were detected in the lungs of mice through day 5 p.i. (Figure 3). One day after inoculation, the amount of SARS-CoV (Urbani) in lungs was ∼106 TCID50/g tissue. Following administration of a lethal dose, MA15 virus was found to replicate to significantly higher titers of ∼109 TCID50/g tissue within 24 h (p = 0.0001; Figure 3). In this study and in previous studies (K. Subbarao, A. Roberts, L. Vogel, E. Lamirande, et al., unpublished data), peak virus titers (∼107.0 TCID50/g tissue) occur at day 2 following inoculation with SARS-CoV (Urbani). In comparison, by day 2 p.i., viral titers in MA15-inoculated mice reached, or persisted at, titers of ∼109 TCID50/g tissue, regardless of inoculating dose (Figure 3). Furthermore, in mice inoculated with a sub-lethal dose of MA15, virus was detected at 107.7 TCID50/g lung on day 5 p.i. By comparison, in SARS-CoV (Urbani)–infected mice, virus was detected at titers of 105.1 TCID50/g lung on day 5 p.i. By 6 d.p.i., virus was no longer consistently detected in mice inoculated with SARS-CoV (Urbani) or with sub-lethal doses of MA15 virus (Figure 3).
Figure 3. Virus Titers in Lungs of BALB/c Mice Inoculated with SARS-CoV or MA15 Virus.

Data represents a compilation of two experiments. In each experiment, groups of four mice were inoculated intranasally with 50 μL of SARS-CoV (Urbani) (105.0 TCID50/mouse, black bars) or MA15 virus at lethal (105.6 TCID50/mouse, white bars) or sub-lethal (103.6 TCID50/mouse, light gray bars) doses. Mice were sacrificed on indicated d.p.i. Mice receiving lethal doses of MA15 virus did not survive beyond day 4. Bars represent mean viral titers; error bars indicate standard error. Asterisks indicate significant differences (p < 0.05) compared with titers in mice receiving lethal doses of MA15 virus. Dotted line indicates lower limit of detection (101.5 TCID50/g).
MA15 Virus Detected in Extrapulmonary Tissues
Following inoculation with a lethal dose of MA15 virus, virus was also recovered from spleen, brain, and liver from day 1 through day 4 p.i. at titers ranging from 101.8 to 102.7 TCID50/g (brain) and from 101.8 to 103.3 TCID50/g (spleen) (Table 3). Virus was detected more frequently in the liver and at higher titers (102.1–104.3 TCID50/g) than in brain or spleen. Virus was not recovered from any of these organs following inoculation with SARS-CoV (Urbani), except in one mouse, sacrificed 5 d.p.i., in which virus was detected in the spleen at a titer just above the limit of detection (Table 3). Although the levels of viral replication detected in these tissues in MA15-infected mice are modest, the number of mice in which virus was detected is remarkable, and both the frequency and the titers are significantly higher than those observed in SARS-CoV (Urbani)–infected mice.
Table 3.
Detection of SARS-CoV and MA15 Infectious Virus

In a separate experiment, infectious virus was also detected in sera of mice infected with MA15 virus or recombinant SARS-CoVs on days 2 and 4 p.i. Data from this experiment suggest that the presence of virus in serum may be facilitated by the mutations in the S and M genes, since virus was detected more frequently and at higher titers in sera from mice infected with MA15, rMA15, or rMA15SM than in sera from mice infected with icSARS-CoV or MA15ORF1ab (Figure S2).
vRNA Detected in Pulmonary and Extrapulmonary Tissues
In addition to recovery of infectious virus from these tissues, total RNA was also isolated from whole blood, brain, kidney, liver, intestine, spleen, thymus, heart, and lungs of BALB/c mice after infection. Although quantitative virology holds biological relevance since it indicates viral burden by measuring infectious virus, RT-PCR of vRNA or sub-genomic mRNAs may be more sensitive assays for the presence of virus. Due to the replication strategy of SARS-CoV, primers that specifically amplify sub-genomic mRNAs allow detection of products of viral transcription. Furthermore, amplification of virus-specific mRNAs allows distinction from vRNA that may represent non-viable virus [6]. RT-PCR amplification of genomic, antigenomic, and sub-genomic mRNA-specific sequences was employed to confirm the presence of SARS-CoV nucleic acid or viral transcription products in different tissues.
Groups of four mice were inoculated with a lethal dose of MA15 virus or with SARS-CoV (Urbani) (105 TCID50/mouse). Mice inoculated with MA15 virus did not survive past day 4 p.i. SARS-CoV (Urbani)–infected mice were followed through day 14 p.i. vRNA, and viral mRNA were amplified from lungs of MA15-infected mice on days 1–4 p.i. In contrast, vRNA was amplified from SARS-CoV (Urbani)–infected mice through day 14 p.i., but mRNA, indicating viral transcription, was consistently amplified only through day 5 p.i. mRNA was detected as late as day 7 p.i. by RT-PCR in one of four of the SARS-CoV (Urbani)–infected mice (Tables 4 and 5).
Table 4.
Detection of SARS-CoV and MA15 vRNA

Table 5.
Detection of SARS-CoV and MA15 mRNA

We were also able to amplify vRNA and mRNA from blood, brain, thymus, heart, and spleen of MA15-infected mice (Tables 4 and 5). Viral mRNA was consistently detected by RT-PCR in the tissues of MA15-inoculated mice from day 2 p.i. through day 4 p.i. and in the thymus and heart of MA15-infected mice as early as day 1 p.i. In contrast, vRNA and viral mRNA were detected transiently in very few SARS-CoV (Urbani)–infected mice and only in the blood, spleen, and thymus. vRNA and viral mRNA were not detected by RT-PCR analysis in liver, intestines, or kidneys from MA15 or SARS-CoV (Urbani)–infected mice. These tissues may contain factors that inhibit RT-PCR amplification of SARS-CoV RNAs, since virus was isolated from liver homogenates of MA15-infected mice but was not detected by RT-PCR, and viral mRNA was detected in intestines by in situ hybridization (unpublished data). In summary, virus or viral-specific RNA was detected in blood, lung, thymus, brain, spleen, liver, and heart from almost all MA15-infected mice. In contrast, virus or viral-specific RNA was detected in the lungs, and only sporadically in blood, spleen, and thymus from SARS-CoV (Urbani)–infected mice (Tables 3–5).
Histopathologic and Immunohistochemical Findings
The lungs of mice infected with SARS-CoV (Urbani) showed a rapid progression from focal, mild, perivascular, and peribronchiolar mononuclear inflammatory cell infiltrates to a diffuse, but transient, interstitial pneumonitis on day 3 p.i. (Figure 4A, 4C, 4E, and 4G). Occasional ciliated columnar epithelial cells of the bronchioles and alveolar pneumocytes stained for viral antigen by immunohistochemistry (IHC) (Figure 5A, 5C, 5E, and 5G). No viral antigens were detected at day 14 p.i., and most mice at this time point showed no significant pulmonary histopathology (unpublished data).
Figure 4. Histopathological Changes in Lungs from SARS-CoV (Urbani)–Infected and MA15-Infected Mice (Hematoxylin and Eosin Stain, Original Magnifications ×25).
