

Abstract
Sympathetic hyperinnervation occurs in human ventricular tissue after myocardial infarction and may contribute to arrhythmias. Aberrant sympathetic sprouting is associated with elevated nerve growth factor (NGF) in many contexts, including ventricular hyperinnervation. However, it is unclear whether cardiomyocytes or other cell types are responsible for increased NGF synthesis. In this study, left coronary arteries were ligated and ventricular tissue examined in rats 1-28 days post-infarction. Infarct and peri-infarct tissue was essentially devoid of sensory and parasympathetic nerves at all time points. However, areas of increased sympathetic nerve density were observed in the peri-infarct zone between post-ligation days 4-14. Hyperinnervation occurred in regions containing accumulations of macrophages and myofibroblasts. To assess whether these inflammatory cells synthesize NGF, sections were processed for NGF in situ hybridization and immunohistochemistry. Both macrophage1 antigen-positive macrophages and α-smooth muscle actin immunoreactive myofibroblasts expressed NGF in areas where they were closely proximate to sympathetic nerves. To investigate whether NGF produced by peri-infarct cells induces sympathetic outgrowth, we co-cultured adult sympathetic ganglia with peri-infarct explants. Neurite outgrowth from sympathetic ganglia was significantly greater at post-ligation days 7-14 as compared to control tissue. Addition of an NGF function-blocking antibody prevented the increased neurite outgrowth induced by peri-infarct tissue. These findings provide evidence that inflammatory cell NGF synthesis plays a causal role in sympathetic hyperinnervation following myocardial infarction.
Section: Disease-Related Neuroscience
Keywords: Myocardial Infarction, Nerve Sprouting, Sympathetic Nervous System, Nerve Growth Factor, Inflammation
1. INTRODUCTION
Arrhythmias are frequent complications of myocardial infarction, occurring most often in the first 30 days following the event (Solomon, et al., 2005). While increased sympathetic drive and altered ventricular conduction are clearly contributory (Schomig, 1990; Du, et al., 1999; Lal et al., 2005), recent evidence suggests that sympathetic axon remodeling in ventricular myocardium may be a major factor. Postmortem analyses of infarcted human myocardium show abnormal increases in numbers of sympathetic axons adjacent to the site of injury (Cao, et al., 2000a). Ischemic injury appears to be the cause of the remodeling, as coronary artery ligation induces hyperinnervation in dogs (Lai, et al., 2000; Zhou, et al., 2004) and rats (Ahonen, et al., 1975; Paessens and Borchard, 1980; Holmgren, et al., 1981; Vracko, et al., 1990; Kaye, et al., 2000; Li, et al., 2004). Through enhanced norepinephrine-mediated myocardial depolarization, abnormally increased sympathetic nerve density may be proarrhythmogenic and represent a significant factor in post-infarct sudden cardiac death.
The cellular and molecular mechanisms leading to post-infarct sympathetic hyperinnervation remain incompletely understood. However, the neurotrophin nerve growth factor (NGF) is strongly implicated. NGF is a potent growth and survival factor for sympathetic neurons (Korsching and Thoenen, 1985; Glebova and Ginty, 2004). Recent evidence indicates that NGF expression is up-regulated in the region of the infarction (Hiltunen, et al., 2001; Zhou, et al., 2004), and infusion of NGF into the stellate ganglion results in cardiac hyperinnervation in canine hearts (Cao, et al., 2000b). However, the cell types involved in post-infarct cardiac NGF production and the neural selectivity of the response are not well defined. Similarly, while exogenous NGF can cause sympathetic sprouting there is no definitive evidence that endogenous NGF is required for cardiac sympathetic sprouting in the post-infarct heart.
The objective of the present study was to assess whether inflammatory cells, which are present in the infarcted myocardium (Vracko and Thorning, 1991; Desmouliére, et al., 1996) and may synthesize NGF in other contexts (Matsuda, et al., 1998; Hasan, et al., 2000; Aloe, 2004; Kawamoto and Matsuda, 2004), serve as a local source of NGF which in turn initiates sympathetic sprouting. Accordingly, we assessed whether inflammatory cells express NGF and whether expression is temporo-spatially consistent with the observed pattern of hyperinnervation. Additionally, the requirement for NGF release by this tissue in inducing sympathetic sprouting was assessed by antibody neutralization explant culture studies. The present study provides novel evidence that inflammatory myofibroblasts and macrophages play a central role in peri-infarct NGF synthesis, and that inflammatory cell-derived NGF is required for and sufficient to induce selective sprouting of cardiac sympathetic axons.
2. MATERIALS AND METHODS
Coronary Artery Ligation
Female Sprague-Dawley rats (60-70 days postnatal, ∼225g, Harlan Breeding Laboratories, Indianapolis, IN) were anesthetized by intraperitoneal injection of 60 mg/kg ketamine, 8 mg/kg xylazine, and 0.4 mg/kg atropine and ovariectomized bilaterally via flank incisions (Zoubina, et al., 2001). Ovariectomy eliminates potentially confounding effects of reproductive hormones which can influence both cardiovascular innervation and wound healing (Gillardon, et al., 1991; Saleh and Connell, 2000; Ashcroft and Ashworth, 2003; Gilliver, et al., 2003). Moreover, it simulates postmenopausal conditions in which post-infarction mortality increases dramatically (Grace, et al., 2004).
Seven days after ovariectomy, rats were anesthetized as above, intubated, respired mechanically, and a left lateral thoracotomy performed. The left coronary artery was ligated (6-0 silk suture with an atraumatic needle) approximately 8 mm distal to its emergence beneath the left atrium (Scheuer and Mifflin, 1998). This elicited a visible infarct corresponding to the ischemic region of the myocardium at post-ligation days (PLD) 1-28 (n=4-7/time point, Figure 1A). Sham surgery (n=5) involved similarly passing a suture around the artery but leaving it untied for a comparable period. Infarct confirmation was carried out under isoflurane anesthesia using in vivo high resolution magnetic resonance imaging (MRI; 9.4 Tesla scanner, Varian, Palo Alto, CA) with a 6 cm inner diameter volume coil and a special surface coil (Bilgen, 2004) oriented longitudinally with respect to the heart. ECG- and respiratory-gated data acquisition was monitored (Model 1025, SA Instruments, Stony Brook, NY) and a trigger delay time incorporated to capture the heart at end-diastole. Images were acquired using spin-echo sequence (repetition time = 2000 ms, echo time= 12 ms for proton density imaging and 34 ms for T2-weighted imaging). MRI of subsets of infarcted rats at PLD1-28 (n=2/randomly chosen group) showed occlusion of the coronary artery (Figure 1B) and a hyperintense region of the myocardium corresponding to the infarction (Figure 1C). For PLD7-28 hearts, tissue was processed only if visible dimensions of the infarct were ∼4x6 mm ± 1mm.
Figure 1.

