

Abstract
Cancer therapeutics are primarily thought to work by inducing apoptosis in tumor cells. However, various tumor suppressors and oncogenes have been shown to regulate senescence in normal cells, and senescence bypass appears to be an important step in the development of cancer. Cellular senescence limits the replicative capacity of cells, thus preventing the proliferation of cells that are at different stages of malignancy. A recent body of evidence suggests that induction of senescence can be exploited as a basis for cancer therapy.
Introduction
In simplistic terms, cancer is a disease of uncontrolled cell proliferation occurring at the wrong place at the wrong time, caused by oncogenic signals. To counter abnormal cell proliferation, a cell can either enter a quiescence-like growth arrest phase, undergo apoptosis, or senesce. These antiproliferative programs are induced by various tumor suppressors in response to potential oncogenic signals. In particular, p53 and pRB tumor suppressors are important mediators of quiescence, apoptosis, and senescence. The senescence phenomenon was first described by Hayflick and Moorhead in human fibroblasts (Hayflick and Moorhead, 1961). Senescent cells in culture are identified by large cell size, flat vacuolated morphology, inability to synthesize DNA, and the presence of the senescence-associated β-galactosidase (SA-β-gal) marker, which is detected by a colorimetric assay using X-gal as a substrate at pH 6.0 (Dimri et al., 1995). Using the SA-β-gal marker, and other senescence and aging biomarkers, several recent studies have demonstrated a role for cellular senescence in aging and cancer (reviewed in Itahana et al., 2004; Campisi, 2005; Lombard et al., 2005). While the exact role of senescence in aging is debatable, its role as a tumor suppressor mechanism is more widely accepted (reviewed in Smith and Pereira-Smith, 1996; Itahana et al., 2004).
The first indication of senescence being a tumor suppressor mechanism was obtained by somatic cell hybridization studies (Smith and Pereira-Smith, 1996). It was shown that a hybrid cell generated by fusion of a tumor (immortal) and a normal (mortal) cell always undergoes senescence in culture. Thus, the senescent phenotype is dominant over immortality, which is a recessive trait. Early studies also identified p53 and pRB as two principal regulators of senescence (Shay et al., 1991; reviewed in Itahana et al., 2004), further supporting the hypothesis of cellular senescence being a tumor suppressor mechanism (discussed below in detail). What are the senescence-initiating signals, and how are these signals transduced to induce a senescent phenotype? As described below, several studies implicate telomeric and nontelomeric signals in the induction of cellular senescence.
Senescence-initiating signals: Telomere is only the tip of the iceberg
The tips of a chromosome consist of telomere repeats, which are capped by telomere binding proteins (reviewed in Smogorzewska and de Lange, 2004). In human cells, progressive telomere shortening appears to be the primary cause of cellular senescence (Harley et al., 1990; reviewed in Kim et al., 2002). In most cases, the enzyme telomerase maintains telomere length. The catalytic subunit of telomerase (TERT), together with its RNA component (TERC), builds telomere repeats at the chromosome ends, which otherwise, owing to asymmetric DNA replication, are progressively lost (reviewed in Smogorzewska and de Lange, 2004; Kim et al., 2002). Most human somatic cells do not contain sufficient telomerase to maintain telomere length (Masutomi et al., 2003), resulting in telomere shortening after each round of cell division. Exogenous expression of telomerase can either increase or stabilize telomere length in normal human cells, and in some cases results in cell immortalization (Bodnar et al., 1998).
It is thought that telomere shortening beyond a certain limit or uncapping of telomere ends triggers a DNA damage response, thereby activating a checkpoint mediated by the p53 pathway, resulting in proliferation arrest. Signals emitted by the telomere dysfunction appear to be similar to double-strand DNA break (DSB)-induced signals (d’Adda di Fagagna et al., 2003; Takai et al., 2003; Gire et al., 2004). Senescent cells are enriched in the nuclear foci of phosphorylated histone H2AX (γ-H2AX), p53 binding protein 53BP1, NBS1, the phosphoS966 form of SMC1, and MDC1 (d’Adda di Fagagna et al., 2003). Moreover, inactivation of CHK1 and CHK2 checkpoint kinases can restore S phase progression in senescent cells (d’Adda di Fagagna et al., 2003; Gire et al., 2004). A more recent study using single cell parameters also suggested that telomere shortening-triggered senescence is a DNA damage response mediated by the ATM/ATR-p53-p21 pathway (Herbig et al., 2004). If cellular senescence induced by telomere dysfunction in human cells is a DNA damage response, then the DNA damaging agents and factors that mimic DNA damage should be able to induce cellular senescence. Indeed, DNA damaging agents and other cellular stresses can trigger a senescence-like phenotype (reviewed in Ben-Porath and Weinberg, 2004, Itahana et al., 2004), characterized by a large, flat morphology and the presence of the SA-β-gal marker (Figure 1).
Figure 1.
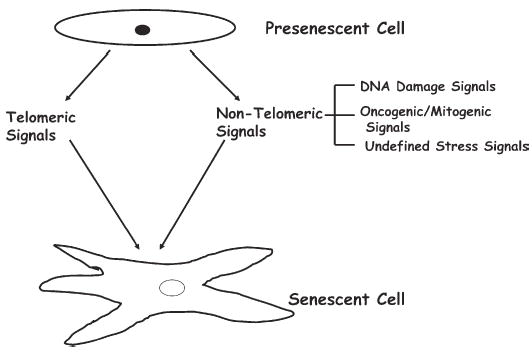
Presenescent cells undergo senescence in response to telomeric and nontelomeric signals
Telomeric signals such as telomere shortening and uncapping of telomere ends, as well as nontelomeric signals such as DNA damaging agents, oncogenic/mitogenic signaling, and undefined stress signals, induce senescence. Undefined stress signals are signals that come from a variety of sources, such as culture media. Senescent cells in culture are often identified by large, flat cell morphology, and stain positively for SA-β-gal marker.
Apart from DNA damage, certain undefined stress-causing signals also induce senescence. For example, senescence in cultured murine cells is thought to be due to stress induced by culture conditions (Sherr and De Pinho, 2000), which can be abrogated by decreasing oxygen concentration used in culturing these cells (Parrinello et al., 2003). In addition, oncogenic and mitogenic signals, such as activated H-RAS, can also induce senescence in primary cells (Figure 1). Thus, senescence-inducing signals can be telomeric or nontelomeric (Figure 1). Senescence induced by nontelomeric signals is termed accelerated senescence, premature senescence, stress- or aberrant signaling-induced senescence (STASIS), or extrinsic senescence (Itahana et al., 2003, 2004). Senescence induced by nontelomeric signals may have evolved to protect organisms from acute signals that may cause cancer or other diseases resulting from faulty DNA replication.