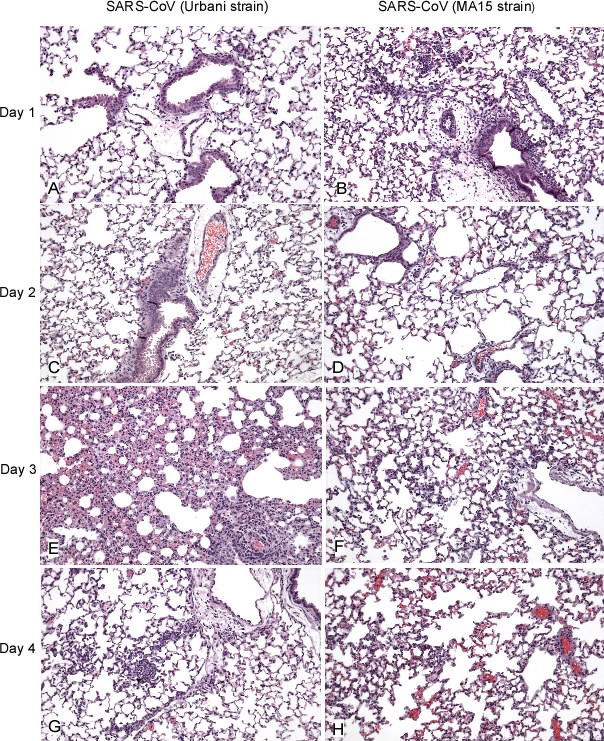
Day 1 p.i. (A) SARS-CoV (Urbani)–infected mice: no significant inflammatory cell infiltrates. (B) MA15-infected mice: foci of perivascular, peribronchiolar, and interstitial inflammatory infiltrates comprised predominantly of mononuclear cells.
Day 2 p.i. (C) Same as in (A). (D) MA15-infected mice: small, mononuclear inflammatory cell focus in the alveolar interstitium.
Day 3 p.i. (E) SARS-CoV (Urbani)–infected mice: extensive and confluent interstitial pneumonitis. (F) MA15-infected mice: mild interstitial inflammation with diffuse pyknosis and karyorrhexis of alveolar pneumocytes.
Day 4 p.i. (G) SARS-CoV (Urbani)–infected mice: small, discrete inflammatory cell infiltrates involving the alveolar interstitium. (H) MA15-infected mice: mild interstitial inflammation and necrotic intraalveolar debris. Mice were inoculated with 105.6 TCID50 MA15 virus/mouse or 105.0 TCID50 SARS-CoV (Urbani)/mouse.
Figure 5. Immunohistochemical Staining for SARS-CoV Antigen in Lungs of SARS-CoV(Urbani)–Infected or MA15-Infected Mice.
Immunohistochemical staining is shown in red.
Day 1 p.i. (A) SARS-CoV (Urbani)–infected mice: antigen present in occasional ciliated respiratory epithelial cells in bronchioles. (B) MA15-infected mice: diffuse and extensive staining of bronchiolar respiratory epithelium and alveolar pneumocytes.
Day 2 p.i. (C) SARS-CoV (Urbani)–infected mice: staining of occasional bronchiolar epithelial cells and rare alveolar pneumocytes. (D) MA15-infected mice: staining predominantly in intact and detached cells in pulmonary alveoli.
Day 3 p.i. (E) SARS-CoV (Urbani)–infected mice: occasional staining of alveolar pneumocytes and bronchiolar epithelium. (F) MA15-infected mice: extensive staining of abundant, intraalveolar, necrotic debris.
Day 4 p.i. (G) SARS-CoV (Urbani)–infected mice: occasional focus of predominantly pneumocyte staining. (H) MA15-infected mice: extensive staining of abundant, intraalveolar, necrotic debris. Primary antibody, rabbit anti-SARS-CoV antibody; secondary antibody conjugated with alkaline phosphatase with naphthol fast-red and hematoxylin counterstain; original magnifications ×25. Mice were inoculated with 105.6 TCID50 MA15 virus/mouse or 105.0 TCID50 SARS-CoV (Urbani)/mouse.
In comparison, the pulmonary pathology of mice infected with MA15 virus showed a rapid progression of inflammatory changes (Figure 4B, 4D, 4F, and 4H), but with more extensive damage to bronchiolar and alveolar epithelial cells (Figure 6A and 6B). These changes were especially evident in pneumocytes, and many detached, pyknotic, and necrotic cells were identified in alveolar spaces (Figure 6A). Inflammatory infiltrates, although consistently identified, were generally similar in intensity to those observed in the lungs of SARS-CoV (Urbani)–infected mice (Figure 4). However, the distribution and amount of viral antigens identified by IHC was far greater in the lungs of MA15-infected mice than in the lungs of mice infected with SARS-CoV (Urbani) (Figure 5). Intracellular SARS-CoV antigens were extensively and abundantly distributed in bronchiolar epithelium, alveolar pneumocytes, and in necrotic debris within the alveoli and bronchiole lumens of MA15-infected mice (Figure 6C and 6D).
Figure 6. Histopathology and Immunohistochemical Localization of SARS-CoV Antigens in the Lungs of Mice Infected with MA15 Virus.

Abundant necrotic cellular debris (arrows) in alveoli (A) and a bronchiole lumen (B) of mice at days 2 and 3 p.i., respectively. Abundant SARS-CoV antigens (arrowheads) within alveolar pneumocytes (C) and in necrotic alveolar and bronchiolar cellular debris in mice at day 2 p.i. (D).
(A and B) Hematoxylin and eosin stain; (C and D) primary antibody, rabbit anti-SARS-CoV antibody; secondary antibody conjugated with alkaline phosphatase with naphthol fast-red and hematoxylin counterstain; original magnifications ×100. Mice were inoculated with 105.6 TCID50 MA15 virus/mouse.
No significant histopathologic changes were identified in extrapulmonary organs, including the liver, spleen, thymus, or brain, of mice infected with SARS-CoV (Urbani) or MA15 virus. Furthermore, SARS-CoV antigens were not detected in any of these tissues by IHC staining.
Changes in Blood Counts and Chemistry in MA15-Infected Mice
Infection of young BALB/c mice with 106.4 TCID50/mouse of MA15 virus resulted in elevated levels of the liver enzyme alkaline phosphatase (ALP) in sera collected on days 1–6 p.i. ALP levels were significantly higher in sera of MA15-inoculated mice than they were in pre-infection sera from the same mice. Serum ALP levels in MA15-inoculated mice were also significantly higher than those of mice inoculated with 106.4 TCID50/mouse of SARS-CoV (p = 0.0045; Table S5). Levels of other liver enzymes, including aspartate aminotransferase, alanine aminotransferase, gamma glutamyl transferase, creatine kinase, urea nitrogen, total bilirubin, and albumin, were not significantly altered following inoculation with MA15 virus (unpublished data). Although moderately elevated levels of creatinine were observed in MA15-inoculated mice, no significant differences were seen between MA15- and SARS-CoV–inoculated mice, suggesting that this was not associated with the lethal phenotype of the MA15 virus. Furthermore, significant lymphopenia and neutrophilia were observed following infection with SARS-CoV and MA15 virus. Although some samples were lost due to coagulation of blood, the alterations in the lymphocyte and neutrophil counts were more severe following infection with MA15 virus than they were following SARS-CoV infection (Table S5).
Primary Infection with SARS-CoV (Urbani) Protects Mice from Lethal Challenge with MA15 Virus
In order to determine whether MA15 virus can be used as a more stringent challenge in evaluating vaccines and antiviral therapy designed for SARS-CoV than could the non-lethal SARS-CoV (Urbani), we inoculated eight mice with 50 μL of SARS-CoV (Urbani) at 105 TCID50/mouse and mock-immunized an additional eight mice. Four weeks after inoculation, the mice were bled for determination of SARS-CoV–specific serum neutralizing antibodies and challenged with 106.9 TCID50 of MA15 virus. Mice were followed daily for signs of morbidity and mortality. Serum neutralizing antibodies were detected at low titers (1:10) in SARS-CoV–immunized mice but not in mock-immunized mice (<1:8). Nevertheless, SARS-CoV–immunized mice were protected from lethal challenge and demonstrated reduced morbidity (∼11% weight loss compared with >20% weight loss in mock-immunized mice) following a high titer challenge with MA15 virus. Mock-immunized mice did not survive past day 4 following challenge with MA15 virus (Figure 7).
