

Abstract
Palytoxin is a novel skin tumor promoter, which has been used to help probe the role of different types of signaling mechanisms in carcinogenesis. The multi-stage mouse skin model indicates that tumor promotion is an early, prolonged, and reversible phase of carcinogenesis. Understanding the molecular mechanisms underlying tumor promotion is therefore important for developing strategies to prevent and treat cancer. Naturally occurring tumor promoters that bind to specific cellular receptors have proven to be useful tools for investigating important biochemical events in multi-stage carcinogenesis. For example, the identification of protein kinase C as the receptor for the prototypical skin tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA) (also called phorbol-12-myristate-13-acetate or PMA) provided key evidence that tumor promotion involves the aberrant modulation of signaling cascades that govern cell fate and function. The subsequent discovery that palytoxin, a marine toxin isolated from zoanthids (genus Palythoa), is a potent skin tumor promoter yet does not activate protein kinase C indicated that investigating palytoxin action could help reveal new aspects of tumor promotion. Interestingly, the putative receptor for palytoxin is the Na+,K+-ATPase. This review focuses on palytoxin-stimulated signaling, and how palytoxin has been used to investigate alternate biochemical mechanisms by which important targets in carcinogenesis can be modulated.
Keywords: mitogen activated protein kinase, dual specificity phosphatase, prostaglandins, Na+, K+-ATPase
INTRODUCTION
Historically, investigating the mechanisms of action of carcinogenic agents classified as tumor promoters has helped to reveal critical biochemical events underlying the process of carcinogenesis (36, 96). In the multi-stage mouse skin model of carcinogenesis, the first stage, called initiation, typically involves activation of the oncogene Ras (8). This is a rapid, irreversible stage that only requires a single application of an initiator, which is typically a mutagenic compound such as 7,12-dimethylbenz[a]anthracene. The second stage, known as tumor promotion, involves the development of benign tumors called papillomas (36, 96). This stage requires repeated application of a tumor promoter over the course of several weeks. Reversing the order of application, such that tumor promoter treatment precedes initiator treatment, does not stimulate tumor development. Likewise, tumor promoter treatment alone is not sufficient to produce tumors. In contrast to initiation, the tumor promotion stage is prolonged and also reversible if tumor promoter treatment is ceased.
Although the multi-stage mouse skin model was developed in the 1940s, it remains a powerful model for understanding multi-stage carcinogenesis in humans (36, 56). For example, activation of Ras through mutation is a frequent early event in human cancers (21). The regulation of Ras activity is also disrupted by over-expression of growth factor receptors during carcinogenesis. The importance of tumor promotion in human cancer is illustrated by the long latency in the development of cancer, and by examples of reversibility, such as decreased lung cancer risk after smoking cessation (67). Tumor promoters are therefore excellent tools for revealing how expression of oncogenic Ras makes cells susceptible to the subsequent action of tumor promoting stimuli, and for developing strategies to prevent tumor development. Certainly the identification of protein kinase C as the receptor for the prototypical skin tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA) (also called phorbol-12-myristate-13-acetate or PMA) helped establish that tumor promotion involves the aberrant modulation of signaling cascades that govern cell fate and function (65). Palytoxin belongs to a class of non-TPA-type tumor promoters that stimulate signaling pathways that do not require protein kinase C (28). The novel properties of palytoxin make it an intriguing tool for probing alternative mechanisms by which important targets in carcinogenesis can be modulated.
Palytoxin was identified as a skin tumor promoter in the 1980s in a screen to identify tumor promoters in the environment that differed in structure from TPA (28–30). Compounds isolated from various sources, including plant extracts, Japanese and Chinese spices, Chinese medicines, extracts of fungi, and marine products, were screened by a series of short-term tests that included irritation of mouse ear, induction of ornithine decarboxylase in mouse skin, and adhesion of human promyelocytic leukemia (HL-60) cells. The compounds were tested for tumor promoting activity in the multi-stage mouse skin model. The tumor promoters were further classified as either TPA-type or non-TPA-type based on their ability to bind to protein kinase C in vitro. This screen revealed that palytoxin, like TPA, was a skin irritant and a tumor promoter in the multi-stage mouse skin assay. In contrast to TPA, however, palytoxin did not induce ornithine decarboxylase in mouse skin and did not induce HL60 cell adhesion. Furthermore, palytoxin did not bind to protein kinase C in vitro and was therefore classified as a non-TPA-type tumor promoter. Subsequent cell culture studies provided further evidence that palytoxin stimulates signaling pathways that do not require protein kinase C (85). The apparent differences between palytoxin and TPA raised the following question: Do palytoxin and TPA induce tumor promotion by independent biochemical pathways, or do the distinct signal transduction pathways activated by palytoxin and TPA converge to modulate common targets that are critical for carcinogenesis? As discussed below, TPA and palytoxin can modulate common targets, although through very different mechanisms (82, 83).
THE Na+,K+-ATPase AS THE PALYTOXIN RECEPTOR
The structure of palytoxin alone suggests that it would interact with a different type of receptor than would phorbol esters (60). Phorbol esters are relatively small, lipophilic molecules that can dissolve in cell membranes relatively easily and therefore interact with an intracellular receptor such as protein kinase C (65). Palytoxin is a large (Mr 2,681), water-soluble polyalcohol isolated from zoanthids (genus Palythoa), which are closely related to sea anemones (60). This type of molecule is more likely to interact with a cell-surface receptor. A broad range of studies indicate that the Na+,K+-ATPase is a high affinity cellular receptor for palytoxin (6, 7, 34, 73). For example, ouabain, which binds to the Na+,K+-ATPase, blocks palytoxin action in a variety of systems, including erythrocytes isolated from different species, human cervical carcinoma cells, and bovine adrenomedullary cells (34). Conversely, palytoxin inhibits ouabain binding (34). Most importantly, palytoxin stimulates potassium efflux in yeast engineered to express the mammalian Na+,K+-ATPase, but does not affect nontransformed yeast or yeast that express just the α subunit or the β subunit of the Na+,K+-ATPase (73). Although the binding sites for ouabain and palytoxin may overlap, these compounds have different effects on the Na+,K+-ATPase. In particular, palytoxin, appears to bind to the Na+,K+-ATPase and transform this pump into an ion channel that transports sodium and potassium, but has low permeability for calcium (6, 7).
Although the interaction of palytoxin with the Na+,K+-ATPase has been studied extensively, it remains possible that palytoxin can also act through other cellular receptors (25, 72). For example, the observation that high concentrations of palytoxin (1 μM) can stimulate the release of norepinephrine in the absence of extracellular sodium in a rat pheochromocytoma cell line, led to speculation that under some conditions palytoxin can directly increase calcium influx (81). The Na+/H+ antiporter has also been investigated as a possible target of palytoxin action (26, 94). Studies conducted in chick cardiac cells indicated, however, that the primary effect of palytoxin is to acidify the cell, which then stimulates that activation of the Na+/H+ antiporter (26). Interestingly, subsequent studies have suggested that the H+,K+-ATPase, which is related to the Na+,K+-ATPase (39), may be a target of palytoxin action in rat colon (72). More detailed pharmacological studies are required to determine how the H+,K+-ATPase contributes to specific palytoxin-stimulated cellular responses.