Visualization of the infarct after coronary artery ligation. A. Stereomicroscopic image of the rat heart on post-ligation day (PLD) 7 shows an infarcted region (INF; dashed line) on the ventral surface of the left ventricle. Non-infarcted myocardium (MYC) lies lateral to the infarct, and a peri-infarct transition zone (PI) surrounds the infarct. Arrow indicates the site of coronary artery ligation. B. Proton density spin echo magnetic resonance image obtained with a surface coil from a PLD4 heart in coronal section demonstrates the left coronary artery and the absence of perfusion below the site of ligation (arrow). C. Volume coil T2-weighted MRI image from a PLD4 heart in axial section shows a hyperintense area, as compared to normal myocardium, reflecting an area of infarction in the ventricular wall (arrowhead).
At PLD 1, 4, 7, 14, and 28, rats were anesthetized (pentobarbital, 60 mg/kg ip) and tissue harvested (n=4-7/time point). In addition to sham-operated rats, a group of unoperated rats (n=5) were included to provide age-matched control data; because no differences between these groups were observed, results are presented as a combined control (CON) data set.
After tissue harvesting, left ventricular myocardium was cut in half through the center of the infarct along the baso-apical axis; one half was used fresh for explant tissue culture and the other half for immunohistochemistry was snap-frozen on dry ice in tissue freezing medium (Triangle Biomedical Sciences, Durham, NC), and stored at -80°C. All experimental manipulations were approved by the Institutional Animal Care and Use Committee of the University of Kansas Medical Center and conformed with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Immunocytochemistry
Myocardium was cryosectioned perpendicular to the ventricular basal-apical axis and 10 sets of consecutive 10 μm sections collected at 500 μm intervals beginning 2 mm basal to the infarct margin and continuing to the apex, yielding ∼24 sections per series. Sections were post-fixed for 5 min at room temperature with fresh 4% paraformaldehyde in PBS (Fisher, Fair Lawn, NJ, USA) then rinsed with PBS containing 0.3% Triton X-100 (PBST; Sigma), blocked (1% bovine serum albumin plus 5% goat or donkey serum in PBST), and incubated overnight at room temperature with primary antibody in antibody diluent (5% goat or donkey serum in PBST). Secondary antibody incubation was for 90 min with visualization for single or double staining by Cy2- or Cy3-labeled secondary antibody fluorophores (1:100, Jackson ImmunoResearch, West Grove, PA), or tyramide signal amplification (TSA, Molecular Probes, Eugene, OR) for NGF. Sympathetic axons were visualized with antibodies to Tyrosine Hydroxylase (TH, mouse monoclonal, 1:100, Immunostar, Hudson, WI), Dopamine β-hydroxylase (DBH, rabbit IgG, 1:400, Immunostar) and Vesicular Monoamine Transporter-2 (VMAT; rabbit IgG, 1:100, Chemicon, Temecula, CA). Parasympathetic nerves were identified by antisera to vesicular acetylcholine transporter (VAChT; goat IgG, 1:100, Chemicon) and peptidergic sensory nociceptor nerves by antisera to calcitonin gene related peptide (CGRP; 1:100, rabbit IgG, Chemicon). Activated macrophages were stained with mouse IgG for the macrophage antigen MAC1 (OX42 or CD11b; 1:100, Serotec, Raleigh, NC) or CD68 (ED1; 1:100, Chemicon). Inflammatory myofibroblasts, a modified fibroblast with contractile properties, were demonstrated with a Cy3-conjugated monoclonal antibody to α-smooth muscle actin (α-SMA; 1:400, Sigma, St. Louis, MO). NGF protein was detected with an antibody to NGF-β (rabbit IgG, 1:100, Chemicon or rabbit IgG, 1:50, Santa Cruz Biotech., Santa Cruz, CA). All incubations were carried out in a humidified chamber and slides were mounted with Fluoromount G (Southern Biotechnology Associates, Inc., Birmingham, AL). Antibody omission, antigen pre-adsorption, and primary antibody substitution with pure immunoglobulin served as negative controls.
Sympathetic innervation quantitation
Initial microscopic inspection of the infarct revealed variable distributions of sympathetic innervation, with large areas of infarction and adjacent myocardium having few or no nerves punctuated by smaller localized regions with obvious hyperinnervation, as noted by others (Cao et. al., 2000b; Zhou et. al., 2004). To assess whether regional maximal axon density following infarction exceeded that of controls and changed as a function of time after ligation, one set of stepped serial sections (10 sections per heart, n=4-5 per time point) was stained for TH-ir and images of regions containing aggregations of sympathetic axons were captured digitally from coded sections by a blinded observer. For each heart, 3 sample fields (0.136 mm2 per field) containing maximum innervation were selected and innervation density measured by threshold discrimination (AnalySIS 3.1, Soft Imaging System, Lakewood, CO) to obtain apparent axon sample field, and these values were averaged for each subject. Statistical analysis was conducted by ANOVA (p≤0.05) on ranked data with post-hoc analysis by the Holm-Sidak method (SigmaStat 3.0, SPSS, Chicago, IL).
NGF in situ hybridization
NGF transcripts were detected by in situ hybridization using a digoxigenin-labeled antisense probe to rat pre-pro-NGF cDNA (Hasan, et al., 2000). Sense probes, RNase A treatment, or probe omissions were carried out as negative controls, with salivary gland as a positive control. Control and experimental sections were processed together to ensure that variations in processing did not contribute to differences in staining intensity.
Tissue explant co-cultures
Peri-infarct tissue (∼1mm of non-infarcted tissue immediately adjacent to the infarct margin) and intact myocardium (3-4 mm lateral to the infarct) were excised and immediately placed in ice-cold DMEM/F12 media (Life Technologies, Grand Island, NY); tissue was also obtained from corresponding regions in CON rats. Superior cervical and stellate ganglia from unoperated rats were desheathed, each cut into 4 pieces measuring approximately 250 μm/side, and embedded in rat tail collagen. One piece of ganglion explant was embedded in 200 μl of freshly neutralized rat tail collagen in the center of a well of a 48 well plate (Nalge Nunc, Rochester, NY). Then, two 1 mm2 pieces of peri-infarct myocardial tissue were placed on opposite sides of the ganglion explant at a distance of 1 mm, and 200 μl of serum-free DMEM/F12 media layered over the solidified gel. Cultures were maintained at 37°C in room air with 5% CO2 as in previous studies (Krizsan-Agbas, et al., 2003). Three-4 replicates were conducted with tissue from 4-5 animals per time point.
Following incubation for 72 hours, cultures were fixed with 4% paraformaldehyde (120 min) then stored at 4°C in PBS. Neurites emanating from ganglion explants were visualized by differential interference contrast microscopy (Nikon Eclipse TE300 inverted microscope). Neurites exceeding a length of 60 μm as measured using an eyepiece reticle were counted by a blinded observer. To asses the role of NGF in ventricle-induced outgrowth, an NGF function-blocking antibody which does not cross-react demonstrably with other known members of the neurotrophin family was added to cultures (anti-rat β-NGF, goat IgG, 0.4 μg/ml, R&D Systems, Minneapolis, MN).