Figure 7. Challenge of SARS-CoV– or Mock-Immunized Mice with the Lethal MA15 Virus.

Groups of eight mice (8 wk old) were immunized intranasally with 50 μL of SARS-CoV (Urbani) (105TCID50/mouse) or L15 tissue culture media. Four weeks after immunization, mice were challenged intranasally with 50 μL MA15 virus (106.9 TCID50/mouse), weighed daily, and observed twice daily for morbidity and mortality. Surviving mice that lost in excess of 20% initial body weight were euthanized. Symbols represent mean values for SARS-CoV–immunized mice (triangles) and mock-immunized mice (circles). Error bars indicate standard error.
Discussion
Serial passage of SARS-CoV (Urbani) in the lungs of BALB/c mice resulted in a mouse-adapted SARS-CoV (MA15 virus) that is lethal for young (6- to 8-wk-old) BALB/c mice. The virulence and lethality of the MA15 virus result from six mutations in the SARS-CoV genome that occurred within 15 passages through BALB/c mice. Introduction of these six mutations into a recombinant SARS-CoV infectious clone, rMA15, conferred a lethal phenotype on the virus for young BALB/c mice. Five of these six mutations (not including the T67A mutation occurring in the non-structural protein [nsp] 9 of ORF 1ab) occur within nsp 5 (two mutations), nsp 13, S, and M. These genes have been reported as ones where sequence evolution occurred during adaptation of SARS-CoV in humans [7]. The mutations in nsp 9, nsp 5 (H133Y and E269A within the main protease, 3CLpro), and nsp 13 (A4V within the helicase protein) do not occur within any known functional domains and do not alter known cleavage sites utilized in the processing of the ORF 1ab polyprotein. Furthermore, the mutations in the MA15 virus do not occur at any specific amino acid positions identified by the Chinese SARS Molecular Epidemiology Consortium [7]. Only the mutation in S (Y436H) occurs within a known functional domain, the receptor-binding motif (RBM). Other reports have indicated that mutations within the RBM may account for increased affinity of the virus for its cellular receptor angiotensin converting enzyme 2 [8]. However, the Y436H mutation in MA15 virus does not occur at previously identified sites of the RBM and angiotensin converting enzyme 2 interaction, and preliminary findings suggest that the Y436H mutation does not increase binding of the SARS-CoV (Tor2) RBD to murine angiotensin–converting enzyme 2 [9].
Reports of adaptation of an influenza A virus for increased virulence in mice indicate that multiple gene products may interact or contribute independently to virulence. In one adaptation of influenza A/FM/1/47 (FM-MA), findings indicated that four viral gene segments contributed to increased virulence [4], but additional analyses indicated that mutations occurring in at least two and likely three gene products acted synergistically to account for the increased virulence [10–12]. Recombinant icSARS-CoV produced mild pneumonia identified by X-ray changes in macaques, similar to the clinical disease noted with wild-type SARS-CoV (Urbani) [13]. Consistent with these findings, recombinant SARS-CoV bearing the six novel mouse-adapted mutations, rMA15, recapitulated a fatal respiratory disease phenotype in mice, demonstrating the ability of the SARS molecular clones to capture complex disease phenotypes. We generated two other recombinant SARS-CoVs, rMA15SM (encoding the two mutations in the S and M genes) and rMA15 ORF1ab (encoding the four mutations in ORF 1ab). Neither of these was lethal (Table 2) in BALB/c mice, indicating that SARS-CoV adaptation for BALB/c mice involves mutations in at least two and possibly three genes (S + ORF 1ab, M + ORF 1ab, or S + M + ORF 1ab). Although recombinant viruses bearing two or four of the mutations were not lethal, each had a different phenotype than that of SARS-CoV (Urbani). It is possible that the mutations in S and M contribute to increased viremia (Figure S2) and that the mutations in ORF 1ab contribute to increased pathogenicity indicated by weight loss (Table 2).
Both quantitative virology and IHC analysis of the lungs of MA15-infected mice indicate extraordinarily high levels of viral replication in pulmonary tissues as early as 24 h.p.i., and the level of replication remains high through day 4 p.i. Unlike SARS-CoV–infected mice, in which viral antigen is detected by IHC staining at modest levels in bronchial epithelium and in alveolar pneumocytes on days 1 and 2 p.i., the bronchial epithelium of MA15-infected mice is replete with viral antigen at day 1 p.i., as are alveolar pneumocytes on days 1 and 2 p.i. By day 3 p.i., viral antigen is rarely detected in the lungs of SARS-CoV–infected mice; in contrast, viral antigen remains abundant in pneumocytes, necrotic debris, and pyknotic cells in MA15-infected mice through day 4 p.i.
In this model, day 3 to day 4 p.i. seems to be a critical time for the outcome of SARS-CoV infection. SARS-CoV (Urbani)–infected mice demonstrate a pronounced but transient interstitial inflammation at day 3 p.i. that is absent in MA15-infected mice. This transient inflammation is not associated with significant weight loss but coincides with the beginning of viral clearance from the lungs. In contrast, by day 3 p.i., MA15 virus–infected mice lose weight, and necrotic cellular debris begins to fill the bronchioles and alveoli. By day 4 p.i., MA15-infected mice have lost in excess of 20% of their initial body weight, and several die without other overt clinical signs such as ataxia, paralysis, hunched posture, etc. Viral titers in lungs of MA15-infected mice are up to 1,000-fold higher than those in SARS-CoV–infected mice by 24 h.p.i. and remain higher than peak levels in SARS-CoV–infected mice through day 4 p.i. In situ hybridization further confirms the intense and persistent signal of MA15 (and rMA15) vRNA in lung tissues at day 4 p.i. when vRNA from SARS-CoV (Urbani) and the non-lethal rMA15SM and rMA15ORF1ab viruses are less abundant (Figure S3). Objective data related to respiratory distress such as plethysmography and measurement of blood gases could not be collected because of practical and logistical constraints on experiments carried out in an Animal Biosafety Level 3 laboratory. However, our findings indicate that MA15 virus–infected mice die as a result of overwhelming pulmonary viral infection and destruction of bronchiolar epithelial cells and alveolar pneumocytes. Viral load in SARS cases was an important determinant of severe disease and death [14], but the mechanism of disease leading to fatal outcome in human cases of SARS may be different than that observed in MA15-infected BALB/c mice. The mechanism of death following SARS-CoV infection in humans, and particularly the relative contribution of virus-induced damage and immunopathology, are not fully understood. The histopathology documented in the mice infected with the MA15 virus includes a rapid progression and extensive damage to bronchiolar and alveolar epithelial cells (Figure 6A and 6B), but it does not show some features such as alveolar edema and hyaline membranes that were reported in many SARS-CoV–infected patients, or in aged mice infected with the Urbani strain of SARS-CoV [15]. This can be explained by the relative virulence of the MA15 strain, and the time p.i. when the lungs were evaluated. Mice infected with the MA15 virus developed severe morbidity and died within 3 to 5 d following infection and did not survive long enough to show progression of the diffuse alveolar damage seen in aged mice or in human patients, who survive for a relatively prolonged length of time before succumbing to complications of the acute infection. In aged mice following SARS-CoV infection, histopathologic changes indicative of progressive diffuse alveolar damage, including proteinaceous deposits around alveolar walls and intraalveolar edema, are seen beginning on day 5 [15].
The rapid progression of infection and the extensive and persistent pulmonary replication of the virus are accompanied by viremia and detection of virus in extrapulmonary sites, suggesting that other factors may contribute to the increased pathogenicity of the MA15 virus. A prolonged viremic state or a secondary viremia is seen in mice infected with MA15 and rMA15 that is not observed following infection with the recombinant SARS-CoVs (Urbani), rMA15SM, or rMA15ORF1ab. The MA15 virus model captures viremia and multi-organ involvement noted in human SARS patients [16]. However, as in all SARS cases, the primary site of infection is the lung.