Research on palytoxin action generally falls into two major areas. One broad class of studies focuses on how palytoxin affects ion flux, which is the immediate effect of this compound on the cell. Another broad class of studies, discussed later, focuses on subsequent cellular effects stimulated by palytoxin that may be related to tumor promotion. Palytoxin stimulates sodium influx and potassium efflux, and thus depolarization of the membrane, in a wide range of systems (34). In excitable systems, palytoxin-stimulated depolarization can modulate calcium channel activity, resulting in a rise in intracellular calcium, which can then stimulate events that are regulated by calcium-dependent pathways. In muscle, depolarization stimulates calcium release and contraction. In vivo, palytoxin-stimulated depolarization can stimulate vasoconstriction, which can be lethal. In other systems, however, palytoxin can stimulate cellular events through mechanisms that do not require a rise in intracellular calcium (11, 86). Furthermore, a rise in intracellular calcium can sometimes be a secondary effect of palytoxin-induced cytotoxicity (5, 86). Extracellular calcium can increase palytoxin binding (34, 44, 87). This could lead to misinterpretation of palytoxin studies that use calcium chelators.
Like other skin tumor promoters, palytoxin stimulates a wide range of cellular responses. The constellation of responses required for tumor promotion and the link between these responses and palytoxin-stimulated changes in ion flux still needs to be established. Among the best studied palytoxin-stimulated responses that are likely to play a role in carcinogenesis are stimulation of arachidonic acid metabolism and the production of prostaglandins (46, 59, 66), modulation of the epidermal growth factor (EGF) receptor (84–86), and modulation of mitogen activated protein (MAP) kinase cascades (38, 45, 82).
ARACHIDONIC ACID METABOLISM AND THE PRODUCTION OF PROSTAGLANDINS
One of the cellular responses to palytoxin that is common to many skin tumor promoters is stimulation of arachidonic acid metabolism and the production of prostaglandins (46, 66). Several lines of evidence, reviewed in (33, 53, 56), indicate that prostaglandins play a role in both multi-stage mouse skin carcinogenesis and human carcinogenesis. Furthermore, non-steroidal anti-inflammatory drugs (NSAIDs), which inhibit prostaglandin synthesis, can block TPA-induced tumor promotion and are being investigated as potential chemopreventive agents to reduce cancer risk in humans (33, 53, 56). Picomolar concentrations of palytoxin stimulate the production of prostaglandins in a wide variety of cell cultures systems, including rat liver cells (46), rat and mouse macrophages (2, 66), squirrel monkey aorta smooth muscle cells (49), bovine smooth muscle cells, porcine and bovine aorta endothelial cells (47), and Balb/c 3T3 mouse fibroblasts (59). Although palytoxin stimulated the production of prostaglandins in primary rat keratinocytes (47), it did not do so in primary mouse epidermal cells (2). It is not clear whether this difference was due to a species-specific effect or whether it was due to a difference in the preparation and purity of the cell types used in the assays.
The observation that palytoxin and TPA have synergistic effects on arachidonic acid metabolism and prostaglandin synthesis was an early indicator that these tumor promoters stimulate different signaling pathways (46, 66). Palytoxin is also synergistic with EGF, transforming growth factor-α, transforming growth factor-β, interleukin-1, insulin, and 1-oleoyl-2-acetyl-glycerol (46, 48). This likewise suggested that palytoxin-stimulates signaling pathways that differ, at least in part, from growth factor-stimulated signaling. Studies with cycloheximide indicated that palytoxin-stimulated prostaglandin production requires new protein synthesis (66). Otherwise, these early studies did not fully explore the nature of the signaling pathways leading from palytoxin binding to arachidonic acid metabolism.
A more recent study investigated the role of prostaglandins in the tumor promoting action of palytoxin through the use of an in vitro Balb/c 3T3 cell transformation model (59). The cells were initiated with 3-methylcholanthrene, and then treated for 2 weeks with palytoxin or other agents. Transformed foci were counted as an indication of tumor promoting activity. Nontoxic concentrations of palytoxin (0.4 – 5.6 pM) increased the number of transformed foci. In this assay, palytoxin and TPA stimulated transformation to a similar extent. Palytoxin also stimulated the production of prostaglandins in this system. The NSAID indomethacin, an inhibitor of prostaglandin synthesis, blocked both the ability of palytoxin to stimulate the production of PGE2 and PGF2α and the ability of palytoxin to increase transformed foci. This study showed that palytoxin stimulated the phosphorylation of extracellular signal regulated kinase (ERK), a member of the MAP kinase family of protein kinases. Indomethacin blocked the ability of palytoxin to activate ERK, which suggests that palytoxin activates ERK in this system through an autocrine mechanism that involves the production of prostaglandins. Altogether, these studies indicate that the Balb/c 3T3 system could be a very useful model for investigating the signaling pathways by which palytoxin stimulates the production of prostaglandins and for establishing the link between the interaction of palytoxin with the Na+,K+-ATPase, or other cellular receptors, and palytoxin-stimulated arachidonic acid metabolism.
MODULATION OF THE EGF RECEPTOR AND PALYTOXIN SIGNALING
Another set of early studies revealed that the EGF receptor is a common target of palytoxin and TPA. The intent of these studies, conducted in Swiss 3T3 fibroblasts, was to use the regulation of the EGF receptor as an endpoint for investigating the signaling pathways stimulated by palytoxin and for comparing the action of palytoxin to TPA-type tumor promoters (84–87). Altogether, these studies supported the idea that palytoxin and TPA can modulate common targets through different mechanisms (85). It had been established that TPA could inhibit EGF binding to a class of high affinity receptors (27). Although palytoxin, like TPA, could stimulate a loss of high affinity EGF binding sites, palytoxin also stimulated a loss of low affinity EGF receptors. Furthermore, in contrast to TPA, palytoxin could modulate EGF binding through signaling pathways that did not require protein kinase C (85).
The observation that palytoxin stimulated ion flux in a variety of excitable and non-excitable systems, together with the implication of the Na+,K+-ATPase as the palytoxin receptor (34), suggested that ion flux might play an important role in triggering palytoxin-stimulated signaling. Picomolar concentrations of palytoxin stimulated a loss of EGF binding under conditions where there was no increase in cytosolic calcium, and thus indicated that a rise in intracellular calcium was not required for the effects of palytoxin on the EGF receptor (86, 87). Although calcium appeared to enhance palytoxin binding, an influx of calcium in this system was typically associated with toxicity. Rather, this set of studies suggested that sodium influx played an important role in the ability of palytoxin to stimulate down modulation of the EGF receptor (84, 86). Palytoxin stimulated sodium influx in a manner that correlated with the loss of EGF binding in Swiss 3T3 fibroblasts. Conversely, ionophores that stimulate sodium influx could mimic palytoxin action. Further studies indicated that the effects of palytoxin on the EGF receptor were not due to common secondary effects of sodium influx, including membrane depolarization, changes in intracellular pH, or inhibition of protein synthesis (84).
The mechanisms by which palytoxin-stimulated changes in ion flux down modulate the EGF receptor have not yet been elucidated. Interestingly, UV light and high osmolarity trigger the clustering and internalization of membrane receptors for EGF, TNF and interleukin-1 (69). It has been speculated that these cellular stresses might affect receptor trafficking by stimulating a perturbation in the plasma membrane or inducing changes in protein conformation. It is possible that palytoxin stimulates the down modulation of the EGF receptor through a mechanism that is similar to that induced by these other cellular stresses. Yet, the observation that palytoxin did not affect PDGF binding in Swiss 3T3 cells indicates that palytoxin does not stimulate the nonspecific endocytosis of membrane receptors (84). Alternatively, palytoxin may trigger the down modulation of the EGF receptor by altering protein-protein interactions that are more specific to this receptor. As discussed below, recent studies have revealed that the Na+,K+-ATPase interacts with several major signaling proteins in the plasma membrane (reviewed in (91) and (92)). It is possible that the interaction of palytoxin with the Na+,K+-ATPase results not only in a change in ion flux, but also in a shift in the association of proteins involved in what has been called the Na+,K+-ATPase signalosome, some of which interact with the EGF receptor (92).