All data are expressed as means ±SEM. Comparisons were performed by one or two-way ANOVA followed by a Student-Neumann-Keuls test (significance at p<0.05).
3. RESULTS
Axons and inflammatory cells in ventricular myocardium following artery ligation
CON
In left ventricles of CON rats TH-immunoreactive (-ir) fibers were present at low density adjacent to cardiomyocytes and blood vessels (Figure 2A, 3), and as larger bundles in the epicardium. TH-ir cells, 8-12 μm diameter and lacking axons, were sometimes encountered near blood vessels (Figure 2B). CGRP-(Figure 4A) and VAChT-ir (Figure 4B) axons were less abundant than TH-ir nerves and were primarily associated with vasculature. α-SMA staining was restricted to vascular smooth muscle (Figure 5A) and cells rarely stained for the macrophage marker MAC-1 (Figure 6A).
Figure 2.
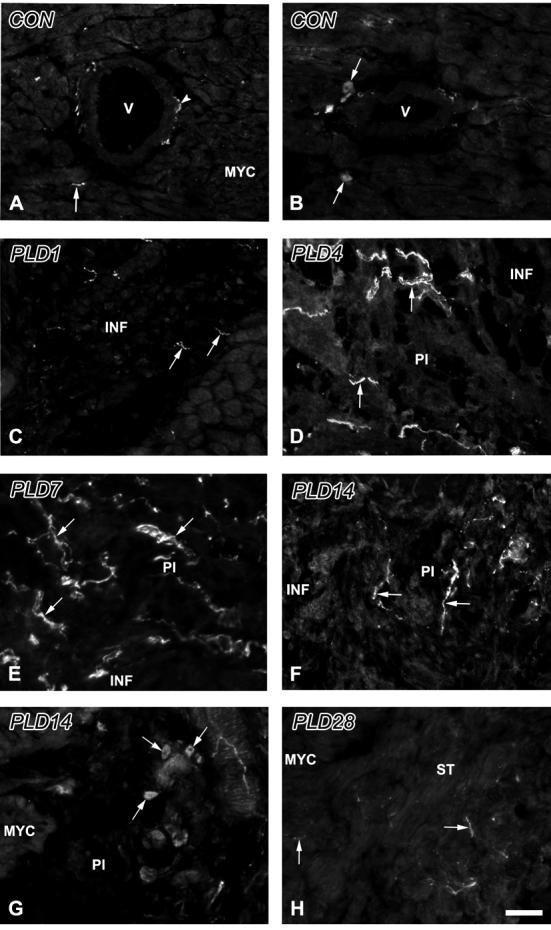
Tyrosine hydroxylase-immunoreactive sympathetic axons and intrinsic neurons within the ventricle. A. Control. Axons (arrowhead) are associated with blood vessels (V), as well as with myocardial cells (arrow). B. Control. Small, axonless cells (arrows) are occasionally observed in conjunction with myocardial blood vessels (V). C. PLD1. The region of infarction (INF) contains several small, irregular axon profiles (arrows). D. PLD4. The peri-infarct (PI) zone lies adjacent to the INF, and shows numerous immunoreactive axons (arrows). E. PLD7. PI tissue contains very high densities of sympathetic axons. F. PLD14. Numbers of nerves (arrows) are reduced relative to PLD7, but remain elevated relative to controls. G. PLD 14. Numbers of small, axonless cells (arrows) appear to be increased in the PI, adjacent to normal myocardium (MYC). H. PLD28. Axon numbers are reduced relative to PLD 14, but fibers (arrows) are still encountered frequently near and within the scar tissue (ST). Scale bar in H is 30 μm.
Figure 3.

Quantitative analysis of maximal regional sympathetic nerve density within peri-infarct myocardium. Area occupied by tyrosine hydroxylase-immunoreactive nerve profiles in peri-infract sample fields (0.136 mm2) is shown for control (CON) and post-ligation days (PLD) 1-28. a, p<0.05 vs. CON. b, p<0.05 vs. PLD 1. c, p<0.05 vs. PLD 28.
Figure 4.
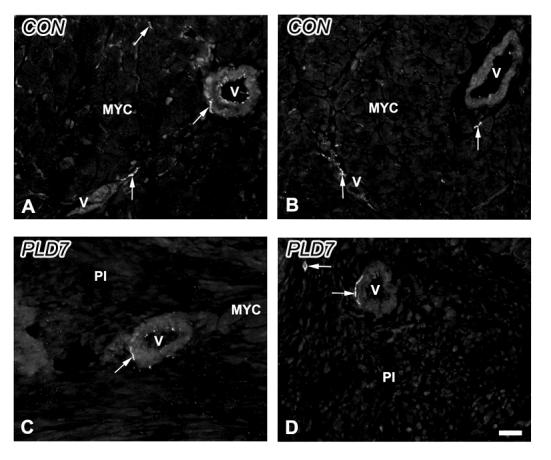
Sensory and parasympathetic nerves in control and ligated ventricular myocardium. A. Control. Presumptive sensory unmyelinated axons immunostained for calcitonin gene-related peptide (arrows) are present freely within the myocardium (MYC) and in association with blood vessels (V). B. Control. Parasympathetic cholinergic nerves immunostained for vesicular acetylcholine transporter (VAChT; arrows) are associated directly with cardiomyocytes and with blood vessels. C. PLD7. Calcitonin gene-related peptide-immunoreactive nerves (arrow) are associated with blood vessels (V) in the peri-infarction (PI) zone. D. PLD7. VAChT-immunoreactive nerves (arrow) in the PI are encountered with low frequency. Scale bar = 30 μm.
Figure 5.

α-Smooth muscle actin immunoreactivity in the control and infarcted ventricle. A. Control. α-smooth muscle actin immunoreactivity (α-SMA-ir) in non-infarcted myocardium is restricted to the smooth muscle walls of blood vessels (V). B. PLD1. α-SMA-ir is present within individual spindle-shaped cells (arrows) as well as in aggregates, and in blood vessels (arrowheads) within the infarct area (INF) adjacent to normal myocardium (MYC). C. PLD4. The edge of the peri-infarct (PI) is delineated by α-SMA-ir myofibroblasts (arrows). D. PLD 7. Expression of α-SMA-ir myofibroblasts (arrows) continues to be robust in the PI accompanied by neovascularizing blood vessels (arrowhead). E. PLD7. α-SMA-ir is observed in blood vessels (arrow) and cardiomyocytes (arrowheads) at the transition between the PI and the adjacent MYC. F. PLD 28. α-SMA-ir myofibroblasts (arrows) are reduced in number, while cardiomyocytes adjacent to scar tissue maintain their expression of this protein (arrowheads). Scale bar in E is 30 μm for panels A-C, E; scale bar in F is 15 μm for panels D, F.
