

The major objective of this lecture is to illustrate how eye cancer research has dramatically affected not only patients with these ocular tumors, but also our fundamental understanding of cancer biology and many other aspects of biomedical research. Retinoblastoma and uveal melanoma, the two most common eye cancers in children and adults, respectively, are very different clinically and biologically. Retinoblastoma is a rare childhood eye cancer that usually occurs before 5 years of age, is often hereditary and bilateral, is caused by the mutation of a single rate-limiting gene, and is rarely fatal if treatment of the eye cancer is successful. Uveal melanoma occurs mostly in older adults, is rarely hereditary or bilateral, is caused by an accumulation of multiple genetic lesions with no single disease-causing gene mutation, and is fatal in up to 50% of patients, even when the eye cancer is successfully treated. Despite these profound differences, retinoblastoma and uveal melanoma have provided unique and complementary insights into the cell cycle, differentiation, development, neoplasia, and other fundamental biological processes. Herein, we will consider some of the highlights in ocular oncology research over the past two decades, starting with the identification of the retinoblastoma gene in 1986. We will examine how the molecular genetics of retinoblastoma has fundamentally altered our view of development, cell cycle and cancer biology. We will then explore the role of the retinoblastoma tumor suppressor pathway in uveal melanoma. Finally, we will review recent exciting research in uveal melanoma that may lead to a significant change in our understanding of that cancer, and we will consider the far-reaching implications of this breakthrough in future research, clinical trials, and patient care.
Retinoblastoma
Identification of the Retinoblastoma Gene
Despite its relative rarity, occurring in only approximately 1 patient per 20,000 live births, retinoblastoma has been at the heart of many of the landmark discoveries in cancer research over the past two decades. Most patients with unilateral, unifocal retinoblastoma do not transmit the disease to their children, nor are they predisposed to second primary cancers elsewhere in the body. In contrast, patients with bilateral retinoblastoma usually have multiple, bilateral tumors; they transmit the disease in an autosomal dominant pattern; and they have an alarmingly high rate of second primary cancers.1 Because of the autosomal dominant inheritance pattern, it was widely assumed for many years that retinoblastoma was caused by a dominantly acting oncogene.2 In 1971, however, Knudson3 proposed the “two-hit hypothesis,” which heralded a major paradigm shift in retinoblastoma and, indeed, all cancer biology. Knudson’s supposition was that the RB gene inhibits tumor formation in a recessive manner and that inactivation of both copies of the RB gene in a susceptible retinal cell leads to retinoblastoma. In nonhereditary retinoblastoma, the mutation of both RB alleles occurs in the same retinoblast. Since the chance of both events occurring in the same cell is very small, this same-cell mutation explains the low incidence and unifocality. In hereditary retinoblastoma, the first RB mutation is passed in the germline and is present in most or all cells in the body. The second mutation then occurs somatically in a retinal cell that already contains one RB mutation. Because only one somatic mutation is required in any susceptible retinal cell, this mode of mutation explains the multifocality and bilaterality of hereditary retinoblastoma. The presence of the RB mutation throughout the body (including the germline) explains the hereditary transmission and the risk of second primary cancers. In 1986, Knudson’s hypothesis was validated by the identification of the RB gene on chromosome 13, region q14, by a Dryja et al.,4 and this finding was confirmed shortly thereafter by other groups.5,6
The Retinoblastoma Gene and Protein