Tumor suppressor pathways and senescence: Meant for each other
Regardless of the senescence-initiating signals (telomeric or nontelomeric), tumor suppressor pathways are critical for genesis and maintenance of the senescent phenotype in human and mouse cells. However, tumor suppressor pathways that pertain to senescence differ in human and mouse cells (Figure 2). In human cells, telomeric signals principally engage the p53-p21‐pRB pathway (Figure 2A), while nontelomeric signals engage both the p53-p21-pRB and p16-pRB pathways (Figure 2A). In mouse cells, the ARF-p53-p21-pRB pathway is the dominant pathway of senescence (Figure 2B). However, pRB function in mouse cells can be substituted by pRB-related proteins p107 and p130 (reviewed in Itahana et al., 2004). It has also been suggested that p21 may not be the sole conduit of p53 during senescence induction in mouse cells (Pantoja and Serrano, 1999). Thus, other p53 targets may also participate in induction of senescence in mouse cells (Figure 2B). It is likely that these other p53 targets also contribute to senescence in human cells. Various other tumor suppressors and oncogenes also impinge upon these pathways of senescence and modulate them accordingly (Figure 2) (described below). Nonetheless, p53 and pRB remain the two main regulators of cellular senescence in human and mouse cells.
Figure 2.
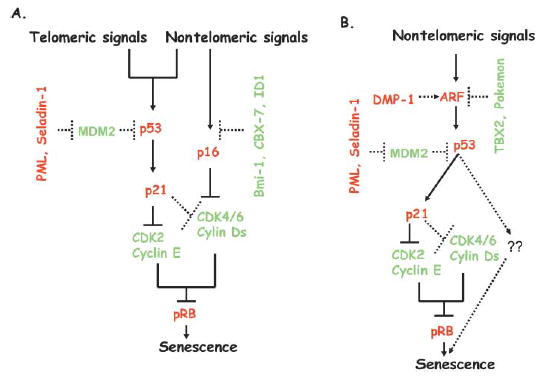
Telomeric and nontelomeric signals induce senescence via tumor suppressor pathways in human and mouse cells
Induction of p21 and p16 by senescence-inducing signals results in inhibition of activity of CDK2 and CDK4/6. Downregulation of activity of these pRB kinases leads to pRB hypophosphorylation, which results in cell cycle arrest during senescence. Regulators of senescence pathways in human and mouse cells include PML, MDM2, ID1, Bmi-1, CBX7, and Seladin-1. In mouse cells, tumor suppressor ARF is negatively regulated by two potential oncogenes, TBX2 and Pokemon, and positively regulated by DMP-1.
A: In human cells, telomeric and nontelomeric signals induce senescence primarily via the p53-p21-pRB pathway. Nontelomeric signals also induce the p16-pRB pathway of senescence in human cells.
B: Mouse cells undergo senescence via the ARF-p53-p21-pRB pathway in response to nontelomeric signals. Mouse cells do not senesce by the telomeric signal-induced pathway of senescence.
Solid lines indicate principal pathways of senescence, while dotted lines indicate auxiliary pathways that can modulate senescence. The red letters indicate tumor suppressors and growth inhibitors, while the green letters indicate oncogenes and growth promoters.
p53, the master regulator of senescence
Depending on the severity of damage to the genome, p53 can activate genetic programs that halt cell proliferation transiently (G1 and G2 cell cycle arrest) or permanently (senescence), or eliminate the cell altogether (apoptosis). Evidence for its role in senescence comes from several studies. First, as described earlier, it has been clearly shown that telomere shortening or dysfunction induces a DNA damage response mediated by p53. Second, abrogation of the p53-p21 pathway by various strategies can bypass senescence in human and mouse cells (Shay et al., 1991; Brown et al., 1997; Dirac and Bernards, 2003; Beausejour et al. 2003). Third, enforced expression of p53 or p21 in certain cell types can induce a senescence-like phenotype (Itahana et al., 2001). Finally, a variety of stimuli induce senescence in a p53/p21-dependent manner (Itahana et al., 2001, 2004).
It is interesting to note that studies using DNA tumor viruses to inactivate p53 suggested that p53 inactivation only extends the replicative life span, and complete abrogation of senescence leading to crisis requires pRB inactivation as well (Shay et al., 1991). However, more recent studies using either somatic cell knockout or an RNAi (RNA interference) approach suggest that p53 and p21 inactivation can lead to complete abrogation of senescence and induction of a crisis-like phenotype (Brown et al., 1997, Dirac and Bernards, 2003, and our unpublished data). How does p53 induction during senescence cause cell cycle arrest? Most studies suggest that p21 induction by p53 inhibits CDK2/Cyclin E activity. Activity of CDK4/Cyclin Ds can also be inhibited by p21. Inhibition of activity of CDKs by p21 results in hypophosphorylation of pRB, which very likely mediates cell cycle arrest during senescence (Figure 2) (Itahana et al., 2004).
The role of p53 regulators in senescence
Tumor suppressor p53 is positively or negatively regulated by a plethora of factors (Figure 2) (reviewed in Vousden, 2002). The E3 ubiquitin ligases MDM2, PIRH2, and COP1 negatively regulate p53 by targeting it for proteosome-mediated degradation (reviewed in Lu, 2005). On the other hand, p53 is positively regulated by ARF (p14ARF in human or p19ARF in mouse), PML, PTEN, NPM, p33ING1, and other potential tumor suppressors, which posttranslationally stabilize p53 (Weber et al., 1999, Leung et al., 2002, Freeman et al., 2003, Bernardi et al., 2004, Kurki et al., 2004). In principle, these positive and negative regulators of p53 can also impact cellular senescence (Figure 2).
Indeed, p33ING1 is known to be overexpressed in senescent cells (Garkavtsev and Riabowol, 1997), and its overexpression can induce senescence in proliferating cells (Goeman et al., 2005). Similarly, nucleophosmin (NPM) overexpression was shown to induce senescence in a p53-dependent manner (Colombo et al., 2002). On the other hand, NPM also interacts with ARF tumor suppressor and inhibits its function (reviewed in Zhang, 2004). Interestingly, NPM is mutated in a large number of cases of acute myelogenous leukemia (AML) in humans (Falini et al., 2005). It is tempting to speculate that NPM mutations in these cases may be related to its possible role in senescence.