Figure 6.

MAC1-immunoreactivity in the control and infarcted ventricle. A. Control. MAC-1 immunoreactive cells (arrowheads) are few and primarily observed around blood vessels (V). B. PLD1. MAC-1 immunoreactivity is present in cells with a macrophage morphology (D) within the infarct (INF). C. PLD4. Numbers of MAC-1 immunoreactive cells (arrowheads) are high within peri-infarct (PI) tissue. D. PLD7. MAC-1 immunoreactivity continues to be abundant within PI tissue, particularly around blood vessels (V). E. PLD28. Staining for MAC-1 is occasional within scar tissue (ST). Scale bar in E is 30 μm for panels A, C-E; scale bar in B is 15 μm for panel B.
PLD1
Myocardial cells within the infarction zone appeared to be undergoing cellular disruption characteristic of necrotic cell death. Infrequently, isolated TH-ir fibers were associated with damaged cells (Figure 2C). TH-ir nerve density in regions adjacent to the infarct resembled that in CON myocardium (Figure 3). Similarly, CGRP- and VAChT-ir fibers were scarce both within and near the infarct area, but comparable to CON in areas adjacent to the infarct (data not shown).
α-SMA-ir was associated with damaged vessels within the wound, and with small vessels in the peri-infarct region which may reflect neovascularization (Figure 5B). Small numbers of spindle-shaped α-SMA-ir myofibroblasts were now observed at the infarct margins (Figure 5B). MAC-1-ir cells were present in the peri-infarct zone in numbers greater than those seen in CON myocardium (Figure 6B).
PLD4
The infarct zone contained a necrotic core composed of amorphous material and inflammatory cells, and was essentially devoid of TH-ir nerves. However, many TH-ir axons were present in the peri-infarct region, exceeding densities normally seen in intact myocardium (p<0.005; Figure 2D, 3). Lateral and basal to the infarct, TH-ir innervation was similar to CON tissue. TH- and DBH-ir cells were occasionally observed in the peri-infarct area (data not shown). CGRP-ir fibers were sometimes observed in areas of TH-ir hyperinnervation while VAChT-ir fibers were absent (data not shown).
In peri-infarct areas with TH-ir hyperinnervation, α-SMA-ir myofibroblasts were prominent, often in sheath-like aggregates (Figure 5C). Numerous MAC-1-ir macrophages (Figure 6C) were present within these regions.
PLD7
TH-ir nerves were extremely abundant in the peri-infarct zone as compared to CON (p<0.001) or PLD1 (p<0.005) myocardium (Figure 2E, 3), but typically did not penetrate the infarction. TH- and DBH-ir cells were observed with greater frequency than at earlier times. CGRP-ir (Figure 4C) and VAChT-ir (Figure 4D) fibers were only rarely encountered.
α-SMA-ir myofibroblasts and MAC-1-ir macrophages were prominent within the peri-infarct tissue (Figure 5D, 6D). α-SMA-ir was now also expressed in some peri-infarct cardiomyocytes (Figure 5E).
PLD14
TH-ir innervation was less abundant than at PLD7 but still greater than CON (p<0.001) or PLD1 (p<0.05) myocardium (Figure 2F, 3), while TH-ir cells continued to be frequently encountered (Figure 2G). CGRP- and VAChT-ir nerves remained sparse.
Both myofibroblasts and macrophages remained prominent residents of the peri-infarct myocardium, and some cardiomyocytes continued to display α-SMA-ir (data not shown).
PLD28
The infarction zone was now characterized by fibrotic tissue, and TH-ir nerves occasionally penetrated deeply into the scar with density slightly greater than CON (p<0.05) but less than PLD7 (p<0.05; Figure 2H, 3). TH-ir cells remained outside the scar. CGRP-ir and VAChT-ir fibers were occasionally encountered.
α-SMA-ir myofibroblasts were less abundant and α-SMA-ir was present occasionally in peri-wound cardiomyocytes (Figure 5F). MAC-1-ir was present, but appeared mainly in cells undergoing dissolution, probably reflecting apoptosis (Figure 6E). TH-ir nerves typically were not associated with inflammatory cells at this time.
Sympathetic axons associate spatially with inflammatory cells
To define the spatial relationships among sympathetic axons, macrophages and myofibroblasts, adjacent sections were immunostained for selective markers. Both MAC-1-ir macrophages and α-SMA-ir myofibroblasts tended to aggregate in overlapping regions of the peri-infarct (Figure 7). While TH-ir sympathetic nerves were seen associated with both cell types alone, the highest densities of TH-ir tended to be in regions containing both inflammatory cell types (Figure 7).
Figure 7.
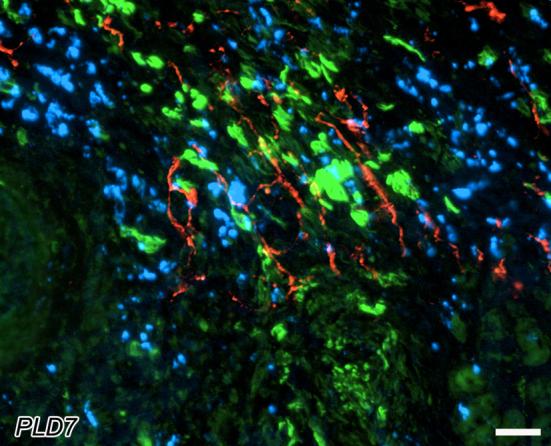
Spatial coexistence of peri-infarct sympathetic nerves with myofibroblasts and macrophages at post-ligation day 7. Tyrosine hydroxylase-immunoreactive nerves (red) are abundant within the peri-infarct region. These fibers associate with accumulations of myofibroblasts (green), as revealed by α-smooth muscle actin immunostaining in the adjacent section. Macrophages are also concentrated in the same regions of the tissue, as indicated by MAC1-immunostaining (blue) in an adjacent section. Note that sympathetic axons are most abundant in regions containing both macrophages and myofibroblasts. Scale bar = 30μm.
Peri-infarct inflammatory cells synthesize NGF
To define sites of NGF synthesis following infarction, we evaluated NGF mRNA and protein in tissue sections from control ventricles, PLD7 peri-infarct tissue when hyperinnervation is maximal, and PLD28 when it has regressed. In CON ventricles, NGF expression was modest, and both mRNA and protein were localized primarily to vascular smooth muscle (Figure 8A, B). At PLD7, NGF transcripts (Figure 8C) were strongly expressed within the peri-infarct in regions corresponding to areas of VMAT-ir sympathetic hyperinnervation (Figure 8D). α-SMA-immunostaining showed that regions of NGF expression corresponded to areas containing large numbers of myofibroblasts (Figure 8E). Additionally, CD68-ir macrophages frequently exhibited NGF-ir (Figure 8F). Catecholaminergic cells within the peri-infarct zone also exhibited NGF mRNA expression (Figures 8G). However, given their relatively low abundance, these cells did not represent a major contribution to overall peri-infarct NGF mRNA. By PLD28, NGF expression in the infarct region was reduced (Figure 8H) but remained greater than that of negative control sections (Figure 8I).