The RB gene contains 27 exons distributed over approximately 200 kb of DNA. Germline RB mutations tend to occur at CpG dinucleotides and are distributed throughout the gene.7 RB mutations are also found in many of the second primary cancers found in patients with retinoblastoma, such as osteosarcoma, soft tissue sarcoma, and melanoma.8,9 In addition, RB mutations are found in some sporadic cancers of the lung, prostate, breast, and other tissues.4,6,10–13
The Rb protein, composed of 928 amino acids, is a nuclear phosphoprotein that contains an N-terminal region, a central pocket domain composed of an A box, and a B box, and a C terminus. In virtually all human cancers in which the RB gene is not mutated, the Rb protein is functionally inactivated, suggesting that Rb is a tumor suppressor of fundamental importance in cancer biology.14 In fact, tumor-causing viruses such as SV40, adenovirus, and human papillomavirus all produce proteins that bind and inhibit Rb, and these oncoproteins are essential for the tumorigenic properties of these viruses.15–17 The Rb “pocket” is formed by interaction of the A and B boxes along an extended interface and is essential for the tumor-suppressor function of Rb, as evidenced by the disruption of the pocket by virtually all tumor-causing mutations.7,18
Assembly of Multimeric Protein Complexes
The tumor suppressor activity of Rb is due, at least in part, to its ability to inhibit cell division by blocking S-phase entry.19 This and most other physiologic effects of Rb rely on its ability to regulate gene expression by assembling multimeric protein complexes at promoters. Rb does this through its multiple binding sites that allow it to interact simultaneously with several proteins (Fig. 1). The B box within the pocket contains a leucine-x-cysteine-x-glutamate (LxCxE) binding site to which bind the viral oncoproteins and many chromatin remodeling proteins that contain an LxCxE motif. A separate binding site formed by a cleft at the interface of the A and B boxes, through which Rb binds to E2F transcription factors (E2Fs).5,20,21 There may also be other binding sites in the pocket, since chromatin remodeling proteins such as BRG1 do not need an LxCxE motif to bind Rb.22 The C terminus contains binding sites for the oncoproteins HDM2 and c-Abl23,24 Thus, Rb is able to assemble a large variety of multimeric complexes through its distinct binding sites.
Figure 1.
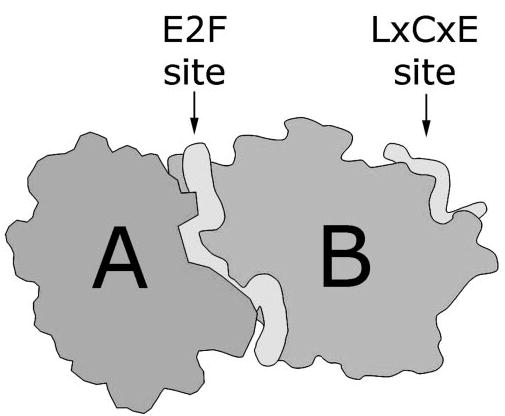
The structure of the Rb pocket domain. Cartoon based on the crystal structure of the highly conserved A and B boxes of Rb, which form a strong intramolecular complex. The tubular structures represent peptide segments from E2F, which binds Rb at a cleft formed between the A and B boxes, and the human papillomavirus E7 protein, which binds Rb at a site in the B box through its LxCxE motif. Adapted, with permission, from Lee C, Chang JH, Lee HS, Cho Y. Structural basis for the recognition of the E2F transactivation domain by the retinoblastoma tumor suppressor. Genes Dev. 2002;16:3199–3212. © 2002 Cold Spring Harbor Laboratory Press.
Transcriptional Repression
Rb and the other pocket proteins p107 and p130 do not contain a DNA binding domain, but they interact with specific DNA elements through binding to E2Fs1 to −5, which do contain sequence-specific DNA binding domains.25–27 E2F sites are present in many genes involved in the cell cycle, in differentiation, and in apoptosis, and Rb represses these genes by at least two mechanisms (Fig. 2). First, Rb binds and physically blocks the E2F transactivation domain.28,29 Second, when Rb is brought to the promoter through interaction with E2Fs, it can actively repress transcription by suppressing the activity of surrounding enhancers on the promoter.30 Rb does this by recruiting chromatin remodeling proteins to promoters, where they alter local chromatin structure and inhibit access by the transcriptional machinery.31,32 Several major classes of chromatin remodeling proteins have been shown to interact with Rb, including histone deacetylases (e.g., HDAC1 to −3), SWI/SNF ATP-dependent nucleosome assembly proteins (e.g., BRG1), polycomb group proteins (e.g., HPC2 and Ring1), DNA methyltransferases (e.g., DNMT1), and histone methyltransferases (e.g., SUV39h).32–46 These interactions with chromatin-remodeling proteins are in turn regulated by specific Rb phosphorylation events.22,47
Figure 2.