MITOGEN ACTIVATED PROTEIN KINASES (MAP KINASES)
A clue to the nature of the biochemical mediators of palytoxin-stimulated signals came with the observation that palytoxin can stimulate the activation of MAP kinases. A common theme that had emerged for the mechanisms of action of structurally diverse tumor promoters was the modulation of protein kinase cascades. TPA activates protein kinase C (65). The non-TPA-type tumor promoter thapsigargin activates ERK (17), and another non-TPA-type tumor promoter okadaic acid modulates protein kinase cascades indirectly by inhibiting serine/threonine protein phosphatases (10, 80). This raised the question of whether palytoxin-stimulated signals could also be transmitted through the action of protein kinases. Initially, a member of the MAP kinase family, c-Jun N-terminal kinase (JNK), also called stress activated protein kinase or SAPK, presented a potential target for palytoxin action (44).
MAP kinases are a family of serine/threonine kinases that play a central role in coordinating the transmission of various types of signals to the nucleus (reviewed in (16, 18)). Once activated, MAP kinases can translocate from the cytoplasm to the nucleus, and modulate gene expression by phosphorylating various transcription factors. The three best-characterized members of the MAP kinase family are ERK, JNK, and p38. Distinct types of signals often activate different members of the MAP kinase family. For example, mitogenic agents, such as EGF and TPA, almost universally activate ERK. Stress-inducing agents, such as UV light and TNF-α, typically activate JNK and p38. The activation of ERK, JNK, and p38 by specific signals, and the role of specific MAP kinase family members in regulating cell fate and function, can differ depending on the cell type, however (22). For example, in neuronal cells, JNK activation, in the absence of ERK activation, stimulates apoptosis (90). In other cell types, activation of JNK and p38 are associated with cell proliferation and protection from apoptosis. For example, activation of JNK is required for EGF-induced stimulation of growth and transformation of a human lung carcinoma cell line (12, 13). In HeLa cells, expression of specific isoforms of p38 inhibits UV light-induced apoptosis (64).
MAP kinases are regulated by the sequential activation of a cascade of protein kinases (16, 18). Generally, a MAP kinase kinase kinase (MAPKK kinase) phosphorylates and activates a MAP kinase kinase (MAPK kinase), which then phosphorylates and activates a MAP kinase, which then phosphorylates cellular substrates. The best-defined MAP kinase cascade is the Raf/MEK/ERK pathway. For example, EGF activates the GTPase Ras, which activates Raf (a MAPKK kinase), which phosphorylates and activates MEK (a MAPK kinase), which phosphorylates and activates ERK (a MAP kinase).
Abnormal regulation of MAP kinases is likely to play a role in carcinogenesis. For example, in the mouse skin model, elevated ERK activity has been detected in TPA-induced papillomas and squamous cell carcinomas (76). TPA-induced tumor promotion is also reduced in ERK1-knockout mice (14). Keratinocytes isolated from ERK-1 knockout mice were characterized by reduced expression of genes involved in cell growth and invasion, as well as reduced growth, and reduced resistance to apoptosis. Elevated ERK activity has also been detected in several human cancers, including lung, colon, breast, melanoma, leukemia, and head and neck squamous carcinoma (3, 19, 37, 43, 71, 77). Furthermore, oral administration of MEK inhibitors blocked ERK activity and tumor growth in mice implanted with human colon or pancreatic tumors (74, 75). JNK and p38 may also play a role in carcinogenesis. Interestingly, depending on the context, JNK and p38 may act as either tumor suppressors or as pro-oncogenic signaling molecules (22).
The idea that palytoxin might activate JNK was based on the observation that this MAP kinase family member could be activated by osmotic stress, and that palytoxin could cause a type of osmotic stress by stimulating a dramatic change in ion flux through its interaction with the Na+,K+-ATPase. Accordingly, picomolar concentrations of palytoxin stimulate sustained activation of JNK in Swiss 3T3 fibroblasts, the same cell culture model that was used to study the regulation of the EGF receptor by palytoxin (44). These initial studies indicated that sodium influx, not calcium, was important in the activation of JNK by palytoxin, which was consistent with the results of the EGF receptor studies (86). Work by another group subsequently indicated, however, that potassium efflux, as opposed to sodium influx, was key to the activation of JNK in rat fibroblasts (38). In Swiss 3T3 cells, phorbol esters predominantly activated ERK, not JNK, while palytoxin predominantly activated JNK, but not ERK (44). These results were consistent with the observation that TPA stimulates mitogenic pathways, while palytoxin stimulates stress-activated pathways. Similar results were obtained in HeLa and COS7 cells, two cell lines that are commonly used to study signal transduction (45). These studies also demonstrated that palytoxin activated JNK through stimulation of upstream protein kinases. Like other MAP kinases, JNK is regulated by protein kinase cascades (40). SEK1 (also called JNKK and MKK4) is a dual specificity kinase that activates JNK through phosphorylation of specific tyrosine and threonine residues (20, 52, 70). Studies conducted in COS7 cells demonstrated that palytoxin activates JNK through a pathway that requires the activation of SEK1 (45).
Many signals that stimulate JNK activation also stimulate the activation of p38, another major stress activated MAP kinase. Accordingly, palytoxin is a potent stimulator of p38 activity in a variety of cell types (50, 83). Interestingly, the pathway by which palytoxin stimulates p38 activity differs depending on the cell type. Three MAPK kinases have been identified that phosphorylate and activate p38. MKK3 and MKK6 specifically phosphorylate and activate p38 (35, 61, 78). Under some conditions SEK1 can also phosphorylate and activate p38 (20, 52). MKK3, MKK6, and SEK1 are expressed in both HeLa and COS7 cells (50). As expected, palytoxin stimulated the activation of all three MAPK kinases in HeLa cells. Surprisingly, palytoxin only stimulated the activation of MKK6 and SEK1 in COS7 cells. In HeLa cells, palytoxin activated p38 through MKK6- and MKK3-dependent protein kinase cascades; SEK1 was not required to activate p38. By contrast, in COS7 cells, palytoxin activated p38 through MKK6- and SEK1-dependent protein kinase cascades. Altogether, these studies indicate that palytoxin-stimulated signals couple to different protein kinase cascades, depending on the cell type.
The initial work that investigated the effects of palytoxin on MAP kinase activity suggested that palytoxin and TPA activate different MAP kinase family members. This was consistent with the observations that palytoxin and TPA initially trigger different types of signals. In COS7, HeLa, and Swiss 3T3 cells, palytoxin predominantly activates JNK and p38, but not ERK, whereas phorbol esters predominantly activate ERK, but not JNK or p38 (44, 45, 51). Although JNK, p38, and ERK are often activated by different signals, the three major MAP kinases can modulate some common targets, such as the transcription factor Elk-1, which regulates the expression of c-Fos, and the AP-1 family of transcription factors, which are dimers made up of various combinations of Jun and Fos family members (4, 32, 41, 68, 88, 89). Significantly, AP-1 appears to play a role in TPA-stimulated tumor promotion (95). This suggested that MAP kinases might act as mediators through which the different signaling pathways stimulated by palytoxin and TPA could converge to regulate common targets.