Figure 8.

NGF expression within control and infarcted ventricular myocardium. A. Control. NGF transcript expression (arrowheads) in control myocardium (MYC) is present primarily within the smooth muscle of blood vessels (V). B. Control. NGF immunoreactivity (arrowheads) is present within vascular smooth muscle in a similar pattern to mRNA. C. PLD7. NGF transcript expression (arrowheads) is strong within cells of the peri-infarct (PI). D. PLD7. VMAT immunoreactive nerves (arrows), in an adjacent section to panel C, are observed in a similar spatial plane to NGF expressing cells. E. PLD7. NGF immunoreactivity (Cy2, green) is co-localized within α-SMA-ir myofibroblasts (Cy3, red) resulting in a yellow coloration (arrowheads). F. PLD7. Immunostaining for NGF protein (Cy3, red) and CD68 macrophages (Cy2, green, arrows) shows colocalization of these proteins (arrowheads). G. PLD7. NGF mRNA expressing cells (arrowheads) are observed in the PI, some associated with a blood vessel (V). H. PLD28. NGF mRNA expression (arrowheads) is low, being present within only occasional cells of the scar tissue (ST). I. Control. Sections processed for in situ hybridization with a control sense probe do not show any demonstrable staining in ventricular tissue. Scale bar in G is 30μm for panels E, G; scale bar in I is 15μm for panels A-D, F, H-I.
NGF released by peri-infarct tissue induces sympathetic axon sprouting
To assess the ability of cardiac tissue to promote sympathetic outgrowth, we co-cultured cardiac explants with sympathetic ganglia. Superior cervical ganglia cultured with left ventricular myocardium from CON rats displayed relatively few neurites after 3d in culture (Figure 9A). However, when cultured with myocardium containing the peri-infarct zone from PLD7 rats, much more robust outgrowth was evident (Figure 9B).
Figure 9.

Explant co-culture of myocardial tissue with sympathetic ganglia. A. Superior cervical ganglion (SCG) explant shows modest neurite outgrowth when co-cultured with control ventricular myocardium. B. Co-culture of SCG with PLD7 peri-infarct tissue resulted in markedly increased neurite outgrowth. C. Addition of an NGF function-blocking antibody limits outgrowth to levels comparable to that of non-infarcted myocardium. Scale bar in C is 50 μm. D. Quantitative analysis of neurite outgrowth from the SCG induced by control ventricular myocardium (CON), peri-infarct myocardium, and non-infarcted regions of the myocardium taken several mms lateral to the infarct. Peri-infarct tissue induces greater neurite outgrowth than control or non-infarcted tissue (p<0.005). E, F. Quantitative analysis of the effect of blockade of NGF function by a selective anti-NGF antibody added to the culture medium. PLD 7 tissue induces greater neurite outgrowth from SCG (E) or stellate ganglion (StG; F) explants than control tissue (* indicates p<0.05). Anti-NGF added to PLD7 tissue cultures reduces neurite outgrowth to a level similar to control tissue.
The time course for the development of the enhanced trophic response of peri-infarct tissue was assessed quantitatively relative to ventricular tissue from CON rats and non-infarcted ventricle taken several mm lateral to the infarct. At PLD1, neurite outgrowth induced by all tissues was comparably low (Figure 9D). However, numbers of neurites induced by peri-infarct tissue were increased at PLD7 relative to CON tissue and non-infarcted ventricular tissue (p<0.01). In comparison to non-infarcted myocardium, peri-infarct tissue induced greater numbers of neurites at all times (p<0.005). However, neurite outgrowth induced by both infarcted and non-infarcted tissue increased after ligation (p<0.001, Figure 9D).
To assess the contribution of NGF synthesized in cardiac tissue towards sympathetic sprouting, we conducted additional cultures in which an NGF function-blocking antibody was added. NGF antibody neutralization reduced outgrowth markedly (Figure 9C), essentially eliminating the neuritogenic effect of the infarcted myocardium (p<0.001, Figure 9E). A similar effect was observed when stellate ganglia were co-cultured with PLD 7 tissue; peri-infarct tissue induced neuritogenesis was increased substantially (315%, p<0.05, Figure 9F) compared to CON myocardium. Addition of anti-NGF to the medium abrogated the peri-infarct tissue induced neurite outgrowth.
4. DISCUSSION
Cardiac sympathetic hyperinnervation as sequela of the inflammatory repair process
Myocardial infarction results in rapid remodeling of ventricular sympathetic innervation. Within 4 days of coronary artery ligation, sympathetic innervation in the peri-infarct zone is substantially greater than that of normal myocardium. Sympathetic hyperinnervation occurs selectively in regions containing abundant myofibroblasts and macrophages. This spatial association is consistent with the idea that these cells provide molecular signals attractive to ingrowing sympathetic axons. Further, these inflammatory cell types appear within the peri-infarct region prior to axonal ingrowth, suggesting that their presence may be requisite to sprouting of sympathetic nerves. The temporo-spatial dynamics of inflammatory macrophages and myofibroblasts are therefore consistent with a role in mediating sympathetic axon ingrowth and proliferation within peri-infarct tissue.
Previous studies in dogs showed that NGF protein increases by 7 days post-infarct, and returns to lower levels by 1 month (Zhou, et al., 2004), temporally paralleling changes in peri-infarct sympathetic innervation. Using a rodent model, we show here that i) both NGF mRNA and protein increase in the peri-infarct zone with a time course similar to increased protein reported for canines; ii) the time frame for increased NGF mRNA and protein parallels macrophage and myofibroblast emigration into the peri-infarct; iii) NGF protein and mRNA is localized primarily within peri-infarct macrophages and myofibroblasts, and iv) peri-infarct regions containing NGF-producing inflammatory cells are preferentially hyperinnervated. Together, these findings support the idea that peri-infarct hyperinnervation occurs as a result of the inflammatory process associated with myocardial repair rather than as a result of increased NGF synthesis in the myocardium itself.