PML and p53 pathway of senescence
PML binds MDM2 and sequesters it into the nucleolus (Bernardi et al., 2004), thus protecting p53 from proteosome-mediated degradation. As a result, overexpression of PML, and accumulation of PML and MDM2 in the nucleolus after DNA damage, results in p53 stabilization (Bernardi et al., 2004). PML is upregulated during cellular senescence (Ferbeyre et al., 2000), and induces premature senescence in response to oncogenic H-RAS by promoting p53 acetylation (Pearson et al., 2000). Moreover, human SIR2, which deacetylates p53, inhibits PML- and p53‐induced premature senescence, further confirming the role of PML as a senescence-regulatory protein (Langley et al., 2002).
More detailed studies have recently suggested that PML isoform IV, when overexpressed, induces senescence in human fibroblasts in a pRB-dependent manner (Mallette et al., 2004; Bischof et al., 2005), and that the cytoplasmic isoform of PML induces cellular senescence in response to TGF-β (Lin et al., 2004). Collectively, these studies suggest that the PML tumor suppressor contributes to cellular senescence in human and mouse cells.
ARF, an upstream regulator of p53 pathway and senescence
The INK4a/ARF locus encodes p16INK4a and ARF, which regulate pRB and p53 pathways of senescence and tumor suppression, respectively (reviewed in Lowe and Sherr, 2003; Sharpless, 2005). As indicated before, the ARF-p53 pathway is the major pathway of senescence in mouse cells. ARF is overexpressed in cultured senescent mouse embryo fibroblasts (MEFs) and upregulated during premature senescence induced by onocogenic signals such as activated H-RAS (Kamijo et al., 1999; reviewed in Lowe and Sherr, 2003; Sharpless, 2005) (described below in detail). Although ARF overexpression can also promote senescence in human cells (Dimri et al., 2000; Wei et al., 2001), it may not be a major regulator of senescence in human cells (Wei et al., 2001; reviewed in Sharpless, 2005).
ARF is thought to antagonize MDM2-mediated ubiquitination of p53 through translocation of MDM2 to the nucleolus, thereby stabilizing p53 (reviewed in Sherr and Weber, 2000). Although it can also inhibit cell proliferation by p53-independent pathways (reviewed in Cleveland and Sherr, 2004), ARF probably promotes senescence by regulating the p53 pathway (Dimri et al., 2000; Wei et al., 2001). The specific transcriptional repressors of ARF include TBX2, which was identified in a senescence bypass screen (Jacobs et al., 2000), and the protooncogene Pokemon, which can bind to the ARF promoter and repress its transcription (Maeda et al., 2005). MEFs lacking Pokemon (Zbtb7) exhibit constitutive upregulation of p19ARF and undergo premature senescence (Maeda et al., 2005). Importantly, TBX2 and Pokemon are aberrantly overexpressed in a subset of breast cancers and lymphomas, respectively (Jacobs et al., 2000; Maeda et al., 2005), suggesting a possible role for these ARF regulators in senescence in human cells as well.
pRB, the second regulator of cellular senescence
In contrast to p53, the role of pRB in cellular senescence is less clear (Itahana et al., 2004). Earlier studies using DNA tumor viruses that bind and inactivate pRB clearly indicate that pRB cooperates with p53 during cellular senescence (Shay et al., 1991). More recent studies suggest that perhaps pRB is, in some instances, as important as p53 in inducing cellular senescence. Using the Cre-Lox system to delete pRB in senescent MEFs, Sage et al. showed that the loss of pRB is sufficient for cell cycle entry and the reversal of cellular senescence (Sage et al., 2003). In human fibroblasts, loss of pRB by targeted disruption of one copy followed by the spontaneous loss of its other allele results in bypass of replicative senescence and a crisislike phenotype similar to that induced by the abrogation of the p53-p21 pathway (Wei et al., 2003). Inactivation of an exogenously introduced temperature-sensitive pRB in senescent SAOS-2 cells also results in S phase reentry (Alexander et al., 2003). Collectively, these data suggest that pRB maintains cell cycle arrest during senescence in human and mouse cells.
It is very well established that pRB remains constitutively hypophosphorylated in senescent cells (Stein et al., 1999), suggesting downregulation of the activity of pRB kinases during senescence. The p16INK4a, an inhibitor of CDK4 and CDK6 activity, is upregulated during senescence in human fibroblasts (Alcorta et al., 1996; Stein et al., 1999) and M0 senescence in human mammary epithelial cells (Wong et al., 1999). Thus, high p16 very likely accounts for the hypophosphorylation of pRB in senescent cells. Consistent with this hypothesis, p16INK4a and its upstream regulators such as Bmi-1, CBX7, ID1, and Ets-1 regulate senescence (Jacobs et al., 1999; Ohtani et al., 2001; Itahana et al., 2003; Gil et al., 2004) in a pRB-dependent manner (Figure 2).
Our recent data suggested that p16 upregulation during senescence is not nearly as universal as previously thought (Itahana et al., 2003). In certain fibroblast strains such as the commonly used WI-38 strain, p16 is clearly upregulated, while in other strains such as BJ fibroblasts, it is only minimally expressed under normal culture conditions (Itahana et al., 2003). The fibroblasts that contain high or low p16 differ in their propensity to reverse senescence by the inactivation of p53 pathway (Beausejour et al., 2003). Senescent WI-38 fibroblasts, which contain high p16, are essentially resistant to inactivation of p53 pathway, and cannot be induced to reenter cell cycle by inactivation of p53. In other fibroblasts, which contain undetectable p16, senescence can by reversed by p53 inactivation (Beausejour et al., 2003). Fibroblast strains that accumulate p16 differentially during senescence also differ in the presence or absence of senescence-associated heterochromatic foci (SAHF) (Narita et al., 2003). SAHF are heterochromatic DNA regions, where pRB was found to be colocalized (Narita et al., 2003). SAHF are present only in human fibroblasts that contain high p16 during senescence, suggesting that p16 plays a role in generating SAHF. Interestingly, senescence in SAHF-containing fibroblasts is irreversible (Beausejour et al., 2003). Thus, SAHF possibly contribute to the stable repression of proliferation‐associated genes mediated by E2F-pRB complexes to enforce the senescent phenotype.
Recently, using a p16 knockdown strategy, it was shown that p16 downregulation may not be functionally equivalent to pRB inactivation (Wei et al., 2003), leaving the possibility that other CDK inhibitors might play a surrogate role in cellular senescence. In support of this hypothesis, overexpression of various CDK inhibitors is known to induce a senescent phenotype (McConnell et al., 1998). Since p21 induction can also lead to the inhibition of pRB phosphorylation by inhibiting CDK2/Cyclin E activity (Figure 2), p16 and p21 are likely to cooperate to keep pRB in a hypophosphorylated form during senescence. Alternatively, it is possible that p21 initially and temporarily keeps pRB in its inhibitory form, while p16 ensures permanent hypophosphorylation of pRB, which practically makes senescence irreversible (Beausejour et al., 2003).