The idea that palytoxin and TPA stimulate signaling pathways that converge to modulate common targets was explored by investigating the signaling pathways by which these two diverse tumor promoters stimulate the expression of matrix metalloproteinase-13 (MMP-13), an enzyme implicated in carcinogenesis (1, 24, 83). These studies were conducted in a mouse keratinocyte cell line called 308 (79). The 308 cell line was derived from initiated mouse skin and expresses activated Ras. It thus represents an excellent model for the likely target cells of skin tumor promoters in vivo. Surprisingly, and in contrast to the results of studies conducted in other cell lines, palytoxin, like TPA, stimulated ERK activity in 308 cells (83, 97). Altogether, the results from these studies indicated that in initiated mouse keratinocytes, as opposed to several other cell types, the distinct signaling pathways stimulated by palytoxin and TPA converge to stimulate ERK activation, which results in the stimulation of several common downstream effects likely to be related to tumor promotion, including c-Fos gene expression, the modulation of AP-1, and the expression of MMP-13 (83, 97). The significance of ERK as a common target for non-TPA-type and TPA-type tumor promoters is underscored by the studies, discussed above, that implicate aberrant regulation of ERK in human carcinogenesis.
The observation that palytoxin action on initiated keratinocytes differed dramatically from its effects on other cell types suggested that investigating the mechanisms by which palytoxin activates ERK in 308 cells might reveal new aspects of tumor promotion. In particular, these studies suggested that initiated keratinocytes express modulators of ERK activity, not expressed in other cell types, that are sensitive to palytoxin action. As discussed below, subsequent studies revealed that the ability of palytoxin to activate ERK is not a unique characteristic of keratinocytes, but rather a characteristic of cells that express oncogenic Ras (82).
The major regulators of ERK activity are kinases and phosphatases. Like other MAP kinases, ERK activity is regulated through reversible phosphorylation of specific tyrosine and threonine residues (15, 18, 23, 42). MEK is the dual specificity MAPK kinase that phosphorylates and activates ERK. A family of dual-specificity phosphatases, called MAP kinase phosphatases (MKPs), which dephosphorylate both tyrosine and threonine residues, specifically dephosphorylate and inactivate MAP kinases, including ERK (15, 23, 42). In contrast to palytoxin-stimulated JNK and p38 activation, which involves activation of upstream MAPK kinases, in 308 cells palytoxin does not activate ERK through direct stimulation of MEK (82). Instead, these studies indicated that palytoxin increases ERK activity in 308 keratinocytes by inducing the down modulation of MKP-3, an MKP family member that is highly selective for desphosphorylating and inactivating ERK (62). Evidence that the mechanism by which palytoxin stimulates ERK activity is related to the expression of oncogenic Ras, as opposed to being specific to mouse keratinocytes, came from the observation that palytoxin stimulated ERK activity in H-ras MCF10A cells, a human breast epithelial cell line engineered to express oncogenic Ras, but did not activate ERK in the parental MCF10A cells, which do not express the Ras oncogene (82). Significantly, H-ras MCF10A cells, like 308 cells, express elevated levels of MKP-3. Interestingly, several studies indicate that the upregulation of MKPs may be characteristic of cells that express activated Ras oncogenes (31, 82, 93).
The observation that MKPs are highly expressed in cells that express oncogenic Ras suggests that cells may compensate for chronic activation of the Ras-stimulated pathways by upregulating phosphatases that constrain those protein kinase cascades (31, 93). This type of compensation mechanism may contribute, in part, to the multi-stage nature of carcinogenesis. The observation that activation of Ras alone is not sufficient to induce tumors may be due, at least in part, to the ability of cells to compensate for the super-activation of Ras-stimulated signal transduction pathways by upregulating phosphatases that act as negative regulators of those pathways. Inactivating phosphatases would weaken this compensatory system and thus be an effective additional step, which would unleash such super-activated signaling cascades. Accordingly, studies suggest that MKP-3 is a tumor suppressor in human pancreatic cancer, where the frequency of Ras mutation approaches 90% (21, 31).
Altogether, these studies suggest that MKP-3 may be a vulnerable target of palytoxin action in cells that express oncogenic Ras. The duration and magnitude of ERK activation has profound effects on cell fate and function (57, 58, 63). For example, in mouse fibroblasts, prolonged ERK activation is associated with c-Fos stabilization and entry into S Phase; these cellular events are not induced by transient ERK activation (63). In PC12 cells, prolonged ERK activation is associated with differentiation, whereas transient ERK activation is associated with cell proliferation (57). The contrasting actions of TPA and palytoxin in initiated keratinocytes illustrate two major mechanisms by which carcinogenic agents and events can shift the balance to favor phosphorylation and activation of ERK during early stages of carcinogenesis that involve oncogenic Ras. TPA represents a well-characterized mechanism, which is to further stimulate protein kinase cascades such that the rate of phosphorylation is greater than the rate of dephosphorylation (9, 97). Palytoxin represents an alternative mechanism, which is to shift the balance toward phosphorylation by blocking the action of dual specificity phosphatases that directly inhibit ERK activity.
In summary, palytoxin appears to be able to modulate MAP kinase signaling pathways by at least three major mechanisms (Fig. 1). First, the Balb/c 3T3 model suggests that palytoxin can activate ERK through an autocrine mechanism that involves the production of prostaglandins (59). Second, palytoxin can activate JNK and p38 through the stimulation of upstream protein kinase cascades (45, 50). Third, palytoxin can increase ERK activity by disrupting the action of protein phosphatases that act as negative regulators of MAP kinases (82). Further research is needed to determine whether palytoxin also affects MKPs that regulate JNK and p38.
Fig. 1.
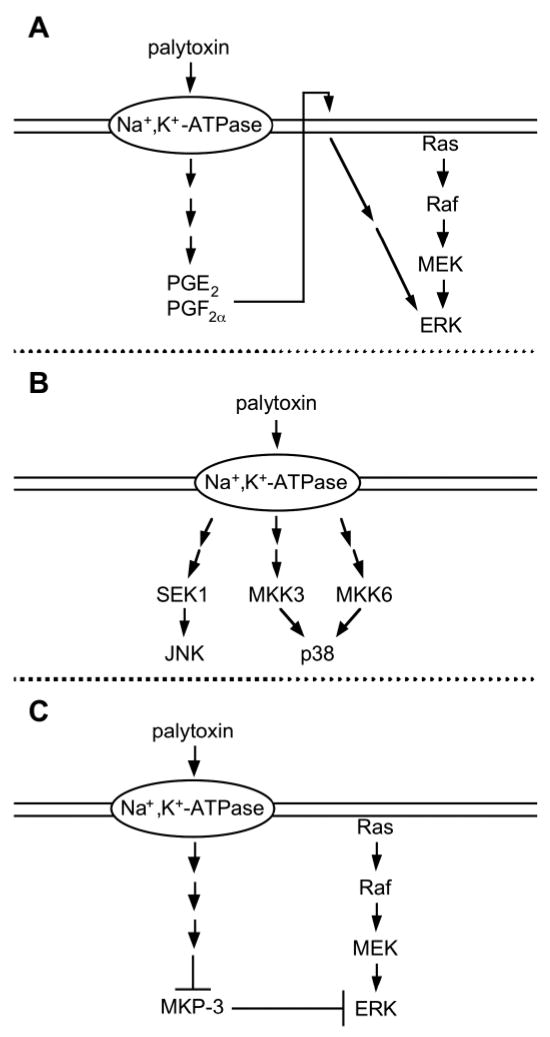
Palytoxin modulates MAP kinase activity by different mechanisms. (A) Palytoxin may activate ERK through an autocrine mechanism that involves the production of prostaglandins. (B) Palytoxin can activate stress activated protein kinases through the stimulation of upstream protein kinase cascades. Palytoxin activates JNK through a mechanism that requires activation of the MAPK kinase SEK1. Depending on the cell type, palytoxin activates p38 through protein kinase cascades that require the MAPK kinases MKK3 and MKK6. Although not depicted in this scheme, in COS cells, palytoxin can also activate p38 through a protein kinase cascade that requires SEK1. (C) In cells that express oncogenic Ras, palytoxin can increase ERK activity by disrupting the action of MKP-3, a dual specificity protein phosphatase that specifically dephosphorylates and inactivates ERK.