Perfusion of tissues harvested from mice in these experiments was not performed, and therefore, we cannot be certain that the detection of vRNA in various extrapulmonary sites in MA15-inoculated mice is not due to virus carried to and remaining in the organs and blood. However, on days 2 through 4 p.i., MA15-specific mRNAs in various tissues are detected at a higher frequency and signal intensity than in whole blood, and the presence of MA15 vRNA in extrapulmonary tissues is detected by in situ hybridization. Furthermore, infectious virus was detected on day 1 p.i. in homogenates of extrapulmonary tissues when RT-PCR of RNA from blood did not amplify any viral specific products (Tables 3–5). Taken together, these observations support a hypothesis that virus is present and replicating at low levels in extrapulmonary tissues.
In BALB/c mice infected with MA15 virus or SARS-CoV, significant lymphopenia and neutrophilia were observed compared with pre-infection levels. MA15 virus–infected mice had significantly greater lymphopenia and neutrophilia than SARS-CoV–infected mice, reflecting hematological evidence of increased disease severity following MA15 virus infection. MA15 virus–infected mice also had significantly higher levels of ALP than uninfected or SARS-CoV–infected mice. Elevated levels of ALP, an enzyme found in all tissues, but especially concentrated in the liver, may indicate cell destruction in the liver, intestines, or other tissues. Reports of elevated ALP levels in SARS patients are rare [17,18], but neutrophilia and lymphopenia have been reported frequently in human cases of SARS [19–25]. Although steroid treatment and secondary bacterial infections may be speculated to be the cause of neutrophilia, some patients with SARS had neutrophilia prior to initiation of steroid treatment and in the absence of bacterial infections [26]. Additionally, elevated absolute neutrophil counts at presentation were independent indicators of severe outcome of SARS in human cases [21,27,28]. The definitive causes of lymphopenia, neutrophilia, and elevated ALP levels have not been identified in human cases of SARS or in this mouse model.
The mouse-adapted SARS-CoV MA15 virus will be a valuable tool in evaluating SARS-CoV vaccines and antiviral therapy. Quantitative virology was the only outcome measure available in young BALB/c mice challenged with SARS-CoV (Urbani). Because the MA15 virus replicates rapidly to high titer and is lethal for young BALB/c mice, this virus provides a more stringent challenge than the SARS-CoV (Urbani) virus in evaluating the efficacy of therapeutic interventions or prevention strategies. Prophylaxis that is able to rescue MA15-infected mice from a lethal outcome must lower viral burden in lungs very rapidly (e.g., within the first 24 h). Prophylaxis that accomplishes such reduction in the burden of MA15 virus may also reduce the severity of immunopathology that follows. A reduction of immunopathology was demonstrated in a SARS-CoV hamster model when a monoclonal antibody specific to SARS-CoV spike protein administered the day after infection was able to arrest a further rise in viral titer in the lungs [29]. If similar protection can be demonstrated against challenge with MA15 virus in BALB/c mice, the efficacy of intervention will be well proven.
Infection of young BALB/c mice with the MA15 virus provides a model that is small and accessible and that can be evaluated extensively at an immunological level. Finally, and most importantly, MA15 virus infection of young BALB/c mice provides many elements that replicate observations in acute (and chronic) cases of SARS infection in humans, including sequence changes during adaptation in several genes (nsp 5, nsp 13, S, and M); viral replication and histopathological changes in lungs of infected animals; viremia; detection of vRNA in extrapulmonary sites, including the intestines; clinical indicators of illness, including mortality; and changes in blood counts, including lymphopenia and neutrophilia. The availability of a molecular clone of the MA15 virus, and the ability to generate additional SARS-CoV recombinant viruses that recapitulate the in vivo phenotype, will allow for detailed probing of the mechanisms mediating SARS-CoV pathogenesis and acute lung damage.
Materials and Methods
All animal and viral experiments were conducted in Biosafety Level 3 laboratories or animal facilities, and all personnel wore personal protective equipment, including Tyvek suits and hoods and positive pressure HEPA-filtered air respirators. All animal protocols employed in these studies have been approved by NIAID's Animal Care and Use Committee.
Serial passage of SARS-CoV in mice.
A dose of 105 50% TCID50 of SARS-CoV (Urbani) was administered intranasally to a lightly anesthetized, 6-wk-old female BALB/c mouse in a total volume of 50 μL [30]. Two days after inoculation, the mouse was euthanized, and its lungs were removed and homogenized with an EX-Gen Omni GLH homogenizer (Omni International, http://www.omni-inc.com) as a 10% w/v suspension in Leibovitz's L15 Medium (Invitrogen, http://www.invitrogen.com), supplemented with the following antibiotics: 0.4 mg/L piperacillin (Sigma, http://www.sigmaaldrich.com), 0.1 mg/L gentamicin (Invitrogen), and 5 mg/L amphotericin B (Quality Biological, http://www.qualitybiological.com). The lung homogenate was clarified by low-speed centrifugation at 2,000 rpm (650g) for 5 min, and the supernatant was administered intranasally to three naïve mice. The process of intranasal inoculation of three female BALB/c mice with pooled, clarified supernatants of 10% lung homogenates collected 2 to 3 d.p.i. was repeated 14 times.
Identification of lethal phenotype and biological cloning of a lethal virus.
The supernatant from P15 lung homogenates was subjected to three rounds of terminal dilution on Vero cells. Then, 5-fold serial dilutions were added to cells in 96-well plates (one dilution/plate). Two to three days later, when cytopathic effect was evident in no more than nine wells per plate, supernatants from infected wells were collected, diluted, and transferred to fresh monolayers of Vero cells. After the third round of terminal dilution, 50 μL of five independent clones were screened for lethality in four 8-wk-old female BALB/c mice. Mortality associated with these five clones was between 50% and 100%. One of these five clones (MA15) that caused 100% lethality within 6 d was expanded by two more passages in Vero cells and used in further studies.
Virus replication in the respiratory tract of mice.
Young (6- to 8-wk-old), female BALB/c mice were lightly anesthetized and inoculated intranasally with 50 μL of serially diluted virus. For SARS-CoV (Urbani), a dose of 105 TCID50/mouse was administered unless noted otherwise. For lethal doses of MA15 virus, the dose administered was equal to105.6 TCID50/mouse; for sub-lethal doses, the dose administered was equal to 103.6 TCID50/mouse. At various time points p.i., mice were euthanized and tissues collected for analyses. For determination of viral titers, tissues were homogenized to a final 10%w/v suspension in Leibovitz's L15 Medium supplemented with antibiotics. Tissue homogenates were clarified by low-speed centrifugation, and virus titers were determined in Vero cells on 24- and 96-well plates as previously described [30]. Virus titers are expressed as TCID50/g of tissue with a lower limit of detection of 101.5 TCID50/g.
Purification of vRNA.
Tissues homogenates were clarified by low-speed centrifugation; supernatants were transferred to Eppendorf tubes (http://www.eppendorf.com) and further clarified by centrifugation at 16,000g for 3 min. vRNA was extracted from 600 μL of supernatant using the QIAamp Viral RNA Mini Kit (Qiagen, http://www1.qiagen.com) eluted in 50 μL of water, quantified by measurement of optical density at 280 nm on a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, http://www.nanodrop.com), and stored at −80 °C.
RT-PCR and sequence analysis.