Oncogenic and mitogenic signals induce senescence: A failsafe mechanism
As senescence is regulated by various tumor suppressors, it could function as a natural barrier to tumorigenesis. This hypothesis could be directly tested by subjecting a normal cell to potential oncogenic or mitogenic stimuli. Indeed, activated H-RAS (V12) was found to induce premature senescence in primary rodent and human cells (Serrano et al., 1997). Depending on the cellular context, induction of senescence by oncogenic signals such as activated H-RAS depends on either or both p53 and p16INK4a tumor suppressor proteins (Palmero et al., 1998; Lin and Lowe, 2001).
Since RAS signaling involves the RAF-MEK-ERK pathway, it is conceivable that other components of this pathway can also induce premature senescence. Indeed, oncogenic RAF and constitutive expression of mitogen-activated protein (MAP) kinase mimic RAS-induced premature senescence in IMR90 fibroblasts (Zhu et al., 1998; Lin et al., 1998). Furthermore, during oncogenic RAS-induced premature senescence, the RAF-MEK-ERK pathway activates p38 MAPK, and inhibition of p38 activity results in a failure to induce premature senescence by activated RAS (Wang et al., 2002). Constitutively active MKK3 and MKK6, which activate p38 MAPK by phosphorylation, can also induce premature senescence by upregulating p53 and p16INK4a in human fibroblasts (Wang et al., 2002).
Apart from p53, PML, ARF, and p16INK4a, other proteins are also likely to mediate the H-RAS response. For example, recently, a genetic suppressor element (GSE) screen identified Seladin-1 as a target gene that is involved in H-RAS‐induced premature senescence (Wu et al., 2004). Interestingly, Seladin-1 encodes an oxidoreductase enzyme involved in cholesterol metabolism (Wu et al., 2004). Further studies on Seladin-1 demonstrated that it is an effector of RAS-induced reactive oxygen species (ROS) signaling (Wu et al., 2004).
A careful analysis of oncogenic, mitogenic, and other hyperproliferative signals is likely to reveal more cases of premature senescence induction by such signals in primary cells, which in all likelihood represents a failsafe mechanism. For example, similar to activated H-RAS expression, E2F1 overexpression, a potent mitogenic signal, leads to premature senescence in normal human fibroblasts (Dimri et al., 2000). Premature senescence induction by E2F1 depends on the p53 status of the cells and is mediated by transcriptional induction of p14ARF by E2F1 (Dimri et al., 2000). Constitutive overexpression of E2F3 also results in induction of senescence in a transgenic mouse model and cultured MEFs (Denchi et al., 2005). In this study, it was found that a sustained E2F activity, which provides a hyperproliferative signal, induced senescence-like features in mouse pituitary gland (Denchi et al., 2005). This report is significant because it indicates that senescence induction by hyperproliferative signals is not merely an in vitro phenomenon.
Recently, it was shown that the overexpression of oncogenic ERBB2 also upregulates p21 and induces premature senescence in MCF-7 cells (Trost et al., 2005). The induction of ERBB2 in this setting causes p53-independent p21 upregulation and premature senescence, which can be reversed by the inhibition of p38 MAPK or functional inactivation of p21 by antisense oligonucleotides (Trost et al., 2005).
Beyond senescence: The road to the cancer highway
Primary cells induce senescence in response to potential oncogenic signals. What happens when the induction of senescence fails to occur due to malfunction of tumor suppressors? Cancer is a multistep process; hence, abrogation of senescence alone does not lead to tumor formation. Nevertheless, the road starts from here. Recent elegant studies from the laboratories of Weinberg and Hahn clearly demonstrate that the first step in creating an in vitro model of human cancer involves the abrogation of cellular senescence (Hahn et al., 1999; reviewed in Boehm and Hahn, 2005). These studies show that a combination of SV40 large T, small t, hTERT, and H-RAS is able to transform a variety of normal human cell types such as fibroblasts, embryonic kidney cells, mammary epithelial cells, ovarian epithelial cells, and endothelial cells (Boehm and Hahn, 2005).
Abrogation of senescence can be achieved by SV40 large T, a combination of HPV oncoproteins E6 and E7, E1A and MDM2 coexpression, or the use of short inhibitory (sh) RNA against pRB and p53 ((Hahn et al., 1999; Seger et al., 2002; Voorhoeve and Agami, 2003; Boehm and Hahn, 2005). Since the INK4a/ARF locus is an upstream regulator of both pRB and p53 (reviewed in Lowe and Sherr, 2003; Sharpless, 2005), various mutations in this locus presumably can also substitute SV40 large T function in transformation assays, albeit at a lower efficiency depending on the nature of the mutation. For example, a combination of hTERT and H-RAS or c-MYC is sufficient to transform Leiden HDFs (human diploid fibroblasts), which bear an INK4a/ARF mutation resulting in p16INK4a deficiency (Drayton et al., 2003). Similarly, a combined knockdown of p16INK4a and p53 by the RNAi approach, together with SV40 small t, hTERT, and H-RAS, causes transformation of normal human fibroblasts (Voorhoeve and Agami, 2003).
Compared to human cells, murine cells are clearly less rigid in terms of requirements for different genetic mutations for transformation (reviewed in Rangarajan and Weinberg, 2003). Nevertheless, bypass of senescence is also essential for murine cell transformation. Established immortal MEF cell lines have often lost p53 or p19ARF. MEFs deficient in p19ARF are highly susceptible to oncogenic transformation and resistant to H-RAS-induced senescence (Sharpless et al., 2004). The H-RAS induced senescence can also be abrogated by other potential oncogenes, such as hDRIL and BCL6 (Peeper et al., 2002; Shvarts et al., 2002), which indirectly affects pRB and p53 pathways of senescence. For example, BCL6 overrides p53 pathway by inducing cyclin D1 expression (Shvarts et al., 2002), while hDRIL targets pRB pathway by binding to E2F1 (Peeper et al., 2002). These potential oncogenes strongly cooperate with H-RAS to transform mouse and human cells. Similarly, downregulation of Seladin-1 expression by sh RNA, together with hTERT and H-RAS, not only overcomes RAS-induced senescence, but also transforms human fibroblasts (Wu et al., 2004). Deficiency of transcription factor DMP-1, which works upstream of ARF, also promotes H-RAS-induced transformation in MEFs (Inoue et al., 2000; Sreeramaneni et al., 2005). Thus, malfunction of senescence and tumor suppressor pathways facilitate transformation by oncogenes in human and mouse cells.