NGF is obligatory for enhanced sympathetic sprouting by peri-infarct tissue
These findings also imply that inflammatory cell NGF synthesis represents a molecular mechanism responsible for sympathetic axon sprouting. However, it is important to note that recent evidence shows that NGF exists normally as multiple isoforms in various tissues (Hasan, et al., 2003), and that some NGF isoforms promote sympathetic degeneration rather than survival and sprouting (Lee, et al., 2001). Therefore, it is critical to confirm that peri-infarct NGF promotes sympathetic neuritogenesis. Our studies show that in tissue culture, peri-infarct myocardium exerts a much more potent effect of sympathetic neuritogenesis than does normal myocardium, implying that NGF produced by inflammatory cells is pro-neuritogenic and therefore is probably the mature isoform. Moreover, addition of an NGF function blocking antibody completely abrogated this effect. These studies provide strong evidence that peri-infarct myofibroblasts and macrophages synthesize biologically active, pro-neuritogenic NGF, and show for the first time that this protein is required for infarcted myocardial tissue to promote sprouting of sympathetic axons.
Selectivity of post-infarct hyperinnervation
One issue not fully resolved by earlier studies is whether peri-infarct noradrenergic hyperinnervation derives entirely from extrinsic sympathetic neurons, or whether intrinsic catecholaminergic neurons (Huang, et al., 1996) also contribute to peri-infarct innervation. While we identified small intrinsic catecholaminergic cells in the normal heart, their numbers were substantially greater in peri-infarct tissue. This observation is of interest in light of recent findings (Drapeau, et al., 2005) showing the presence of presumptive neural stem cells in myocardial infarct tissue that can differentiate into catecholaminergic neurons in culture. Although the function of these cells is unclear, intrinsic catecholaminergic neurons normally lack axons (Huang, et al., 1996), as is apparently the case in the peri-wound environment as well. They are therefore unlikely to contribute appreciably to peri-infarct noradrenergic hyperinnervation. Another possible role for these cells is NGF production within the peri-infarct. Our findings show that, similar to cells derived from neuronal precursors in culture (Drapeau, et al., 2005), these cells exhibit NGF mRNA and protein, as is true for other peripheral noradrenergic neurons (Hasan, et al., 2003). However, because the numbers of these cells are relatively small, it seems unlikely that they could contribute substantially to overall NGF synthesis within the peri-infarct.
Although the present study confirms peri-infarct hyperinnervation (Cao, et al., 2000a; Cao, et al., 2000b), other studies have reported reduced cardiac sympathetic innervation after infarction (Barber, et al., 1983; Igawa, et al., 2000; Li, et al., 2004). These differences may be related to the extent of infarction as larger infarcts can result in frank congestive heart failure, and this has been linked to cardiac NGF depletion and diminished sympathetic axon density (Kaye, et al., 2000; Qin, et al., 2002). Similarly, reduced NGF levels have been noted in patients with acute coronary symptoms as well as in atherosclerotic coronary vascular tissue (Chaldakov, et al., 2004; Manni, et al., 2005). Attenuation of NGF levels in cardiovascular dysfunctions therefore result in sympathetic denervation. In our model, distal ligation resulted in limited infarctions which, based on culture-NGF neutralization studies, did not deplete peri-infarct NGF. It would therefore appear that the extent of damage and the resulting cardiovascular dysfunction may be a significant variable in determining effects on both neurotrophin expression and sympathetic nerve remodeling.
Aside from infarct size and CHF status, regional heterogeneity in sympathetic innervation may also occur in different infarction models. Following ischemia-reperfusion, for example, an acute neurochemical change occurs in sympathetic innervation as a consequence of inflammatory cytokine release (Li, et al., 2004). Denervation in the apex of the heart after ischemia-reperfusion can also occur as the infarct may disrupt passage of sympathetic nerves (Barber, et al., 1983). After occlusion, infusion of NGF into coronary arteries can prevent this postischemic denervation (Abe, et al., 1997). Denervation is heterogeneous however; indeed seven days after ischemia-reperfusion, sympathetic hyperinnervation foci along with denervated myocardium are also present in the peri-infarct region (Li, et al., 2004). In ischemia-reperfusion models therefore, in parallel with denervation, hyperinnervation may also occur. We suspect that local synthesis of NGF, possibly with a similar mechanism to that in our study, is responsible for these regions of hyperinnervation following ischemia-reperfusion.
Hyperinnervation and its relationship to cardiac wound healing
Cellular changes following myocardial infarction are largely similar to those occurring in cutaneous wound healing. In both cases, initial injury is followed by early peri-wound increases in inflammatory macrophages, proliferation of myofibroblasts, and to some extent, dedifferentiation of resident skeletal and cardiac muscle (Hasan, et al., 2000). As the wound heals, numbers of inflammatory cells diminish in concert with formation of a stable scar (Hasan, et al., 2000). In addition, both cutaneous wounds and myocardial infarction display peri-wound hyperinnervation (Reynolds and Fitzgerald, 1995; Liu, et al., 1999), suggesting that this is a common feature associated with inflammatory cell proliferation during wound healing. However, cutaneous wounds exhibit hyperinnervation by sensory nociceptor CGRP-ir axons whereas sympathetic nerves avoid the wound (Reynolds and Fitzgerald, 1995; Liu, et al., 1999). In contrast, cardiac peri-infarct tissue is hyperinnervated by sympathetic nerves but sensory and parasympathetic axons are scarce. The reasons that selective hyperinnervation occurs differentially in different types of wounds is unclear. However, in cultured peri-infarct tissue, NGF appears to be essential for enhanced sympathetic sprouting, whereas similar explant studies showed that sensory axon sprouting induced by wounded skin from neonates occurs largely independently of NGF (Reynolds, et al., 1997). Thus, the nature of wound hyperinnervation apparently varies with tissue type and as a function of which neurotrophic factors are produced. In the case of cutaneous wounds, sensory hyperinnervation plays an essential role in promoting optimal wound healing by regulating dynamics of mitosis and apoptosis (Smith and Liu, 2002). Whether sympathetic nerves play comparable roles in cardiac scar formation remains to be addressed.
Summary
These findings support the idea that post-infarction inflammatory processes associated with myocardial repair result in substantial local increases in NGF synthesis and release by inflammatory cells, including macrophages and myofibroblasts, within the peri-infarct tissue. Increased NGF expression and release, in turn, leads to and is required for sympathetic nerve sprouting in the region of the infarction. The higher density of these excitatory nerves is presumed to contribute to arrhythmias which are a common cause of sudden death in the first month following myocardial infarction. Accordingly, modification of the post-infarct cardiac inflammatory process could influence sympathetic hyperinnervation, and may provide new strategies for reducing post-infarction sudden cardiac death.
ACKNOWLEDGEMENTS
Supported by NIH HL079652 with core support by RR016475 and HD02528. We thank Dr. Deborah Scheuer for help with coronary artery ligations, Dr. Dora Krizsan-Agbas and Ms. Alison Ting for help with surgeries and for critical reading of the manuscript, and Dr. Donald Warn of the MRRC Integrative Imaging Core for assistance with imaging.