First-strand cDNAs were generated by reverse transcription from RNA purified from clarified cell supernatants of Vero cells infected with either SARS-CoV MA15, SARS-CoV (Urbani) (P0), or from the clarified supernatants of P15 lung homogenates using reagents from the Brilliant qRT-PCR Plus Core Reagent Kit (Stratagene, http://www.stratagene.com). In brief, 11 μL of vRNA was heat denatured at 95 °C for 2 min and quenched on ice before addition of 10X Core RT Buffer, 0.2 μg random primers, 0.08 μmol dNTP mixture, and 20U StrataScript reverse transcriptase for a 20-μL reaction mixture. Reverse transcription was performed with an initial incubation of 25 °C for 10 min, followed by a 30-min incubation at 45 °C and a 3-min denaturation at 95 °C. First-strand cDNAs were amplified in overlapping PCR products spanning the entire genome. Three microliters of each cDNA reaction were amplified by PCR using the Advantage-HF PCR Kit (BD Biosciences, http://www.bdbiosciences.com) or Herculase Enhanced DNA polymerase (Stratagene) as per manufacturers' protocols. Fifteen pairs of primers were used to generate the overlapping PCR products (Tables S1 and S2). Amplification products were visualized on agarose gels and purified by use of the QIAquick Gel Extraction Kit (Qiagen). PCR products were sequenced in both forward and reverse directions, using 69 forward and 62 reverse primers (Table S3). Automated sequencing was performed utilizing the BigDye Terminator version 3.1 Cycle Sequencing Kit (Applied Biosystems) as per manufacturer's instructions on an ABI Prism 3730 DNA Analyzer (Applied Biosystems). Sequences were assembled and analyzed with Vector NTI and Auto Assembler DNA Sequence Assembly Software (ABI Prism; Applied Biosystems). When a mutation was identified in comparison with the SARS-CoV (Urbani) published sequence, independent RT-PCR reactions were run and subsequent RT-PCR products were sequenced through the region containing the putative mutation to confirm the mutation.
Detection of SARS-CoV by RT-PCR.
Total RNA was isolated from various tissues or whole blood and purified using an RNeasy Mini Kit (Qiagen) with an on-column DNase digestion (RNase-Free DNase Set; Qiagen), as per manufacturer's protocol. Approximately 0.8 g of tissue, cut into pieces no larger than 0.5 cm on any one side, was collected into 1 mL of RNAlater (Ambion, http://www.ambion.com) and stored at room temperature for 24 h and subsequently at 4 °C until processed. Any unused tissue remaining after one month was moved to −20 °C. Approximately one-half or 0.2–0.4 g of tissue was homogenized with a disposable probe (Omni International) in 900 μL of RLT buffer (RNeasy Mini Kit; Qiagen), supplemented with 1% (v/v) β-mercaptoethanol (Sigma). RLT suspensions were transferred to 1.8-mL Eppendorf microcentrifuge tubes and centrifuged at 4 °C for 3 min at 16,000g. RNA was purified further as per manufacturer's protocol. RNA from blood (∼0.5 mL per sample) was purified using a QIAamp RNA Blood Mini Kit (Qiagen) as per manufacturer's protocol with the additional DNase on-column digestion. RNAs were quantified and 100 ng were reverse transcribed and amplified as described above. In brief, total RNA was heat denatured at 95 °C for 2 min and quenched on ice before addition of 10X First Strand Buffer, 20U RNase Block, 0.3 μg random primers, 0.08 μmol dNTP mixture, 25U StrataScript reverse transcriptase, and water for a 20-μL reaction. Reverse transcription was performed with an initial incubation at 25 °C for 10 min, followed by a 30-min incubation at 45 °C and a 3-min denaturation at 95 °C. Three microliters from each RT reaction were subsequently used in a 50-μL PCR with SARS-CoV–specific primers for amplification of a 314-bp sequence of the nucleocapsid gene (SARS-CoV forward (5′-ggtgacggcaaaatgaaagagc-3′), SARS-CoV reverse (5′-ggagaatttcccctactg-3′)). β2 microglobulin (β2M) primers were used in separate reactions as RNA quantity and PCR controls (β2M forward (5′-atgggaagccgaacatactg-3′), β2M reverse (5′-cagtctcagtgggggtgaat-3′)). PCR reactions were performed for a total of 35 cycles with 50 X Advantage cDNA polymerase (Clontech, http://www.clontech.com), 0.04 μmol dNTP mixture (Stratagene), and 10X cDNA PCR reaction buffer (Clontech) or Opti-Prime 10X Buffer 4 (Stratagene) supplemented with 0.02 μmol MgCl2 (Quality Biological) for RNA amplification of β2M and SARS-CoV, respectively (Table S4).
Construction of SARS-CoV cDNA plasmids for reconstructing the mouse-adapted recombinant virus rMA15.
Six codon changes (10384T>A, 10793A>C, 16177C>T, 12814A>G, 22797T>C, and 26428E>K) identified in MA15 compared with the published SARS-CoV (Urbani) sequence, were inserted into cDNA clones and used to construct and rescue infectious recombinant clones of SARS-CoV (Urbani) as described previously [31]. In brief, for each mutation two overlapping amplicons were generated by PCR, joined at primer-introduced BsmBI sites, and ligated into a cDNA of icSARS-CoV using two unique restriction sites that flanked the mutation of interest. PCR reactions were performed with Expand Long Taq (Roche Applied Sciences, http://www.roche-applied-science.com) in 30 cycles of 94 °C for 30 s, 55 °C for 30 s, and extensions at 68 °C for 1 min using the following primers on plasmids encoding portions of the icSARS-CoV genome: for the mutation at nucleotide position 10384, primer pairs 5′-10124 (5′-CATGT CATTT GCACA GCAG) and 3′-10364C (5′-ATTAG GTCTC ATGGC ACAC) and 5′-10384T>A (5′-AGACC TAATT ATACC ATTAA AG) and 3′-11091C (5′-CAAGC ACAAG AATGC GTGC) were used. A second PCR amplification using primers 5′-10124 and 3′-11091C produced a PCR product that was digested with the appropriate restriction enzymes (BglII, Mfe1) and ligated into the icSARS-CoV cDNA plasmid. Similarly, the 10793 mutation was constructed using primer pairs 5′-10124 and 3′-10770C (5′-CTTTC AAAGC AGCAC ACATA TC) and 5′-10793A>C (5′-GAAAG CGCTG CTGCA GAATG) and 3′-11091C, followed by amplification with primers 5′-10124 and 3′-11091C. This PCR product was digested with the appropriate restriction enzymes (BglII and Mfe1) and ligated into the icSARS-CoV cDNA plasmid. The 12814 mutation was similarly constructed using the primer pairs 5′-M13R3 and 3′-SARS D mu1(−) (5′-NNNCG TCTCG TTCCA GTTCT GCGTA AATTG TACCT GTACC) and 5′ SARS Dmu1(+) (5′-NNNCG TCTCT GGAAC CACCT TGTAG GTTTG) and 3′ D1500(−) (5′-CCCTG TAGAC GACAT CAGTAC). The two resulting amplicons were joined following digestion (with BsmBI) and purification (QIAquick PCR Purification Kit; Qiagen). The product DNA was digested with BamHI and AclI and inserted into an icSARS-CoV cDNA. The 16177 mutation was constructed using primers 5′-16004 (5′-CATCCTAATCAGGAGTATGC) and 3′-16157C (5′-CACCTACAGCCTGCAAGAC) and primers 5′-16177C>T (5′-GCTGTAGGTGTTTGTGTATTG) and 3′-18044C (CTTTATATCAACGCTGAGGTG). Primers 5′-16004 and 3′-18044C were used to amplify the appropriate fragment that was purified and digested with PflMI and BbvCI, and ligated into an icSARS-CoV cDNA. The 22797 mutation was constructed using primer pairs 5′ #38 (5′-AGAGG AACTG CTGTA ATGTC TC) and 3′ SMAS MuS(−) (5′-NNNCG TCTCT ATGAT TACCA GTTGA AGTAG CATC) and 5′ SMAS SMuS(+) (5′-NNNCG TCTCA TCATAA TTATA AATAT AGGTA TCTTA GACAT GG) and 3′ SARS E 4592(−) (5′-CTAGC ACAAA TGCCA GCTCC). The two resulting amplicons were joined following digestion (with BsmBI) and purification and ligation. The full-length product was digested and inserted into an icSARS-CoV cDNA utilizing restriction endonuclease sites AgeI and SalI. The 26428 mutation was constructed using primer sets 5′ #44 (5′-TGATCCTCTGCAACCTGAGC) and 3′ SMAS MuM(−) (5′-NNNCGTCTCACGGTAATAGTACCGTTGTCTGC) and 5′ SMAS MuM(+) (5′-NNNCGTCTCTACCGTTGAGAAGCTTAAACAACTCC) and 3′ SARS X5(−) (5′-NNNNNTTAATTAATTAATTTGTTCGTTTATTTAAAACAACA). Resulting amplicons were ligated following digestion with BsmBI. The mutation-containing product was digested and inserted into an icSARS-CoV cDNA at SwaI and NdeI restriction sites. In all cases, the final plasmids and the mutations in the plaque-purified viruses were verified by RT-PCR and sequencing.