Senescence in cancer treatment: Putting a roadblock in the cancer highway
So far, most studies have suggested that the failure of senescence-induction pathways in conjunction with activated oncogenes possibly leads to tumor formation in vivo. However, an important question is, can senescence still be induced in tumors to stop further growth? Indeed, several recent studies have shown that chemotherapeutic drugs and radiation can induce senescence in tumor cells (reviewed in Roninson, 2003). Quite often, tumors develop resistance to chemotherapeutic drug-induced apoptosis. Senescence induction in such cases could serve as a backup plan to inhibit the growth of tumor cells. Indeed, recently, it was reported that a combined treatment of cells with pan caspase inhibitor (Q-VD-OPH) and doxorubicin greatly accelerates senescence and leads to the reversal of drug resistance in several tumor cell lines (Zheng et al., 2004).
The induction of senescence in cultured tumor cells by DNA-damaging agents is encouraging, but the most important question is, does senescence response to chemotherapeutic drugs occur in vivo, and if so, does the therapy-induced senescence contribute sufficiently to the therapeutic efficiency? Recent studies provide compelling evidence that cellular senescence can indeed be induced in vivo by chemotherapeutic drugs. In the first study, te Poele et al. stained newly sectioned archival breast tumors from patients who had undergone a chemotherapy regimen for the SA-β-gal marker and p53 and p16INK4a proteins (te Poele et al., 2002). While normal tissues adjacent to the tumors were devoid of SA-β-gal, importantly, 15 out of 36 (41%) tumors stained positive for SA-β-gal marker (te Poele et al., 2002). Authors also showed that the tumor sections from the patients that did not receive chemotherapy stained positively for SA-β-gal only in 10% of the cases, and this staining was in few isolated cells compared to many intense patches of staining present in treated tumor sections. Furthermore, intense SA-β-gal staining was correlated with high p16INK4a, a protein known to be upregulated during senescence. Although authors did not attempt to correlate SA-β-gal staining to survival, it was speculated that senescence induction by chemotherapy results in a stable disease rather than the regression of tumor, a situation often noticed during treatment by cytotoxic drugs in patients (te Poele et al., 2002). The induction of accelerated or premature senescence during chemotherapy treatment of human lung cancer in vivo was also recently demonstrated (Roberson et al., 2005). Although the sample size in this study was very small, it was found that two of the three patients treated with carboplatin and taxol intensely expressed SA-β-gal, while another three samples from untreated patients showed no significant SA-β-gal staining (Roberson et al., 2005).
A study by Schmitt et al. also provides a clear evidence for the role of senescence in cancer chemotherapy in a transgenic mouse model (Schmitt et al., 2002). The authors showed that CTX (cyclophosphamide), a chemotherapy drug, is able to engage a senescence program when apoptosis is inhibited by Bcl2 overexpression during Eμ-Myc-induced lymphoma in wildtype p53-containing transgenic mice. As a result, Bcl2 and wildtype p53-expressing lymphoma did not progress, and the mice had a better prognosis after CTX treatment (Schmitt et al., 2002). Induction of the senescence-like phenotype was also observed in rat mammary tumors undergoing treatment with chemopreventive agents (Christov et al., 2003). Thus, chemotherapeutic drugs can induce a senescence-like stage in vivo and in vitro by upregulating p53 and/or p16INK4a.
More than 90% of tumors contain readily detectable telomerase activity (Kim et al., 1994). Telomerase, and various telomerase-regulatory and telomere binding proteins, are thought to regulate telomere length in a cell (Smogorzewska and de Lange, 2004). In principle, telomerase and telomerase-regulatory proteins can be targeted to cause telomere dysfunction and induce apoptosis or senescence in precancerous and tumor cells (Shay and Wright, 2002). Indeed, several telomerase inhibitors are known to induce senescence or apoptosis in tumor cells (Damm et al., 2001, Seimiya et al., 2002, Riou et al., 2002, Kim et al., 2003; Preto et al., 2004). Recently, inhibition of tankyrase 1, which poly (ADP-ribosyl)ates TRF1, was shown to cooperate with a telomerase inhibitor MST-312 to cause telomere shortening and rapid cell death (Seimiya et al., 2005). Combined treatment of cells with telomerase inhibitors and other regulators of telomerase may be helpful when tumors develop resistance to telomerase inhibitors alone (Seimiya et al., 2005).
Senescence may promote tumorigenesis: An unwanted side effect
An interesting question is what could be the side effect of the induction of senescence by chemotherapeutic drugs, and what could compromise the efficacy of treatment by these drugs? It has been proposed that senescence in some settings may actually promote tumor progression; possibly by secreting certain matrix metalloproteases, growth factors, and cytokines (Krtolica et al., 2001; reviewed in Campisi, 2005). In particular, senescent fibroblasts were shown to facilitate tumorigenesis by immortal premalignant epithelial cells (Krtolica et al., 2001).
It is important to note that in a therapeutic setting, both normal host and tumors cells are being treated with senescence-inducing agents. Although induction of senescence in the tumor cell itself is unlikely to promote any more tumorigenicity, its induction in normal cells by chemotherapeutic drugs may facilitate tumorigenesis by cells that are not yet fully tumorigenic. Based on a recent study by te Poele et al., normal cells adjacent to tumors appear to be less receptive to senescence induction by chemotherapeutic drugs (te Poele et al., 2002). Clearly, more studies are needed to address this question of facilitation of tumorigenesis by senescent cells during chemotherapy of tumors in patients.
Concluding remarks
In this review, I have highlighted the role of senescence in cancer. Senescence is not only a normal physiological response to accrued cell divisions in culture, but is also very likely a response to potential oncogenic events that a cell might encounter. Thus, a senescence response is elicited by DNA damage and oncogenic and mitogenic signals. In this scenario, senescence acts as a failsafe mechanism. Mutations in p53 and/or pRB are a common occurrence in various cancers. INK4a/ARF locus, which is an upstream regulator of p53 and pRB, is also a target of many somatic and genetic mutations. These mutations often cause a bypass of cellular senescence and cooperate with other oncogenes in transformation assays, thus attesting to the importance of senescence in cancer. The take-home message is that a tumor will not come into existence unless it has bypassed senescence. The fascinating part here is that despite the fact that tumors have bypassed the common pathway(s) of senescence, they retain the ability to undergo senescence in response to treatment with a variety of therapeutic agents.