CONCLUSIONS
Many questions remain regarding palytoxin-stimulated signal transduction and tumor promotion. One major challenge is to define the pathway that links the interaction of palytoxin with the Na+,K+-ATPase to the modulation of MAP kinase protein kinase cascades. In terms of the modulation of MKP-3, palytoxin stimulates a dramatic loss of MKP-3 within one hour (82). MKP-3 is a relatively unstable protein (54, 55), and thus a particularly vulnerable target for agents that block the production of this phosphatase. In rat fibroblasts, palytoxin appears to inhibit protein synthesis through a mechanism that requires potassium efflux (38). Such a block in translation could explain how palytoxin induces the loss of MKP-3. Several MAPKK kinases have been identified that regulate the JNK and p38 signaling pathways (18, 40). The role of specific MAPKK kinases and how their regulation might be coupled to the interaction of palytoxin with the Na+,K+-ATPase remains to be determined. Studies conducted in HeLa cells indicate that UV light and osmotic stress activate JNK by stimulating the clustering, activation, and internalization of growth factor and cytokine receptors (69). Palytoxin activates JNK through a pathway that does not require Ras (51). Therefore, if palytoxin does stimulate JNK activation via the EGF receptor, it exploits an EGF receptor-coupled pathway that does involve Ras.
Recent work on the signaling function of the Na+,K+-ATPase raises an intriguing question about the mechanisms by which the interaction of palytoxin with the Na+,K+-ATPase modulates signaling pathways. In addition to acting as an ion pump, the Na+,K+-ATPase appears to interact with several major signaling proteins in the plasma membrane (reviewed in (91) and (92)). Binding of ouabain to the Na+,K+-ATPase results in the stimulation of the tyrosine kinase Src, transactivation of the EGF receptor, and increased production of reactive oxygen species. This, in turn, activates ERK, p38, phospholipase C and protein kinase C. Ouabain binding appears to alter the binding of the Na+,K+-ATPase to signaling partners, such that stimulation of the signal transduction pathways is independent of changes in ion flux. Although ion flux has been implicated in palytoxin-stimulated signaling, it is possible that palytoxin can also modulate the signaling capacity of the Na+,K+-ATPase. There are some similarities between the signaling stimulated by palytoxin and ouabain, as well as some important differences. For example, in immortalized human bronchial epithelial cells (BEAS-2B), palytoxin stimulated increased DNA synthesis, whereas ouabain caused a decrease in DNA synthesis (11). In this cell type, both ouabain and palytoxin stimulated the expression of c-myc, although with significantly different time courses, suggesting that they modulate gene expression through different mechanisms. Ouabain stimulated a rapid (within 2 hours), prolonged increase in c-myc gene expression, whereas palytoxin induced c-myc gene expression after a delay of over 16 hours. Ouabain, like palytoxin, can activate JNK through a SEK1-dependent, Ras-independent pathway in HeLa cells (51). In HeLa cells, palytoxin could stimulate JNK and p38 activation to a greater extent than ouabain, whereas ouabain could stimulate ERK activation to a greater extent than palytoxin. Although palytoxin does not completely mimic ouabain action, it remains possible that palytoxin-stimulated signaling depends, at least in part, on modulating the signaling function of the Na+,K+-ATPase.
In conclusion, palytoxin has proven to be a useful tool for exploring alternative mechanisms of modulating key signal transduction pathways in carcinogenesis. The major challenge for future research is to establish the major steps that link the interaction of palytoxin with the Na+,K+-ATPase to stimulation of MAP kinase cascades and other signaling pathways and cellular effects related to carcinogenesis.
Acknowledgments
Many thanks to Binks Wattenberg for critical reading of this manuscript.
Footnotes
GRANTS
This work was supported by National Cancer Institute grant CA-104609.
References
- 1.Airola K, Johansson N, Kariniemi AL, Kahari VM, Saarialho-Kere UK. Human collagenase-3 is expressed in malignant squamous epithelium of the skin. J Invest Dermatol. 1997;109:225–231. doi: 10.1111/1523-1747.ep12319441. [DOI] [PubMed] [Google Scholar]
- 2.Aizu E, Yamamoto S, Nakadate T, Kato R. Differential effects of various skin tumor-promoting agents on prostaglandin E2 release from primary cultures of mouse epidermal cells. Eur J Pharmacol. 1990;182:19–28. doi: 10.1016/0014-2999(90)90489-s. [DOI] [PubMed] [Google Scholar]
- 3.Albanell J, Codony-Servat J, Rojo F, Del Campo JM, Sauleda S, Anido J, Raspall G, Giralt J, Rosello J, Nicholson RI, Mendelsohn J, Baselga J. Activated extracellular signal-regulated kinases: association with epidermal growth factor receptor/transforming growth factor alpha expression in head and neck squamous carcinoma and inhibition by anti-epidermal growth factor receptor treatments. Cancer Res. 2001;61:6500–6510. [PubMed] [Google Scholar]
- 4.Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 5.Ares IR, Louzao MC, Vieytes MR, Yasumoto T, Botana LM. Actin cytoskeleton of rabbit intestinal cells is a target for potent marine phycotoxins. J Exp Biol. 2005;208:4345–4354. doi: 10.1242/jeb.01897. [DOI] [PubMed] [Google Scholar]
- 6.Artigas P, Gadsby DC. Large diameter of palytoxin-induced Na/K pump channels and modulation of palytoxin interaction by Na/K pump ligands. J Gen Physiol. 2004;123:357–376. doi: 10.1085/jgp.200308964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Artigas P, Gadsby DC. Na+/K+-pump ligands modulate gating of palytoxin-induced ion channels. Proc Natl Acad Sci U S A. 2003;100:501–505. doi: 10.1073/pnas.0135849100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balmain A, Pragnell IB. Mouse skin carcinomas induced in vivo by chemical carcinogens have a transforming Harvey-ras oncogene. Nature. 1983;303:72–74. doi: 10.1038/303072a0. [DOI] [PubMed] [Google Scholar]
- 9.Barnard D, Diaz B, Clawson D, Marshall M. Oncogenes, growth factors and phorbol esters regulate Raf-1 through common mechanisms. Oncogene. 1998;17:1539–1547. doi: 10.1038/sj.onc.1202061. [DOI] [PubMed] [Google Scholar]
- 10.Bialojan C, Takai A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem J. 1988;256:283–290. doi: 10.1042/bj2560283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonnard C, Lechner JF, Gerwin BI, Fujiki H, Harris CC. Effects of palytoxin or ouabain on growth and squamous differentiation of human bronchial epithelial cells in vitro. Carcinogenesis. 1988;9:2245–2249. doi: 10.1093/carcin/9.12.2245. [DOI] [PubMed] [Google Scholar]