Assembly of full-length cDNAs and recovery of recombinant viruses.
The full-length cDNAs of wild-type SARS-CoV (icSARS-CoV) or mouse-adapted SARS-CoV recombinants were produced as previously described [31]. RNA transcripts from full-length cDNAs were added to 800 μl of the Vero E6 cell suspension (8.0 × 106) in an electroporation cuvette and four electrical pulses of 450 V at 50 μF were given with a Gene Pulser II electroporator (Bio-Rad, http://www.bio-rad.com) similar to protocols previously described [32,33]. The presence of full-length cDNAs and transcripts was verified by separation on agarose gels and visualization by UV light. The transfected Vero E6 cells were seeded in a 75-cm2 flask and incubated at 37 °C for 2 d. Viruses were plaque purified in Vero E6 cells.
In vitro growth of recombinant viruses.
SARS-CoV (Urbani), icSARS-CoV (Urbani infectious clone), rMA15 (icSARS-CoV with all six mutations found in MA15), rMA15ORF1ab (icSARS-CoV with the four mutations found in the replicase genes of MA15 ORF 1ab), and rMA15SM (icSARS-CoV with the two mutations found in the structural genes of MA15 S and M) viruses were propagated on Vero E6 cells in Eagle's MEM supplemented with 10% fetal calf serum, kanamycin (0.25 μg/ml), and gentamicin (0.05 μg/ml) at 37 °C in a humidified CO2 incubator. Cultures of Vero E6 cells were infected in duplicate at an MOI of 0.1 for 1 h. Cell monolayers were washed twice with 2 mL of PBS and overlaid with complete MEM. Supernatants were collected at various times p.i. and virus was quantified by plaque assay on Vero E6 cells in 60 mm2 dishes. Plaques were visualized by neutral red staining and counted at 48 h.
Northern blot analysis.
Cultures of Vero E6 cells were inoculated with SARS-CoV viruses at an MOI of 1 and incubated for 1 h at 37 °C. At 10.5 h.p.i., intracellular RNA was isolated using TRIzol Reagent (Invitrogen) as directed by the manufacturer, and 0.1 μg of total mRNA was treated with glyoxal and separated on agarose gels using NorthernMax-Gly according to the manufacturer's directions (Ambion). The RNA was transferred to BrightStar-Plus membrane (Ambion) for 3.5 h and then cross-linked to the membrane by UV light. The blot was prehybridized and probed with an N gene–specific oligodeoxynucleotide probe (5′-cttgact gccgcct ctgct b t b ccct b ct b gc b-3′), where biotinylated nucleotides are designated with a subscript b. Blots were hybridized overnight, and washed with low and high stringency buffers as recommended by the manufacturer. Filters were incubated with strepavidin-AP, washed, and then incubated with chemiluminescent substrate CDP-STAR (Ambion). The blots were overlaid with film and developed.
Western blot analysis.
Ten hours p.i. with SARS-CoV (Urbani), MA15 (the biologically derived clone), icSARS-CoV, rMA15, rMA15ORF1ab, or rMA15SM cells were washed once in PBS and lysed in buffer containing 20 mM Tris-HCL (pH 7.6), 150 mM NaCl, 0.5% deoxycholine, 1% nonidet-p-40, and 0.1% sodium dodecyl sulphate (SDS). Supernatants clarified of nuclei were added to an equal volume of 5 mM EDTA/0.9% SDS, resulting in a final SDS concentration of 0.5%, and samples were heat inactivated for 30 min at 90 °C prior to transfer to Biosafety Level 2. After transfer to Biosafety Level 2, samples were again heat inactivated for 30 min at 90 °C before use. Equivalent sample volumes were loaded onto 7.5% Ready Gels (Bio-Rad) and transferred to a PVDF membrane (Bio-Rad). For detecting SARS-CoV antigens, blots were probed with polyclonal mouse antisera directed against Venezuelan equine encephalitis virus replicon particles (VRPs) that expressed the SARS-CoV ORF 3a (VRP-ORF3a), S (VRP-S), or N (VRP-N) proteins diluted 1:200 for SARS-CoV ORF 3a and 1:500 for S and N antisera. Dilutions were done in 5% blotto in PBS/0.5% Tween 20 and developed using ECL chemiluminescence reagents (Amersham, http://www.amershambiosciences.com).
In situ hybridization.
Paraffin-embedded sections, 5-mm thick, were probed with 35S UTP-labeled riboprobes complementary to the N gene of SARS-CoV (Urbani) or the Sindbis virus genome, as a negative control, using previously described methods [34]. In brief, following treatment to prevent nonspecific probe binding, the tissues were incubated overnight with either probe at 5 × 104 cpm/μL in hybridization buffer at 42 °C. The slides were then washed, dehydrated, and coated with NBT emulsion (Eastman Kodak, http://www.kodak.com), and incubated at −80 °C for 5 d prior to development. Positive signal, as determined by silver grain deposition, was evaluated by light microscopy.
Statistics.
Log-transformed virus titers were compared in a Mann-Whitney U test, and statistical significance was assigned to differences with p-values <0.05.
Histopathology and immunohistochemistry.
Lungs, liver, spleen, thymus, and brain were obtained from individual mice euthanized at various time points, fixed in 10% neutral buffered formalin, and processed for histopathologic and IHC examination as described [35,36]. Tissue sections (3 μm) were stained with hematoxylin and eosin and by using an immunoalkaline phosphatase technique with a hyperimmune rabbit anti-SARS-CoV nucleocapsid protein antibody at a dilution of 1/1000.
Supporting Information
Cultures of Vero E6 cells were infected at an MOI of 0.1 with indicated viruses in duplicate. Supernatants were collected at 1, 5, 13, 23, and 36 h.p.i., and virus was quantified by plaque assay in Vero E6 cells. Plaques were visualized by neutral red staining and counted at 48 h. SARS-CoV (Urbani) and rMA15SM replicate similarly to icSARS-CoV (unpublished data). Data points represent means of samples measured in duplicate. A repeat experiment demonstrated similar growth kinetics (unpublished data). Diamonds, icSARS-CoV; squares, MA15; circles, rMA15; and triangles, rMA15ORF1ab.