Several in vitro studies suggest that chemotherapeutic drugs can induce a senescence-like phenotype. A limited number of in vivo studies in the human and mouse models provide proof of principle for the concept of induction of accelerated senescence by chemotherapeutic drugs. Clearly, more studies are needed to substantiate these findings, particularly in human cancers. Further caution is also warranted, because senescence induction in normal cells can facilitate tumor progression under certain circumstances. Thus, a critical degree of senescence may be the prerequisite for successful treatment using chemotherapeutic drugs. Although it is still unclear how much therapy-induced senescence can contribute to the therapeutic efficiency, it is likely to result in a stable disease rather than the regression of tumors. Apart from chemotherapeutic drugs, senescence induction can also be achieved by small molecules that directly target senescence-regulatory genes and under- or overexpression of various genes involved in senescence. In summary, senescence has a lot to do with cancer development, and a better understanding of the senescence induction pathways will greatly contribute to the development of effective cancer treatment strategies.
Acknowledgments
I regret that the original work of several of my colleagues could not be cited due to space constraints. I am thankful to Drs. V. Band and J. Campisi for their continued support. My laboratory is funded by the National Cancer Institute grant RO1 CA 094150 and the Department of Defense grant DAMD17-02-1-0509.
References
- Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16INK4a in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci USA. 1996;93:13742–13747. doi: 10.1073/pnas.93.24.13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander K, Yang HS, Hinds PW. pRb inactivation in senescent cells leads to an E2F-dependent apoptosis requiring p73. Mol Cancer Res. 2003;1:716–728. [PubMed] [Google Scholar]
- Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Porath I, Weinberg RA. When cells get stressed: An integrative view of cellular senescence. J Clin Invest. 2004;113:8–13. doi: 10.1172/JCI200420663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi R, Scaglioni PP, Bergmann S, Horn HF, Vousden KH, Pandolfi PP. PML regulates p53 stability by sequestering Mdm2 to the nucleolus. Nat Cell Biol. 2004;6:665–672. doi: 10.1038/ncb1147. [DOI] [PubMed] [Google Scholar]
- Bischof O, Nacerddine K, Dejean A. Human papillomavirus oncoprotein E7 targets the promyelocytic leukemia protein and circumvents cellular senescence via the Rb and p53 tumor suppressor pathways. Mol Cell Biol. 2005;25:1013–1024. doi: 10.1128/MCB.25.3.1013-1024.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Boehm JS, Hahn WC. Understanding transformation: Progress and gaps. Curr Opin Genet Dev. 2005;15:13–17. doi: 10.1016/j.gde.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Brown JP, Wei W, Sedivy JM. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science. 1997;277:831–834. doi: 10.1126/science.277.5327.831. [DOI] [PubMed] [Google Scholar]
- Campisi J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Christov KT, Shilkaitis AL, Kim ES, Steele VE, Lubet RA. Chemopreventive agents induce a senescence-like phenotype in rat mammary tumours. Eur J Cancer. 2003;39:230–239. doi: 10.1016/s0959-8049(02)00497-5. [DOI] [PubMed] [Google Scholar]
- Cleveland JL, Sherr CJ. Antagonism of Myc functions by Arf. Cancer Cell. 2004;6:309–311. doi: 10.1016/j.ccr.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Colombo E, Marine JC, Danovi D, Falini B, Pelicci PG. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat Cell Biol. 2002;4:529–533. doi: 10.1038/ncb814. [DOI] [PubMed] [Google Scholar]
- d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- Damm K, Hemmann U, Garin-Chesa P, Hauel N, Kauffmann I, Priepke H, Niestroj C, Daiber C, Enenkel B, Guilliard B, et al. A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J. 2001;20:6958–6968. doi: 10.1093/emboj/20.24.6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denchi EL, Attwooll C, Pasini D, Helin K. Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Mol Cell Biol. 2005;25:2660–2672. doi: 10.1128/MCB.25.7.2660-2672.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Campisi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Itahana K, Acosta M, Campisi J. Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Mol Cell Biol. 2000;20:273–285. doi: 10.1128/mcb.20.1.273-285.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirac AM, Bernards R. Reversal of senescence in mouse fibroblasts through lentiviral suppression of p53. J Biol Chem. 2003;278:11731–11734. doi: 10.1074/jbc.C300023200. [DOI] [PubMed] [Google Scholar]
- Drayton S, Rowe J, Jones R, Vatcheva R, Cuthbert-Heavens D, Marshall J, Fried M, Peters G. Tumor suppressor p16INK4a determines sensitivity of human cells to transformation by cooperating cellular oncogenes. Cancer Cell. 2003;4:301–310. doi: 10.1016/s1535-6108(03)00242-3. [DOI] [PubMed] [Google Scholar]
- Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, La Starza R, Diverio D, Colombo E, Santucci A, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 2000;14:2015–2027. [PMC free article] [PubMed] [Google Scholar]
- Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, Whale AD, Martinez-Diaz H, Rozengurt N, Cardiff RD, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3:117–130. doi: 10.1016/s1535-6108(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Garkavtsev I, Riabowol K. Extension of the replicative life span of human diploid fibroblasts by inhibition of the p33ING1 candidate tumor suppressor. Mol Cell Biol. 1997;17:2014–2019. doi: 10.1128/mcb.17.4.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil J, Bernard D, Martinez D, Beach D. Polycomb CBX7 has a unifying role in cellular lifespan. Nat Cell Biol. 2004;6:67–72. doi: 10.1038/ncb1077. [DOI] [PubMed] [Google Scholar]
- Gire V, Roux P, Wynford-Thomas D, Brondello JM, Dulic V. DNA damage checkpoint kinase Chk2 triggers replicative senescence. EMBO J. 2004;23:2554–2563. doi: 10.1038/sj.emboj.7600259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeman F, Thormeyer D, Abad M, Serrano M, Schmidt O, Palmero I, Baniahmad A. Growth inhibition by the tumor suppressor p33ING1 in immortalized and primary cells: Involvement of two silencing domains and effect of Ras. Mol Cell Biol. 2005;25:422–431. doi: 10.1128/MCB.25.1.422-431.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumor cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- Hayflick L, Moorhead PS. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a) Mol Cell. 2004;14:501–513. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- Inoue K, Wen R, Rehg JE, Adachi M, Cleveland JL, Roussel MF, Sherr CJ. Disruption of the ARF transcriptional activator DMP1 facilitates cell immortalization, Ras transformation, and tumorigenesis. Genes Dev. 2000;14:1797–1809. [PMC free article] [PubMed] [Google Scholar]