- 12.Bost F, McKay R, Bost M, Potapova O, Dean NM, Mercola D. The Jun kinase 2 isoform is preferentially required for epidermal growth factor-induced transformation of human A549 lung carcinoma cells. Mol Cell Biol. 1999;19:1938–1949. doi: 10.1128/mcb.19.3.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bost F, McKay R, Dean N, Mercola D. The JUN kinase/stress-activated protein kinase pathway is required for epidermal growth factor stimulation of growth of human A549 lung carcinoma cells. J Biol Chem. 1997;272:33422–33429. doi: 10.1074/jbc.272.52.33422. [DOI] [PubMed] [Google Scholar]
- 14.Bourcier C, Jacquel A, Hess J, Peyrottes I, Angel P, Hofman P, Auberger P, Pouyssegur J, Pages G. p44 mitogen-activated protein kinase (extracellular signal-regulated kinase 1)-dependent signaling contributes to epithelial skin carcinogenesis. Cancer Res. 2006;66:2700–2707. doi: 10.1158/0008-5472.CAN-05-3129. [DOI] [PubMed] [Google Scholar]
- 15.Camps M, Nichols A, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. Faseb J. 2000;14:6–16. [PubMed] [Google Scholar]
- 16.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 17.Chao TS, Byron KL, Lee KM, Villereal M, Rosner MR. Activation of MAP kinases by calcium-dependent and calcium-independent pathways. Stimulation by thapsigargin and epidermal growth factor. J Biol Chem. 1992;267:19876–19883. [PubMed] [Google Scholar]
- 18.Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH. MAP kinases. Chem Rev. 2001;101:2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- 19.Cohen C, Zavala-Pompa A, Sequeira JH, Shoji M, Sexton DG, Cotsonis G, Cerimele F, Govindarajan B, Macaron N, Arbiser JL. Mitogen-actived protein kinase activation is an early event in melanoma progression. Clin Cancer Res. 2002;8:3728–3733. [PubMed] [Google Scholar]
- 20.Derijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ, Davis RJ. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- 21.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 22.Engelberg D. Stress-activated protein kinases-tumor suppressors or tumor initiators? Semin Cancer Biol. 2004;14:271–282. doi: 10.1016/j.semcancer.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 23.Farooq A, Zhou MM. Structure and regulation of MAPK phosphatases. Cell Signal. 2004;16:769–779. doi: 10.1016/j.cellsig.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 24.Freije JM, Diez-Itza I, Balbin M, Sanchez LM, Blasco R, Tolivia J, Lopez-Otin C. Molecular cloning and expression of collagenase-3, a novel human matrix metalloproteinase produced by breast carcinomas. J Biol Chem. 1994;269:16766–16773. [PubMed] [Google Scholar]
- 25.Frelin C, Van Renterghem C. Palytoxin. Recent electrophysiological and pharmacological evidence for several mechanisms of action. Gen Pharmacol. 1995;26:33–37. doi: 10.1016/0306-3623(94)00133-8. [DOI] [PubMed] [Google Scholar]
- 26.Frelin C, Vigne P, Breittmayer JP. Palytoxin acidifies chick cardiac cells and activates the Na+/H+ antiporter. FEBS Letters. 1990;264:63–66. doi: 10.1016/0014-5793(90)80765-b. [DOI] [PubMed] [Google Scholar]
- 27.Friedman B, Frackelton AR, Jr, Ross AH, Connors JM, Fujiki H, Sugimura T, Rosner MR. Tumor promoters block tyrosine-specific phosphorylation of the epidermal growth factor receptor. Proc Natl Acad Sci U S A. 1984;81:3034–3038. doi: 10.1073/pnas.81.10.3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujiki H, Suganuma M, Nakayasu M, Hakii H, Horiuchi T, Takayama S, Sugimura T. Palytoxin is a non-12-O-tetradecanoylphorbol-13-acetate type tumor promoter in two-stage mouse skin carcinogenesis. Carcinogenesis. 1986;7:707–710. doi: 10.1093/carcin/7.5.707. [DOI] [PubMed] [Google Scholar]
- 29.Fujiki H, Suganuma M, Suguri H, Yoshizawa S, Hirota M, Takagi K, Sugimura T. Diversity in the chemical nature and mechanism of response to tumor promoters. Prog Clin Biol Res. 1989;298:281–291. [PubMed] [Google Scholar]
- 30.Fujiki H, Suganuma M, Tahira T, Yoshioka A, Nakayasu M, Endo Y, Shudo K, Takayama S, Moore RE, Sugimura T. Nakahara memorial lecture. New classes of tumor promoters: teleocidin, aplysiatoxin, and palytoxin. Princess Takamatsu Symp. 1983;14:37–45. [PubMed] [Google Scholar]
- 31.Furukawa T, Sunamura M, Motoi F, Matsuno S, Horii A. Potential tumor suppressive pathway involving DUSP6/MKP-3 in pancreatic cancer. Am J Pathol. 2003;162:1807–1815. doi: 10.1016/S0002-9440(10)64315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gille H, Strahl T, Shaw PE. Activation of ternary complex factor Elk-1 by stress-activated protein kinases. Curr Biol. 1995;5:1191–1200. doi: 10.1016/s0960-9822(95)00235-1. [DOI] [PubMed] [Google Scholar]
- 33.Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer. 2001;1:11–21. doi: 10.1038/35094017. [DOI] [PubMed] [Google Scholar]
- 34.Habermann E. Palytoxin acts through Na+,K+-ATPase. Toxicon. 1989;27:1171–1187. doi: 10.1016/0041-0101(89)90026-3. [DOI] [PubMed] [Google Scholar]
- 35.Han J, Lee JD, Jiang Y, Li Z, Feng L, Ulevitch RJ. Characterization of the structure and function of a novel MAP kinase kinase (MKK6) J Biol Chem. 1996;271:2886–2891. doi: 10.1074/jbc.271.6.2886. [DOI] [PubMed] [Google Scholar]
- 36.Hennings H, Glick AB, Greenhalgh DA, Morgan DL, Strickland JE, Tennenbaum T, Yuspa SH. Critical aspects of initiation, promotion, and progression in multistage epidermal carcinogenesis. Proc Soc Exp Biol Med. 1993;202:1–8. doi: 10.3181/00379727-202-43511a. [DOI] [PubMed] [Google Scholar]
- 37.Hoshino R, Chatani Y, Yamori T, Tsuruo T, Oka H, Yoshida O, Shimada Y, Ari-i S, Wada H, Fujimoto J, Kohno M. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene. 1999;18:813–822. doi: 10.1038/sj.onc.1202367. [DOI] [PubMed] [Google Scholar]
- 38.Iordanov MS, Magun BE. Loss of cellular K+ mimics ribotoxic stress. Inhibition of protein synthesis and activation of the stress kinases SEK1/MKK4, stress-activated protein kinase/c-Jun NH2-terminal kinase 1, and p38/HOG1 by palytoxin. J Biol Chem. 1998;273:3528–3534. doi: 10.1074/jbc.273.6.3528. [DOI] [PubMed] [Google Scholar]
- 39.Jaisser F, Beggah AT. The nongastric H+-K+-ATPases: molecular and functional properties. Am J Physiol. 1999;276:F812–824. doi: 10.1152/ajprenal.1999.276.6.F812. [DOI] [PubMed] [Google Scholar]
- 40.Karin M, Gallagher E. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life. 2005;57:283–295. doi: 10.1080/15216540500097111. [DOI] [PubMed] [Google Scholar]