(28 KB PDF)
Groups of four mice were inoculated intranasally with 50 μL of recombinant viruses icSARS-CoV, rMA15SM, rMA15ORF1ab, rMA15, or MA15 and sacrificed on (A) day 2 p.i. or (B) day 4 p.i. Lungs and sera were collected for viral titration. Virus titers are expressed as log10 pfu/g lung (circles) or log10 pfu/mL serum (squares) from individual mice; bars represent the geometric mean for the group. Limits of detection for viral titers from lung and sera are 250 pfu/mL and 50 pfu/mL, respectively.
(35 KB PDF)
BALB/c mice were infected with SAR-CoV (Urbani), rMA15SM, rMA15ORF1ab, rMA15, or MA15 virus. Lungs were harvested on days 2 and 4 p.i., and in situ hybridization was performed as described in Materials and Methods. No specific signal was observed with a control riboprobe at any time point, while SARS-CoV–N specific signal was readily apparent within the lungs for all five viruses at day 2 p.i. In contrast, at day 4 p.i., abundant SARS-CoV–specific in situ signal was observed in lungs of mice infected with the lethal MA15 virus or the lethal recombinant rMA15, but not in lungs of mice infected with the non-lethal sub-clones or SARS-CoV (Urbani).
(6.7 MB PDF)
(35 KB DOC)
(46 KB DOC)
(44 KB DOC)
(36 KB DOC)
(149 KB DOC)
Accession Numbers
The GenBank (http://www.ncbi.nlm.nih.gov/Genbank) accession numbers for the viruses and sequences discussed in this paper are mouse-adapted SARS-CoV MA15 sequence (DQ497008) and SARS-Cov (Urbani) (AY278741).
Acknowledgments
We would like to thank Jadon Jackson for his expertise and assistance in all animal studies carried out at NIAID. We would like to thank Maria Giovanni (Microbial Genomics, NIAID) and Elodie Ghedin (The Institute for Genomic Research) for contributions in confirming MA15 mutations. We would like to thank other members of the Subbarao lab for their support.
Abbreviations
- β2M
β2 microglobulin
- ALP
alkaline phosphatase
- d.p.i.
days post-infection
- icSARS-CoV
SARS-CoV infectious clone
- IHC
immunohistochemistry
- LD50
50% lethal dose
- MOI
multiplicity of infection
- NIAID
National Institute of Allergy and Infectious Diseases
- nsp
non-structural protein
- ORF
open reading frame
- P[number]
passage [number]
- p.i.
post-infection
- RBM
receptor-binding motif
- RT-PCR
reverse transcriptase PCR
- SARS
severe acute respiratory syndrome
- SARS-CoV
SARS-coronavirus
- SDS
sodium dodecyl sulphate
- TCID50
50% tissue culture infectious dose
- vRNA
viral RNA
Footnotes
Competing interests. The authors have declared that no competing interests exist.
Author contributions. AR performed passage of the SARS-CoV through BALB/c mice, and all other experimental work except generation of recombinant viruses and in situ analysis of lungs. AR generated related figures, materials and methods, figure legends, and interpretation of findings, and wrote and revised the primary manuscript. DD contributed generation and characterization of recombinant clones, generation of related figures, recording of data, and data interpretation. DD made written contributions to figure legends, materials and methods, and review of primary manuscript. CDP performed all histopathological processing and evaluation of SARS-CoV– and MA15-infected tissues, interpretation of findings, generation of related figures, materials and methods, and figure legends, as well as review of primary manuscript. AC performed partial sequencing of MA15 genome, evaluation of mouse tissues for SARS-CoV mRNAs and vRNAs by RT/PCR, generated supporting tables S1–S5, and wrote related materials and methods, and reviewed the primary manuscript. BY contributed to the generation and characterization of recombinant clones, generation of figures, recording of data, and data interpretation. BY contributed to the writing of materials and methods and the review of primary manuscript. LV performed passage of the SARS-CoV through BALB/c mice and all other experimental work except generation of recombinant viruses and in situ analysis of lungs. LV's written contributions include data recording, generation of materials and methods, and review and editing of primary manuscript. BDH performed partial sequencing of SARS-CoV, P15, and MA15 genomes, and primary manuscript review and editing. TS contributed preparation of SARS-CoV–infected and recombinant virus–infected tissues for in situ hybridization analysis. TS contributed to the writing of materials and methods and the review of primary manuscript. MH contributed experimental design and evaluation of SARS-CoV–infected and recombinant virus–infected tissues by in situ hybridization techniques. MH made written contributions that include generation of materials and methods, the Figure S3 legend, and a review of primary manuscript. GLG performed all preparation of SARS-CoV– and MA15-infected tissues for histopathological processing and evaluation, generation of related figures, materials and methods, figure legends, and interpretation of findings, as well as review of the primary manuscript. SRZ contributed review of all histopathological evaluation of SARS-CoV– and MA15-infected tissues, generation of related figures, materials and methods, figure legends, and interpretation of findings, as well as review of the primary manuscript. RB contributed experimental design, characterization of recombinant clones, generation of related data and figures, and data evaluation and interpretation. RB made written contributions to figure legends, materials and methods, and the drafting and review of the primary manuscript. KS provided data interpretation and review and editing of the manuscript.
Funding. This research was supported in part by the Intramural Research Program of the US National Institutes of Health (NIH), NIAID, and in part by NIH/NIAID grants AI059136 and AI059443.
References
- Li W, Shi Z, Yu M, Ren W, Smith C, et al. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- Roberts A, Wood J, Subbarao K, Ferguson M, Wood D, et al. Animal models and antibody assays for evaluating candidate SARS vaccines: Summary of a technical meeting 25–26 August 2005, London, UK. Vaccine. 2006;24:7056–7065. doi: 10.1016/j.vaccine.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbarao K, Roberts A. Is there an ideal animal model for SARS? Trends Microbiol. 2006;14:299–303. doi: 10.1016/j.tim.2006.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EG. Increased virulence of a mouse-adapted variant of influenza A/FM/1/47 virus is controlled by mutations in genome segments 4, 5, 7, and 8. J Virol. 1990;64:4523–4533. doi: 10.1128/jvi.64.9.4523-4533.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullers JA, Hoffmann E, Huber VC, Nickerson AD. A single amino acid change in the C-terminal domain of the matrix protein M1 of influenza B virus confers mouse adaptation and virulence. Virology. 2005;336:318–326. doi: 10.1016/j.virol.2005.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel V, Ivanov KA, Putics A, Hertzig T, Schelle B, et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J Gen Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- Chinese SARS Molecular Epidemiology Consortium. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004;303:1666–1669. doi: 10.1126/science.1092002. [DOI] [PubMed] [Google Scholar]
- Li W, Zhang C, Sui J, Kuhn JH, Moore MJ, et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24:1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- Brown EG, Bailly JE. Genetic analysis of mouse-adapted influenza A virus identifies roles for the NA, PB1, and PB2 genes in virulence. Virus Res. 1999;61:63–76. doi: 10.1016/s0168-1702(99)00027-1. [DOI] [PubMed] [Google Scholar]
- Smeenk CA, Brown EG. The influenza virus variant A/FM/1/47-MA possesses single amino acid replacements in the hemagglutinin, controlling virulence, and in the matrix protein, controlling virulence as well as growth. J Virol. 1994;68:530–534. doi: 10.1128/jvi.68.1.530-534.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeenk CA, Wright KE, Burns BF, Thaker AJ, Brown EG. Mutations in the hemagglutinin and matrix genes of a virulent influenza virus variant, A/FM/1/47-MA, control different stages in pathogenesis. Virus Res. 1996;44:79–95. doi: 10.1016/0168-1702(96)01329-9. [DOI] [PubMed] [Google Scholar]