- Itahana K, Dimri G, Campisi J. Regulation of cellular senescence by p53. Eur J Biochem. 2001;268:2784–2791. doi: 10.1046/j.1432-1327.2001.02228.x. [DOI] [PubMed] [Google Scholar]
- Itahana K, Zou Y, Itahana Y, Martinez JL, Beausejour C, Jacobs JJL, van Lohuizen M, Band V, Campisi J, Dimri GP. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol Cell Biol. 2003;23:389–401. doi: 10.1128/MCB.23.1.389-401.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itahana K, Campisi J, Dimri GP. Mechanisms of cellular senescence in human and mouse cells. Biogerontology. 2004;5:1–10. doi: 10.1023/b:bgen.0000017682.96395.10. [DOI] [PubMed] [Google Scholar]
- Kamijo T, van de Kamp E, Chong MJ, Zindy F, Diehl JA, Sherr CJ, McKinnon PJ. Loss of the ARF tumor suppressor reverses premature replicative arrest but not radiation hypersensitivity arising from disabled atm function. Cancer Res. 1999;59:2464–2469. [PubMed] [Google Scholar]
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- Kim SH, Kaminker P, Campisi J. Telomeres, aging and cancer: In search of a happy ending. Oncogene. 2002;21:503–511. doi: 10.1038/sj.onc.1205077. [DOI] [PubMed] [Google Scholar]
- Kim JH, Kim JH, Lee GE, Kim SW, Chung IK. Identification of a quinoxaline derivative that is a potent telomerase inhibitor leading to cellular senescence of human cancer cells. Biochem J. 2003;373:523–529. doi: 10.1042/BJ20030363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc Natl Acad Sci USA. 2001;98:12072–12077. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurki S, Peltonen K, Latonen L, Kiviharju TM, Ojala PM, Meek D, Laiho M. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell. 2004;5:465–475. doi: 10.1016/s1535-6108(04)00110-2. [DOI] [PubMed] [Google Scholar]
- Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- Jacobs JJ, Keblusek P, Robanus-Maandag E, Kristel P, Lingbeek M, Nederlof PM, van Welsem T, van de Vijver MJ, Koh EY, Daley GQ, et al. Senescence bypass screen identifies TBX2, which represses Cdkn2a (p19(ARF)) and is amplified in a subset of human breast cancers. Nat Genet. 2000;26:291–299. doi: 10.1038/81583. [DOI] [PubMed] [Google Scholar]
- Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG, Kouzarides T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002;21:2383–2396. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KM, Po LS, Tsang FC, Siu WY, Lau A, Ho HT, Poon RY. The candidate tumor suppressor ING1b can stabilize p53 by disrupting the regulation of p53 by MDM2. Cancer Res. 2002;62:4890–4893. [PubMed] [Google Scholar]
- Lin AW, Lowe SW. Oncogenic ras activates the ARF-p53 pathway to suppress epithelial cell transformation. Proc Natl Acad Sci USA. 2001;98:5025–5030. doi: 10.1073/pnas.091100298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998;12:3008–3019. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HK, Bergmann S, Pandolfi PP. Cytoplasmic PML function in TGF-beta signalling. Nature. 2004;431:205–211. doi: 10.1038/nature02783. [DOI] [PubMed] [Google Scholar]
- Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA Repair, Genome Stability, and Aging. Cell. 2005;120:497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: Progress and puzzles. Curr Opin Genet Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- Lu X. p53: A heavily dictated dictator of life and death. Curr Opin Genet Dev. 2005;15:27–33. doi: 10.1016/j.gde.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Maeda T, Hobbs RM, Merghoub T, Guernah I, Zelent A, Cordon-Cardo C, Teruya-Feldstein J, Pandolfi PP. Role of the protooncogene Pokemon in cellular transformation and ARF repression. Nature. 2005;433:278–285. doi: 10.1038/nature03203. [DOI] [PubMed] [Google Scholar]
- Mallette FA, Goumard S, Gaumont-Leclerc MF, Moiseeva O, Ferbeyre G. Human fibroblasts require the Rb family of tumor suppressors, but not p53, for PML-induced senescence. Oncogene. 2004;23:91–99. doi: 10.1038/sj.onc.1206886. [DOI] [PubMed] [Google Scholar]
- Masutomi K, Yu EY, Khurts S, Ben-Porath I, Currier JL, Metz GB, Brooks MW, Kaneko S, Murakami S, DeCaprio JA, et al. Telomerase maintains telomere structure in normal human cells. Cell. 2003;114:241–253. doi: 10.1016/s0092-8674(03)00550-6. [DOI] [PubMed] [Google Scholar]
- McConnell BB, Starborg M, Brookes S, Peters G. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr Biol. 1998;8:351–354. doi: 10.1016/s0960-9822(98)70137-x. [DOI] [PubMed] [Google Scholar]
- Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- Ohtani N, Zebedee Z, Huot TJ, Stinson JA, Sugimoto M, Ohashi Y, Sharrocks AD, Peters G, Hara E. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature. 2001;409:1067–1070. doi: 10.1038/35059131. [DOI] [PubMed] [Google Scholar]
- Palmero I, Pantoja C, Serrano M. p19ARF links the tumour suppressor p53 to Ras. Nature. 1998;395:125–126. doi: 10.1038/25870. [DOI] [PubMed] [Google Scholar]
- Pantoja C, Serrano M. Murine fibroblasts lacking p21 undergo senescence and are resistant to transformation by oncogenic Ras. Oncogene. 1999;18:4974–4982. doi: 10.1038/sj.onc.1202880. [DOI] [PubMed] [Google Scholar]
- Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741–747. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, Pelicci PG. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406:207–210. doi: 10.1038/35018127. [DOI] [PubMed] [Google Scholar]
- Peeper DS, Shvarts A, Brummelkamp T, Douma S, Koh EY, Daley GQ, Bernards R. A functional screen identifies hDRIL1 as an oncogene that rescues RAS-induced senescence. Nat Cell Biol. 2002;4:148–153. doi: 10.1038/ncb742. [DOI] [PubMed] [Google Scholar]
- Preto A, Singhrao SK, Haughton MF, Kipling D, Wynford-Thomas D, Jones CJ. Telomere erosion triggers growth arrest but not cell death in human cancer cells retaining wild-type p53: Implications for antitelomerase therapy. Oncogene. 2004;23:4136–4145. doi: 10.1038/sj.onc.1207564. [DOI] [PubMed] [Google Scholar]
- Rangarajan A, Weinberg RA. Opinion: Comparative biology of mouse versus human cells: Modelling human cancer in mice. Nat Rev Cancer. 2003;3:952–959. doi: 10.1038/nrc1235. [DOI] [PubMed] [Google Scholar]