- 41.Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 42.Keyse SM. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr Opin Cell Biol. 2000;12:186–192. doi: 10.1016/s0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 43.Kim SC, Hahn JS, Min YH, Yoo NC, Ko YW, Lee WJ. Constitutive activation of extracellular signal-regulated kinase in human acute leukemias: combined role of activation of MEK, hyperexpression of extracellular signal-regulated kinase, and downregulation of a phosphatase, PAC1. Blood. 1999;93:3893–3899. [PubMed] [Google Scholar]
- 44.Kuroki DW, Bignami GS, Wattenberg EV. Activation of stress-activator protein kinase/c-Jun N-terminal kinase by the non-TPA-type tumor promoter palytoxin. Cancer Res. 1996;56:637–644. [PubMed] [Google Scholar]
- 45.Kuroki DW, Minden A, Sanchez I, Wattenberg EV. Regulation of a c-Jun amino-terminal kinase/stress-activated protein kinase cascade by a sodium-dependent signal transduction pathway. J Biol Chem. 1997;272:23905–23911. doi: 10.1074/jbc.272.38.23905. [DOI] [PubMed] [Google Scholar]
- 46.Levine L, Fujiki H. Stimulation of arachidonic acid metabolism by different types of tumor promoters. Carcinogenesis. 1985;6:1631–1634. doi: 10.1093/carcin/6.11.1631. [DOI] [PubMed] [Google Scholar]
- 47.Levine L, Fujiki H, Gjika HB, Van Vunakis H. Production of antibodies to palytoxin: neutralization of several biological properties of palytoxin. Toxicon. 1987;25:1273–1282. doi: 10.1016/0041-0101(87)90005-5. [DOI] [PubMed] [Google Scholar]
- 48.Levine L, Xiao D, Fujiki H. A combination of palytoxin with 1-oleoyl-2-acetyl-glycerol (OAG) or insulin or interleukin-1 synergistically stimulates arachidonic acid metabolism, but combinations of 12-O-tetradecanoylphorbol-13-acetate (TPA)-type tumor promoters with OAG do not. Carcinogenesis. 1986;7:99–103. doi: 10.1093/carcin/7.1.99. [DOI] [PubMed] [Google Scholar]
- 49.Levine L, Xiao DM, Fujiki H. Combinations of palytoxin or 12-O-tetradecanoylphorbol-13-acetate and recombinant human insulin growth factor-I or insulin synergistically stimulate prostaglandin production in cultured rat liver cells and squirrel monkey aorta smooth muscle cells. Prostaglandins. 1986;31:669–681. doi: 10.1016/0090-6980(86)90173-5. [DOI] [PubMed] [Google Scholar]
- 50.Li S, Wattenberg EV. Cell-type-specific activation of p38 protein kinase cascades by the novel tumor promoter palytoxin. Toxicol Appl Pharmacol. 1999;160:109–119. doi: 10.1006/taap.1999.8754. [DOI] [PubMed] [Google Scholar]
- 51.Li S, Wattenberg EV. Differential activation of mitogen-activated protein kinases by palytoxin and ouabain, two ligands for the Na+,K+-ATPase. Toxicol Appl Pharmacol. 1998;151:377–384. doi: 10.1006/taap.1998.8471. [DOI] [PubMed] [Google Scholar]
- 52.Lin A, Minden A, Martinetto H, Claret FX, Lange-Carter C, Mercurio F, Johnson GL, Karin M. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science. 1995;268:286–290. doi: 10.1126/science.7716521. [DOI] [PubMed] [Google Scholar]
- 53.Lupulescu A. Prostaglandins, their inhibitors and cancer. Prostaglandins Leukot Essent Fatty Acids. 1996;54:83–94. doi: 10.1016/s0952-3278(96)90064-2. [DOI] [PubMed] [Google Scholar]
- 54.Marchetti S, Gimond C, Chambard JC, Touboul T, Roux D, Pouyssegur J, Pages G. Extracellular signal-regulated kinases phosphorylate mitogen-activated protein kinase phosphatase 3/DUSP6 at serines 159 and 197, two sites critical for its proteasomal degradation. Mol Cell Biol. 2005;25:854–864. doi: 10.1128/MCB.25.2.854-864.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marchetti S, Gimond C, Roux D, Gothie E, Pouyssegur J, Pages G. Inducible expression of a MAP kinase phosphatase-3-GFP chimera specifically blunts fibroblast growth and ras-dependent tumor formation in nude mice. J Cell Physiol. 2004;199:441–450. doi: 10.1002/jcp.10465. [DOI] [PubMed] [Google Scholar]
- 56.Marks F, Furstenberger G. Cancer chemoprevention through interruption of multistage carcinogenesis. The lessons learnt by comparing mouse skin carcinogenesis and human large bowel cancer. Eur J Cancer. 2000;36:314–329. doi: 10.1016/s0959-8049(99)00318-4. [DOI] [PubMed] [Google Scholar]
- 57.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 58.McCawley LJ, Li S, Wattenberg EV, Hudson LG. Sustained activation of the mitogen-activated protein kinase pathway. A mechanism underlying receptor tyrosine kinase specificity for matrix metalloproteinase-9 induction and cell migration. J Biol Chem. 1999;274:4347–4353. doi: 10.1074/jbc.274.7.4347. [DOI] [PubMed] [Google Scholar]
- 59.Miura D, Kobayashi M, Kakiuchi S, Kasahara Y, Kondo S. Enhancement of Transformed Foci and Induction of Prostaglandins in Balb/c 3T3 Cells by Palytoxin: In Vitro Model Reproduces Carcinogenic Responses in Animal Models Regarding the Inhibitory Effect of Indomethacin and Reversal of Indomethacin's Effect by Exogenous Prostaglandins. Toxicol Sci. 2006;89:154–163. doi: 10.1093/toxsci/kfi342. [DOI] [PubMed] [Google Scholar]
- 60.Moore RE, Bartolini G. Structure of palytoxin. J Am Chem Soc. 1981;103:2491–2494. [Google Scholar]
- 61.Moriguchi T, Toyoshima F, Gotoh Y, Iwamatsu A, Irie K, Mori E, Kuroyanagi N, Hagiwara M, Matsumoto K, Nishida E. Purification and identification of a major activator for p38 from osmotically shocked cells. Activation of mitogen-activated protein kinase kinase 6 by osmotic shock, tumor necrosis factor-alpha, and H2O2. J Biol Chem. 1996;271:26981–26988. doi: 10.1074/jbc.271.43.26981. [DOI] [PubMed] [Google Scholar]
- 62.Muda M, Boschert U, Dickinson R, Martinou JC, Martinou I, Camps M, Schlegel W, Arkinstall S. MKP-3, a novel cytosolic protein-tyrosine phosphatase that exemplifies a new class of mitogen-activated protein kinase phosphatase. J Biol Chem. 1996;271:4319–4326. doi: 10.1074/jbc.271.8.4319. [DOI] [PubMed] [Google Scholar]
- 63.Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol. 2002;4:556–564. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- 64.Nemoto S, Xiang J, Huang S, Lin A. Induction of apoptosis by SB202190 through inhibition of p38beta mitogen-activated protein kinase. J Biol Chem. 1998;273:16415–16420. doi: 10.1074/jbc.273.26.16415. [DOI] [PubMed] [Google Scholar]
- 65.Nishizuka Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature. 1984;308:693–698. doi: 10.1038/308693a0. [DOI] [PubMed] [Google Scholar]
- 66.Ohuchi K, Watanabe M, Yoshizawa K, Tsurufuji S, Fujiki H, Suganuma M, Sugimura T, Levine L. Stimulation of prostaglandin E2 production by 12-O-tetradecanoylphorbol 13-acetate (TPA)-type and non-TPA-type tumor promoters in macrophages and its inhibition by cycloheximide. Biochim Biophys Acta. 1985;834:42–47. doi: 10.1016/0005-2760(85)90174-2. [DOI] [PubMed] [Google Scholar]
- 67.Pitot HC, Dragan YP. The instability of tumor promotion in relation to human cancer risk. Prog Clin Biol Res. 1995;391:21–38. [PubMed] [Google Scholar]