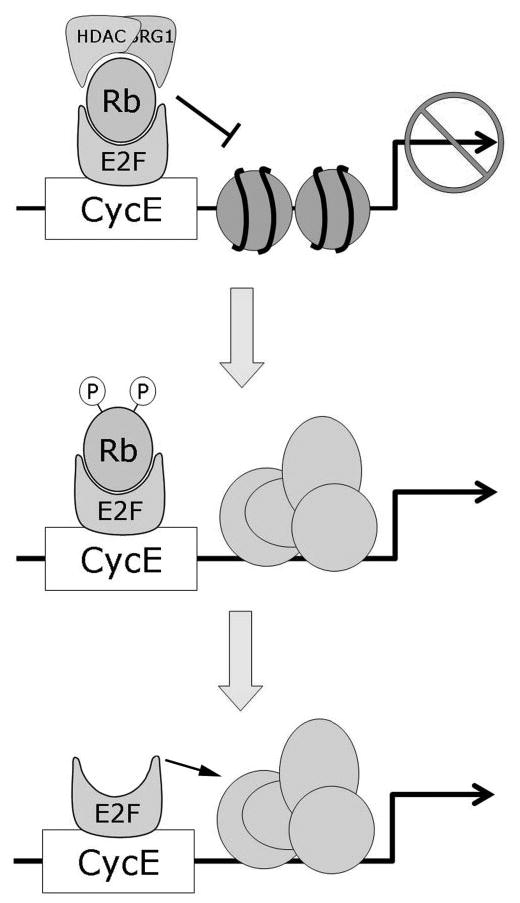
Mechanisms of transcriptional repression by Rb. Rb represses transcription by recruiting chromatin remodeling proteins such as histone deacetylases (HDAC) and BRG1 to DNA regulatory elements containing E2F sites through its interaction with E2F proteins. The resulting alterations in chromatin structure prevent access by the transcriptional machinery. Phosphorylation of Rb by cyclin dependent kinases removes these chromatin remodeling proteins from the multimeric complex, thereby allowing expression of genes involved in cell division. More complete phosphorylation or mutational loss of Rb releases the transactivation domain of E2F to directly activate genes, especially those involved in apoptosis.
Stepwise Regulation of Rb by Phosphorylation
Rb is regulated by phosphorylation and can be phosphorylated at up to 16 serine/threonineproline recognition sites by cyclin-dependent kinases (CDKs). CDKs are activated by interaction with their cyclin-binding partners and inactivated by CDK inhibitors such as the tumor suppressor p16Ink4a.48–51 Early investigators envisaged a binary model of Rb activity in which the protein is active when hypophosphorylated and inactive when hyperphosphorylated.48,52,53 However, this model has been found to be overly simplistic. In reality, Rb is phosphorylated in multiple steps throughout the cell cycle. Initial phosphorylation of Rb is catalyzed by cyclin D-CDK4 in early G1, then by cyclin E-CDK2 late in G1 and later by cyclin A-CDK2 inthe S phase.14 These multiple phosphorylation events are necessary for complete hyperphosphorylation and inactivation of Rb.50 Further, phosphorylation of specific sites appears to regulate distinct Rb functions, suggesting a complex mechanism for regulating of Rb by sequential phosphorylation events.54,55
In 1999, we provided a molecular model for understanding this complex regulation of Rb activity.47 Rb is phosphorylated in a stepwise series of increasingly energetically unfavorable reactions that are enabled by conformational changes induced by the previous phosphorylation event (Fig. 3). Similar sequential phosphorylation mechanisms have been shown for other proteins such as c-Fos.56 The initial phosphorylation of Rb at sites in the C terminus by cyclin D-CDK4 triggers an intramolecular conformational change in which the negatively charged C terminus interacts with a positively charged lysine patch in the B box. This interaction displaces LxCxE proteins such as HDACs from their binding site, thereby partially inactivating Rb. This phosphorylation event is probably sufficient to abrogate the ability of Rb to block the G1-to-S transition. This