REFERENCES
- Abe T, Morgan DA, Gutterman DD. Protective role of nerve growth factor against postischemic dysfunction of sympathetic coronary innervation. Circulation. 1997;95:213–20. doi: 10.1161/01.cir.95.1.213. [DOI] [PubMed] [Google Scholar]
- Ahonen A, Harkonen M, Juntunen J, Kormano M, Penttila A. Effects of myocardial infarction on adrenergic nerves of the rat heart muscle, a histochemical study. Acta Physiol. Scand. 1975;93:336–344. doi: 10.1111/j.1748-1716.1975.tb05822.x. [DOI] [PubMed] [Google Scholar]
- Aloe L. Nerve growth factor, human skin ulcers and vascularization. Our experience. Prog. Brain. Res. 2004;146:515–22. doi: 10.1016/S0079-6123(03)46032-9. [DOI] [PubMed] [Google Scholar]
- Ashcroft GS, Ashworth JJ. Potential role of estrogens in wound healing. Am. J. Clin. Dermatol. 2003;4:737–743. doi: 10.2165/00128071-200304110-00002. [DOI] [PubMed] [Google Scholar]
- Barber MJ, Mueller TM, Henry DP, Felten SY, Zipes DP. Transmural myocardial infarction in the dog produces sympathectomy in noninfarcted myocardium. Circulation. 1983;67:787–796. doi: 10.1161/01.cir.67.4.787. [DOI] [PubMed] [Google Scholar]
- Bilgen M. Simple, low-cost multipurpose RF coil for MR microscopy at 9.4 T. Magn. Reson. Med. 2004;52:937–940. doi: 10.1002/mrm.20228. [DOI] [PubMed] [Google Scholar]
- Cao JM, Fishbein MC, Han JB, Lai WW, Lai AC, Wu TJ, Czer L, Wolf PL, Denton TA, Shintaku IP, Chen PS, Chen LS. Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation. 2000a;101:1960–1969. doi: 10.1161/01.cir.101.16.1960. [DOI] [PubMed] [Google Scholar]
- Cao JM, Chen LS, KenKnight BH, Ohara T, Lee MH, Tsai J, Lai WW, Karagueuzian HS, Wolf PL, Fishbein MC, Chen PS. Nerve sprouting and sudden cardiac death. Circ. Res. 2000b;86:816–821. doi: 10.1161/01.res.86.7.816. [DOI] [PubMed] [Google Scholar]
- Chaldakov GN, Fiore M, Stankulov IS, Manni L, Hristova MG, Antonelli A, Ghenev PI, Aloe L. Neurotrophin presence in human coronary atherosclerosis and metabolic syndrome: a role for NGF and BDNF in cardiovascular disease? Prog. Brain. Res. 2004;146:279–89. doi: 10.1016/S0079-6123(03)46018-4. [DOI] [PubMed] [Google Scholar]
- Desmouliére A, Gabbiani G, Clark RAF. The molecular and cellular biology of wound repair. Plenum Press; New York: 1996. The role of the myofibroblast in wound healing; pp. 391–423. [Google Scholar]
- Drapeau J, El-Helou V, Clement R, Bel-Hadj S, Gosselin H, Trudeau LE, Villeneuve L, Calderone A. Nestin-expressing neural stem cells identified in the scar following myocardial infarction. J. Cell Physiol. 2005;204:51–62. doi: 10.1002/jcp.20264. [DOI] [PubMed] [Google Scholar]
- Du XJ, Cox HS, Dart AM, Esler MD. Sympathetic activation triggers ventricular arrhythmias in rat heart with chronic infarction and failure. Cardiovasc. Res. 1999;43:919–929. doi: 10.1016/s0008-6363(99)00139-x. [DOI] [PubMed] [Google Scholar]
- Gillardon F, Morano I, Ganten U, Zimmermann M. Regulation of calcitonin gene-related peptide mRNA expression in the hearts of spontaneously hypertensive rats by testosterone. Neurosci. Lett. 1991;125:77–80. doi: 10.1016/0304-3940(91)90135-g. [DOI] [PubMed] [Google Scholar]
- Gilliver SC, Wu F, Ashcroft GS. Regulatory roles of androgens in cutaneous wound healing. Thromb. Haemost. 2003;90:978–985. doi: 10.1160/TH03-05-0302. [DOI] [PubMed] [Google Scholar]
- Glebova NO, Ginty DD. Heterogeneous requirement of NGF for sympathetic target innervation in vivo. J. Neurosci. 2004;24:743–751. doi: 10.1523/JNEUROSCI.4523-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace SL, Fry R, Cheung A, Stewart DE. Cardiovascular Disease. BMC Womens Health. 2004;25:S15. doi: 10.1186/1472-6874-4-S1-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan W, Pedchenko T, Krizsan-Agbas D, Baum L, Smith PG. Sympathetic neurons synthesize and secrete pro-nerve growth factor protein. J. Neurobiol. 2003;57:38–53. doi: 10.1002/neu.10250. [DOI] [PubMed] [Google Scholar]
- Hasan W, Zhang R, Liu M, Warn JD, Smith PG. Coordinate expression of Nerve Growth Factor and α-smooth muscle actin mRNA in cutaneous wound tissue of developing and adult rats. Cell Tissue Res. 2000;300:97–109. doi: 10.1007/s004410000175. [DOI] [PubMed] [Google Scholar]
- Hiltunen JO, Laurikainen A, Vakeva A, Meri S, Saarma M. Nerve growth factor and brain-derived neurotrophic factor mRNAs are regulated in distinct cell populations of rat heart after ischaemia and reperfusion. J. Pathol. 2001;194:247–253. doi: 10.1002/path.878. [DOI] [PubMed] [Google Scholar]
- Holmgren S, Abrahamsson T, Almgren O, Eriksson BM. Effect of ischaemic on the adrenergic neurons of the rat heart: a fluorescence histochemical and biochemical study. Cardiovasc. Res. 1981;15:680–689. doi: 10.1093/cvr/15.12.680. [DOI] [PubMed] [Google Scholar]
- Huang MH, Friend DS, Sunday ME, Singh K, Haley K, Austen KF, Kelly RA, Smith TW. An intrinsic adrenergic system in mammalian heart. J. Clin. Invest. 1996;98:1298–1303. doi: 10.1172/JCI118916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igawa A, Nozawa T, Yoshida N, Fujii N, Inoue M, Tazawa S, Asanoi H, Inoue H. Heterogeneous cardiac sympathetic innervation in heart failure after myocardial infarction of rats. Am. J. Physiol. Heart Circ. Physiol. 2000;278:H1134–1141. doi: 10.1152/ajpheart.2000.278.4.H1134. [DOI] [PubMed] [Google Scholar]
- Kawamoto K, Matsuda H. Nerve growth factor and wound healing. Prog. Brain Res. 2004;146:369–84. doi: 10.1016/S0079-6123(03)46023-8. [DOI] [PubMed] [Google Scholar]