- Riou JF, Guitat L, Mailliet P, Laoui A, Renou E, Petitegenet O, Megnin-Chanet F, Helene C, Mergny JL. Cell senescence and telomere shortening induced by a new series of specific G-quadruplex DNA ligands. Proc Natl Acad Sci USA. 2002;99:2672–2677. doi: 10.1073/pnas.052698099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson RS, Kussick SJ, Vallieres E, Chen SY, Wu DY. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005;65:2795–2803. doi: 10.1158/0008-5472.CAN-04-1270. [DOI] [PubMed] [Google Scholar]
- Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63:2705–2715. [PubMed] [Google Scholar]
- Sage J, Miller A, Perez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle reentry. Nature. 2003;424:223–228. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, Lowe SW. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109:335–346. doi: 10.1016/s0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]
- Seimiya H, Oh-hara T, Suzuki T, Naasani I, Shimazaki T, Tsuchiya K, Tsuruo T. Telomere shortening and growth inhibition of human cancer cells by novel synthetic telomerase inhibitors, MST-312, MST-295, and MST-199. Mol Cancer Ther. 2002;1:657–665. [PubMed] [Google Scholar]
- Seimiya H, Muramatsu Y, Ohishi T, Tsuruo T. Tankyrase 1 as a target for telomere-directed molecular cancer therapeutics. Cancer Cell. 2005;7:25–37. doi: 10.1016/j.ccr.2004.11.021. [DOI] [PubMed] [Google Scholar]
- Seger YR, Garcia-Cao M, Piccinin S, Cunsolo CL, Doglioni C, Blasco MA, Hannon GJ, Maestro R. Transformation of normal human cells in the absence of telomerase activation. Cancer Cell. 2002;2:401–413. doi: 10.1016/s1535-6108(02)00183-6. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Sharpless NE. INK4a/ARF: A multifunctional tumor suppressor locus. Mutat Res. 2005 doi: 10.1016/j.mrfmmm.2004.08.021. Published online May 6, 2005. 10.1016/j.mrfm‐mm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, Ramsey MR, Balasubramanian P, Castrillon DH, DePinho RA. The differential impact of p16(INK4a) or p19(ARF) deficiency on cell growth and tumorigenesis. Oncogene. 2004;23:379–385. doi: 10.1038/sj.onc.1207074. [DOI] [PubMed] [Google Scholar]
- Shay JW, Wright WE. Telomerase: A target for cancer therapeutics. Cancer Cell. 2002;2:257–265. doi: 10.1016/s1535-6108(02)00159-9. [DOI] [PubMed] [Google Scholar]
- Shay JW, Pereira-Smith OM, Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res. 1991;196:33–39. doi: 10.1016/0014-4827(91)90453-2. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, DePinho RA. Cellular senescence: Mitotic clock or culture shock? Cell. 2000;102:407–410. doi: 10.1016/s0092-8674(00)00046-5. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Weber JD. The ARF/p53 pathway. Curr Opin Genet Dev. 2000;10:94–99. doi: 10.1016/s0959-437x(99)00038-6. [DOI] [PubMed] [Google Scholar]
- Shvarts A, Brummelkamp TR, Scheeren F, Koh E, Daley GQ, Spits H, Bernards R. A senescence rescue screen identifies BCL6 as an inhibitor of anti-proliferative p19(ARF)-p53 signaling. Genes Dev. 2002;16:681–686. doi: 10.1101/gad.929302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JR, Pereira-Smith OM. Replicative senescence: Implications for in vivo aging and tumor suppression. Science. 1996;273:63–67. doi: 10.1126/science.273.5271.63. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A, de Lange T. Regulation of telomerase by telomeric proteins. Annu Rev Biochem. 2004;73:177–208. doi: 10.1146/annurev.biochem.73.071403.160049. [DOI] [PubMed] [Google Scholar]
- Sreeramaneni R, Chaudhry A, McMahon M, Sherr CJ, Inoue K. Ras-Raf-Arf signaling critically depends on the Dmp1 transcription factor. Mol Cell Biol. 2005;25:220–232. doi: 10.1128/MCB.25.1.220-232.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein GH, Drullinger LF, Soulard A, Dulic V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol. 1999;19:2109–2117. doi: 10.1128/mcb.19.3.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002;62:1876–1883. [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, And de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–1556. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- Trost TM, Lausch EU, Fees SA, Schmitt S, Enklaar T, Reutzel D, Brixel LR, Schmidtke P, Maringer M, Schiffer IB, et al. Premature Senescence Is a Primary Fail-safe Mechanism of ERBB2-Driven Tumorigenesis in Breast Carcinoma Cells. Cancer Res. 2005;65:840–849. [PubMed] [Google Scholar]
- Voorhoeve PM, Agami R. The tumor-suppressive functions of the human INK4A locus. Cancer Cell. 2003;4:311–319. doi: 10.1016/s1535-6108(03)00223-x. [DOI] [PubMed] [Google Scholar]
- Vousden K. Activation of p53 tumor suppressor protein. Biochim Biophys Acta. 2002;1602:47–59. doi: 10.1016/s0304-419x(02)00035-5. [DOI] [PubMed] [Google Scholar]
- Wang W, Chen JX, Liao R, Deng Q, Zhou JJ, Huang S, Sun P. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol Cell Biol. 2002;22:3389–3403. doi: 10.1128/MCB.22.10.3389-3403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1:20–26. doi: 10.1038/8991. [DOI] [PubMed] [Google Scholar]
- Wei W, Hemmer RM, Sedivy JM. Role of p14(ARF) in replicative and induced senescence of human fibroblasts. Mol Cell Biol. 2001;21:6748–6757. doi: 10.1128/MCB.21.20.6748-6757.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Herbig U, Wei S, Dutriaux A, Sedivy JM. Loss of retinoblastoma but not p16 function allows bypass of replicative senescence in human fibroblasts. EMBO Rep. 2003;4:1061–1065. doi: 10.1038/sj.embor.7400001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DJ, Foster SA, Galloway DA, Reid BJ. Progressive region-specific de novo methylation of the p16 CpG island in primary human mammary epithelial cell strains during escape from M(0) growth arrest. Mol Cell Biol. 1999;19:5642–5651. doi: 10.1128/mcb.19.8.5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Miloslavskaya I, Demontis S, Maestro R, Galaktionov K. Regulation of cellular response to oncogenic and oxidative stress by Seladin-1. Nature. 2004;432:640–645. doi: 10.1038/nature03173. [DOI] [PubMed] [Google Scholar]
- Zhang Y. The ARF-B23 connection: Implications for growth control and cancer treatment. Cell Cycle. 2004;3:259–262. [PubMed] [Google Scholar]
- Zheng X, Chou PM, Mirkin BL, Rebbaa A. Senescence-initiated reversal of drug resistance: Specific role of cathepsin L. Cancer Res. 2004;64:1773–1780. doi: 10.1158/0008-5472.can-03-0820. [DOI] [PubMed] [Google Scholar]
- Zhu J, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998;12:2997–3007. doi: 10.1101/gad.12.19.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]