- 68.Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rosette C, Karin M. Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science. 1996;274:1194–1197. doi: 10.1126/science.274.5290.1194. [DOI] [PubMed] [Google Scholar]
- 70.Sanchez I, Hughes RT, Mayer BJ, Yee K, Woodgett JR, Avruch J, Kyriakis JM, Zon LI. Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature. 1994;372:794–798. doi: 10.1038/372794a0. [DOI] [PubMed] [Google Scholar]
- 71.Santen RJ, Song RX, McPherson R, Kumar R, Adam L, Jeng MH, Yue W. The role of mitogen-activated protein (MAP) kinase in breast cancer. J Steroid Biochem Mol Biol. 2002;80:239–256. doi: 10.1016/s0960-0760(01)00189-3. [DOI] [PubMed] [Google Scholar]
- 72.Scheiner-Bobis G, Hubschle T, Diener M. Action of palytoxin on apical H+/K+-ATPase in rat colon. Eur J Biochem. 2002;269:3905–3911. doi: 10.1046/j.1432-1033.2002.03056.x. [DOI] [PubMed] [Google Scholar]
- 73.Scheiner-Bobis G, Meyer zu Heringdorf D, Christ M, Habermann E. Palytoxin induces K+ efflux from yeast cells expressing the mammalian sodium pump. Mol Pharmacol. 1994;45:1132–1136. [PubMed] [Google Scholar]
- 74.Sebolt-Leopold JS. Development of anticancer drugs targeting the MAP kinase pathway. Oncogene. 2000;19:6594–6599. doi: 10.1038/sj.onc.1204083. [DOI] [PubMed] [Google Scholar]
- 75.Sebolt-Leopold JS, Dudley DT, Herrera R, Van Becelaere K, Wiland A, Gowan RC, Tecle H, Barrett SD, Bridges A, Przybranowski S, Leopold WR, Saltiel AR. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med. 1999;5:810–816. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- 76.Segrelles C, Ruiz S, Perez P, Murga C, Santos M, Budunova IV, Martinez J, Larcher F, Slaga TJ, Gutkind JS, Jorcano JL, Paramio JM. Functional roles of Akt signaling in mouse skin tumorigenesis. Oncogene. 2002;21:53–64. doi: 10.1038/sj.onc.1205032. [DOI] [PubMed] [Google Scholar]
- 77.Smalley KS. A pivotal role for ERK in the oncogenic behaviour of malignant melanoma? Int J Cancer. 2003;104:527–532. doi: 10.1002/ijc.10978. [DOI] [PubMed] [Google Scholar]
- 78.Stein B, Brady H, Yang MX, Young DB, Barbosa MS. Cloning and characterization of MEK6, a novel member of the mitogen-activated protein kinase kinase cascade. J Biol Chem. 1996;271:11427–11433. doi: 10.1074/jbc.271.19.11427. [DOI] [PubMed] [Google Scholar]
- 79.Strickland JE, Greenhalgh DA, Koceva-Chyla A, Hennings H, Restrepo C, Balaschak M, Yuspa SH. Development of murine epidermal cell lines which contain an activated rasHa oncogene and form papillomas in skin grafts on athymic nude mouse hosts. Cancer Res. 1988;48:165–169. [PubMed] [Google Scholar]
- 80.Suganuma M, Fujiki H, Okabe S, Nishiwaki S, Brautigan D, Ingebritsen TS, Rosner MR. Structurally different members of the okadaic acid class selectively inhibit protein serine/threonine but not tyrosine phosphatase activity. Toxicon. 1992;30:873–878. doi: 10.1016/0041-0101(92)90385-i. [DOI] [PubMed] [Google Scholar]
- 81.Tatsumi M, Takahashi M, Ohizumi Y. Mechanism of palytoxin-induced [3H]norepinephrine release from a rat pheochromocytoma cell line. Mol Pharmacol. 1984;25:379–383. [PubMed] [Google Scholar]
- 82.Warmka JK, Mauro LJ, Wattenberg EV. Mitogen-activated protein kinase phosphatase-3 is a tumor promoter target in initiated cells that express oncogenic Ras. J Biol Chem. 2004;279:33085–33092. doi: 10.1074/jbc.M403120200. [DOI] [PubMed] [Google Scholar]
- 83.Warmka JK, Winston SE, Zeliadt NA, Wattenberg EV. Extracellular signal-regulated kinase transmits palytoxin-stimulated signals leading to altered gene expression in mouse keratinocytes. Toxicol Appl Pharmacol. 2002;185:8–17. doi: 10.1006/taap.2002.9519. [DOI] [PubMed] [Google Scholar]
- 84.Wattenberg EV, Byron KL, Villereal ML, Fujiki H, Rosner MR. Sodium as a mediator of non-phorbol tumor promoter action. Down-modulation of the epidermal growth factor receptor by palytoxin. J Biol Chem. 1989;264:14668–14673. [PubMed] [Google Scholar]
- 85.Wattenberg EV, Fujiki H, Rosner MR. Heterologous regulation of the epidermal growth factor receptor by palytoxin, a non-12-O-tetradecanoylphorbol-13-acetate-type tumor promoter. Cancer Res. 1987;47:4618–4622. [PubMed] [Google Scholar]
- 86.Wattenberg EV, McNeil PL, Fujiki H, Rosner MR. Palytoxin down-modulates the epidermal growth factor receptor through a sodium-dependent pathway. J Biol Chem. 1989;264:213–219. [PubMed] [Google Scholar]
- 87.Wattenberg EV, Uemura D, Byron KL, Villereal ML, Fujiki H, Rosner MR. Structure-activity studies of the nonphorbol tumor promoter palytoxin in Swiss 3T3 cells. Cancer Res. 1989;49:5837–5842. [PubMed] [Google Scholar]
- 88.Whitmarsh AJ, Davis RJ. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J Mol Med. 1996;74:589–607. doi: 10.1007/s001090050063. [DOI] [PubMed] [Google Scholar]
- 89.Whitmarsh AJ, Shore P, Sharrocks AD, Davis RJ. Integration of MAP kinase signal transduction pathways at the serum response element. Science. 1995;269:403–407. doi: 10.1126/science.7618106. [DOI] [PubMed] [Google Scholar]
- 90.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 91.Xie Z. Molecular mechanisms of Na/K-ATPase-mediated signal transduction. Ann N Y Acad Sci. 2003;986:497–503. doi: 10.1111/j.1749-6632.2003.tb07234.x. [DOI] [PubMed] [Google Scholar]
- 92.Xie Z, Cai T. Na+-K+--ATPase-mediated signal transduction: from protein interaction to cellular function. Mol Interv. 2003;3:157–168. doi: 10.1124/mi.3.3.157. [DOI] [PubMed] [Google Scholar]
- 93.Yip-Schneider MT, Lin A, Marshall MS. Pancreatic tumor cells with mutant K-ras suppress ERK activity by MEK-dependent induction of MAP kinase phosphatase-2. Biochem Biophys Res Commun. 2001;280:992–997. doi: 10.1006/bbrc.2001.4243. [DOI] [PubMed] [Google Scholar]
- 94.Yoshizumi M, Houchi H, Ishimura Y, Masuda Y, Morita K, Oka M. Mechanism of palytoxin-induced Na+ influx into cultured bovine adrenal chromaffin cells: possible involvement of Na+/H+ exchange system. Neurosci Lett. 1991;130:103–106. doi: 10.1016/0304-3940(91)90238-o. [DOI] [PubMed] [Google Scholar]
- 95.Young MR, Li JJ, Rincon M, Flavell RA, Sathyanarayana BK, Hunziker R, Colburn N. Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion. Proc Natl Acad Sci U S A. 1999;96:9827–9832. doi: 10.1073/pnas.96.17.9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yuspa SH. The pathogenesis of squamous cell cancer: lessons learned from studies of skin carcinogenesis. J Dermatol Sci. 1998;17:1–7. doi: 10.1016/s0923-1811(97)00071-6. [DOI] [PubMed] [Google Scholar]
- 97.Zeliadt NA, Warmka JK, Winston SE, Kahler R, Westendorf JJ, Mauro LJ, Wattenberg EV. Tumor promoter-induced MMP-13 gene expression in a model of initiated epidermis. Biochem Biophys Res Commun. 2004;317:570–577. doi: 10.1016/j.bbrc.2004.03.081. [DOI] [PubMed] [Google Scholar]