- Kaye DM, Vaddadi G, Gruskin SL, Du XJ, Esler MD. Reduced myocardial nerve growth factor expression in human and experimental heart failure. Circ. Res. 2000;86:E80–84. doi: 10.1161/01.res.86.7.e80. [DOI] [PubMed] [Google Scholar]
- Korsching S, Thoenen H. Nerve growth factor supply for sensory neurons: site of origin and competition with the sympathetic nervous system. Neurosci. Lett. 1985;54:201–205. doi: 10.1016/s0304-3940(85)80079-3. [DOI] [PubMed] [Google Scholar]
- Krizsan-Agbas D, Pedchenko T, Hasan W, Smith PG. Oestrogen regulates sympathetic neurite outgrowth by modulating brain derived neurotrophic factor synthesis and release by the rodent uterus. Eur. J. Neurosci. 2003;18:2760–2768. doi: 10.1111/j.1460-9568.2003.03029.x. [DOI] [PubMed] [Google Scholar]
- Lai AC, Wallner K, Cao JM, Chen LS, Karagueuzian HS, Fishbein MC, Chen PS, Sharifi BG. Colocalization of tenascin and sympathetic nerves in a canine model of nerve sprouting and sudden cardiac death. J. Cardiovasc. Electrophysiol. 2000;11:1345–1351. doi: 10.1046/j.1540-8167.2000.01345.x. [DOI] [PubMed] [Google Scholar]
- Lal A, Veinot JP, Ganten D, Leenen FH. Prevention of cardiac remodeling after myocardial infarction in transgenic rats deficient in brain angiotensinogen. J. Mol. Cell Cardiol. 2005;39:521–9. doi: 10.1016/j.yjmcc.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948. doi: 10.1126/science.1065057. [DOI] [PubMed] [Google Scholar]
- Li W, Knowlton D, Van Winkle DM, Habecker BA. Infarction alters both the distribution and noradrenergic properties of cardiac sympathetic neurons. Am. J. Physiol. Heart Circ. Physiol. 2004;286:H2229–2236. doi: 10.1152/ajpheart.00768.2003. [DOI] [PubMed] [Google Scholar]
- Liu M, Warn JD, Fan Q, Smith PG. Relationships between nerves and myofibroblasts during cutaneous wound healing in the developing rat. Cell Tissue Res. 1999;297:423–433. doi: 10.1007/s004410051369. [DOI] [PubMed] [Google Scholar]
- Manni L, Nikolova V, Vyagova D, Chaldakov GN, Aloe L. Reduced plasma levels of NGF and BDNF in patients with acute coronary syndromes. Int. J. Cardiol. 2005;102:169–71. doi: 10.1016/j.ijcard.2004.10.041. [DOI] [PubMed] [Google Scholar]
- Matsuda H, Koyama H, Sato H, Sawada J, Itakura A, Tanaka A, Matsumoto M, Konno K, Ushio H, Matsuda K. Role of nerve growth factor in cutaneous wound healing: accelerating effects in normal and healing-impaired diabetic mice. J. Exp. Med. 1998;187:297–306. doi: 10.1084/jem.187.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paessens R, Borchard F. Morphology of cardiac nerves in experimental infarction of rat hearts. I. Fluorescence microscopical findings. Virchows Arch. A, Pathol. Anat. Histol. 1980;386:265–278. doi: 10.1007/BF00427297. [DOI] [PubMed] [Google Scholar]
- Qin F, Vulapalli RS, Stevens SY, Liang CS. Loss of cardiac sympathetic neurotransmitters in heart failure and NE infusion is associated with reduced NGF. Am. J. Physiol. Heart Circ. Physiol. 2002;282:H363–371. doi: 10.1152/ajpheart.00319.2001. [DOI] [PubMed] [Google Scholar]
- Reynolds M, Alvares D, Middleton J, Fitzgerald M. Neonatally wounded skin induces NGF-independent sensory neurite outgrowth in vitro. Brain Res. Dev. Brain Res. 1997;102:275–283. doi: 10.1016/s0165-3806(97)00105-3. [DOI] [PubMed] [Google Scholar]
- Reynolds ML, Fitzgerald M. Long-term sensory hyperinnervation following neonatal skin wounds. J. Comp. Neurol. 1995;358:487–498. doi: 10.1002/cne.903580403. [DOI] [PubMed] [Google Scholar]
- Saleh TM, Connell BJ. 17beta-estradiol modulates baroreflex sensitivity and autonomic tone of female rats. J. Auton. Nerv. Syst. 2000;80:148–161. doi: 10.1016/s0165-1838(00)00087-4. [DOI] [PubMed] [Google Scholar]
- Scheuer DA, Mifflin SW. Repeated intermittent stress exacerbates myocardial ischemia-reperfusion injury. Am. J. Physiol. 1998;274:R470–475. doi: 10.1152/ajpregu.1998.274.2.R470. [DOI] [PubMed] [Google Scholar]
- Schomig A. Catecholamines in myocardial ischemia. Systemic and cardiac release. Circulation. 1990;82:II13–22. [PubMed] [Google Scholar]
- Smith PG, Liu M. Impaired cutaneous wound healing after sensory denervation in developing rats: effects on cell proliferation and apoptosis. Cell Tissue Res. 2002;307:281–291. doi: 10.1007/s00441-001-0477-8. [DOI] [PubMed] [Google Scholar]
- Solomon SD, Zelenkofske S, McMurray JJ, Finn PV, Velazquez E, Ertl G, Harsanyi A, Rouleau JL, Maggioni A, Kober L, White H, Van de Werf F, Pieper K, Califf RM, Pfeffer MA. Sudden death in patients with myocardial infarction and left ventricular dysfunction, heart failure, or both. N. Engl. J. Med. 2005;352:2581–2588. doi: 10.1056/NEJMoa043938. [DOI] [PubMed] [Google Scholar]
- Vracko R, Thorning D. Contractile cells in rat myocardial scar tissue. Lab. Invest. 1991;65:214–227. [PubMed] [Google Scholar]
- Vracko R, Thorning D, Frederickson RG. Fate of nerve fibers in necrotic, healing, and healed rat myocardium. Lab. Invest. 1990;63:490–501. [PubMed] [Google Scholar]
- Zhou S, Chen LS, Miyauchi Y, Miyauchi M, Kar S, Kangavari S, Fishbein MC, Sharifi B, Chen PS. Mechanisms of cardiac nerve sprouting after myocardial infarction in dogs. Circ. Res. 2004;95:76–83. doi: 10.1161/01.RES.0000133678.22968.e3. [DOI] [PubMed] [Google Scholar]
- Zoubina EV, Mize AL, Alper RH, Smith PG. Acute and chronic estrogen supplementation decreases uterine sympathetic innervation in ovariectomized adult virgin rats. Histol. Histopathol. 2001;16:989–96. doi: 10.14670/HH-16.989. [DOI] [PubMed] [Google Scholar]