- Lawler JV, Endy TP, Hensley LE, Garrison A, Fritz EA, et al. Cynomolgus macaque as an animal model for severe acute respiratory syndrome. PLoS Med. 2006;3:e149. doi: 10.1371/journal.pmed.0030149. doi: 10.1371/journal.pmed.0030149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CM, Poon LL, Cheng VC, Chan KS, Hung IF, et al. Initial viral load and the outcomes of SARS. CMAJ. 2004;171:1349–1352. doi: 10.1503/cmaj.1040398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A, Paddock C, Vogel L, Butler E, Zaki S, et al. Aged BALB/c mice as a model for increased severity of severe acute respiratory syndrome in elderly humans. J Virol. 2005;79:5833–5838. doi: 10.1128/JVI.79.9.5833-5838.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farcas GA, Poutanen SM, Mazzulli T, Willey BM, Butany J, et al. Fatal severe acute respiratory syndrome is associated with multiorgan involvement by coronavirus. J Infect Dis. 2005;191:193–197. doi: 10.1086/426870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan HL, Kwan AC, To KF, Lai ST, Chan PK, et al. Clinical significance of hepatic derangement in severe acute respiratory syndrome. World J Gastroenterol. 2005;11:2148–2153. doi: 10.3748/wjg.v11.i14.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung IF, Cheng VC, Wu AK, Tang BS, Chan KH, et al. Viral loads in clinical specimens and SARS manifestations. Emerg Infect Dis. 2004;10:1550–1557. doi: 10.3201/eid1009.040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PL, Chau PH, Yip PS, Ooi GC, Khong PL, et al. A prediction rule for clinical diagnosis of severe acute respiratory syndrome. Eur Respir J. 2005;26:474–479. doi: 10.1183/09031936.05.1076704. [DOI] [PubMed] [Google Scholar]
- Lau YL, Peiris JS. Pathogenesis of severe acute respiratory syndrome. Curr Opin Immunol. 2005;17:404–410. doi: 10.1016/j.coi.2005.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, Hui D, Wu A, Chan P, Cameron P, et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- Li MH, Li XH, Li XW, Ma L, Yi W, et al. Difference and significance of T-lymphocyte subsets in differential diagnosis between severe acute respiratory syndrome and common atypical pneumonia. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi. 2004;18:137–141. [PubMed] [Google Scholar]
- Liu CL, Lu YT, Peng MJ, Chen PJ, Lin RL, et al. Clinical and laboratory features of severe acute respiratory syndrome vis-a-vis onset of fever. Chest. 2004;126:509–517. doi: 10.1378/chest.126.2.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RS, Wu A, To KF, Lee N, Lam CW, et al. Haematological manifestations in patients with severe acute respiratory syndrome: Retrospective analysis. BMJ. 2003;326:1358–1362. doi: 10.1136/bmj.326.7403.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WM, Ho JC, Ooi GC, Mok T, Chan J, et al. Temporal patterns of hepatic dysfunction and disease severity in patients with SARS. JAMA. 2003;290:2663–2665. doi: 10.1001/jama.290.20.2663. [DOI] [PubMed] [Google Scholar]
- Leung CW, Kwan YW, Ko PW, Chiu SS, Loung PY, et al. Severe acute respiratory syndrome among children. Pediatrics. 2004;113:e535–e543. doi: 10.1542/peds.113.6.e535. [DOI] [PubMed] [Google Scholar]
- Manocha S, Walley KR, Russell JA. Severe acute respiratory distress syndrome (SARS): A critical care perspective. Crit Care Med. 2003;31:2684–2692. doi: 10.1097/01.CCM.0000091929.51288.5F. [DOI] [PubMed] [Google Scholar]
- Roberts A, Thomas WD, Guarner J, Lamirande EW, Babcock GJ, et al. Therapy with a severe acute respiratory syndrome–associated coronavirus-neutralizing human monoclonal antibody reduces disease severity and viral burden in golden Syrian hamsters. J Infect Dis. 2006;193:685–692. doi: 10.1086/500143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbarao K, McAuliffe J, Vogel L, Fahle G, Fischer S, et al. Prior infection and passive transfer of neutralizing antibody prevent replication of severe acute respiratory syndrome coronavirus in the respiratory tract of mice. J Virol. 2004;78:3572–3577. doi: 10.1128/JVI.78.7.3572-3577.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yount B, Roberts RS, Sims AC, Deming D, Frieman MB, et al. Severe acute respiratory syndrome coronavirus group–specific open reading frames encode nonessential functions for replication in cell cultures and mice. J Virol. 2005;79:14909–14922. doi: 10.1128/JVI.79.23.14909-14922.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims AC, Yount B, Burkett SE, Baric RS, Pickles RJ. SARS CoV replication and pathogenesis in human airway epithelial cultures. Adv Exp Med Biol. 2006;581:535–538. doi: 10.1007/978-0-387-33012-9_97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yount B, Curtis KM, Fritz EA, Hensley LE, Jahrling PB, et al. Reverse genetics with a full-length infectious cDNA of severe acute respiratory syndrome coronavirus. Proc Natl Acad Sci U S A. 2003;100:12995–13000. doi: 10.1073/pnas.1735582100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise MT, Simpson DA, Johnston RE. A single amino acid change in nsP1 attenuates neurovirulence of the Sindbis-group alphavirus S.A.AR86. J Virol. 2000;74:4207–4213. doi: 10.1128/jvi.74.9.4207-4213.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuliffe J, Vogel L, Roberts A, Fahle G, Fischer S, et al. Replication of SARS coronavirus administered into the respiratory tract of African Green, rhesus and cynomolgus monkeys. Virology. 2004;330:8–15. doi: 10.1016/j.virol.2004.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh WJ, Hsiao CH, Paddock C, Guarner J, Mueller L, et al. Immunohistochemical, in situ hybridization, and ultrastructural localization of SARS-associated coronavirus in a fatal case of severe acute respiratory syndrome in Taiwan. Human Pathol. 2004;36:303–309. doi: 10.1016/j.humpath.2004.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cultures of Vero E6 cells were infected at an MOI of 0.1 with indicated viruses in duplicate. Supernatants were collected at 1, 5, 13, 23, and 36 h.p.i., and virus was quantified by plaque assay in Vero E6 cells. Plaques were visualized by neutral red staining and counted at 48 h. SARS-CoV (Urbani) and rMA15SM replicate similarly to icSARS-CoV (unpublished data). Data points represent means of samples measured in duplicate. A repeat experiment demonstrated similar growth kinetics (unpublished data). Diamonds, icSARS-CoV; squares, MA15; circles, rMA15; and triangles, rMA15ORF1ab.
(28 KB PDF)
Groups of four mice were inoculated intranasally with 50 μL of recombinant viruses icSARS-CoV, rMA15SM, rMA15ORF1ab, rMA15, or MA15 and sacrificed on (A) day 2 p.i. or (B) day 4 p.i. Lungs and sera were collected for viral titration. Virus titers are expressed as log10 pfu/g lung (circles) or log10 pfu/mL serum (squares) from individual mice; bars represent the geometric mean for the group. Limits of detection for viral titers from lung and sera are 250 pfu/mL and 50 pfu/mL, respectively.
(35 KB PDF)
BALB/c mice were infected with SAR-CoV (Urbani), rMA15SM, rMA15ORF1ab, rMA15, or MA15 virus. Lungs were harvested on days 2 and 4 p.i., and in situ hybridization was performed as described in Materials and Methods. No specific signal was observed with a control riboprobe at any time point, while SARS-CoV–N specific signal was readily apparent within the lungs for all five viruses at day 2 p.i. In contrast, at day 4 p.i., abundant SARS-CoV–specific in situ signal was observed in lungs of mice infected with the lethal MA15 virus or the lethal recombinant rMA15, but not in lungs of mice infected with the non-lethal sub-clones or SARS-CoV (Urbani).
(6.7 MB PDF)
(35 KB DOC)
(46 KB DOC)
(44 KB DOC)
(36 KB DOC)
(149 KB DOC)