intramolecular interaction also brings CDK2 docking sites in the C terminus into proximity of additional phosphorylation sites in the pocket, thereby promoting the phosphorylation of these sites by cyclin E-CDK2. These phosphorylation events cause additional conformational changes that provide access by cyclin-CDK complexes to Ser567, which is otherwise buried within the A–B interface and inaccessible to phosphorylation. Phosphorylation of Ser567 grossly disrupts the tertiary structure of Rb, releasing E2Fs. In support of this model, crystallographic studies of Rb have predicted that phosphorylation of the C terminus would trigger an intramolecular interaction with the pocket, that Ser567 mediates critical contacts between the A and B boxes, and that phosphorylation of Ser567 would destabilize the pocket structure and eliminate the E2F binding site.5,21
Figure 3.
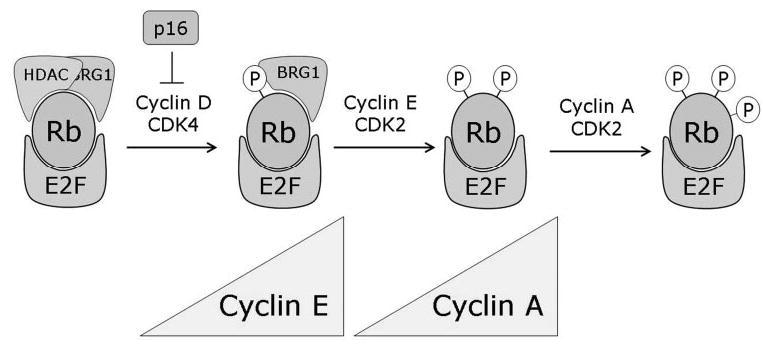
Progressive phosphorylation of Rb. Rb is progressively phosphorylated by successive cyclin-CDK complexes, which allows the consecutive derepression of cyclins during cell cycle progression. Thus, Rb may serve to regulate the orderly cell cycle progression, which could explain why Rb-null cells are prone to chromosomal instability.
To provide a physiologic context for these findings, we later showed that Rb represses cyclin E and other G1-phase cell cycle genes when it is fully active through multimeric complexes containing HDAC and BRG1.22 After initial phosphorylation by cyclin D-CDK4, Rb dissociates from HDAC but remains bound to BRG1 and E2Fs, in which state it continues to repress cyclin A and other S-phase genes. After cells enter the S-phase, the increasing levels of cyclin E and consequent activation of CDK2 leads to further phosphorylation of Rb, releasing BRG1 and derepressing the cyclin A gene, which then allows cyclin A-kinase complexes to accumulate and maintain Rb in a phosphorylated state during cell cycle progression through the S phase. Thus, the sequential phosphorylation and progressive inactivation of Rb may allow orderly cell cycle progression through the precisely timed activation of cyclins (Fig. 3). However, the role of Ser567 phosphorylation remained unclear for several years.
The Role of Rb in Apoptosis
Loss of Rb leads to apoptosis in the retina, lens, and many other tissues through a potent, multifaceted death response, suggesting that there is an inherent pressure for organisms to eliminate aberrant cells that lack Rb.57– 61 Loss of Rb leads to apoptosis through two major mechanisms. First, since Rb binds the transactivation domain of E2Fs, it directly blocks the ability of proapoptotic E2F1 to transactivate genes involved in apoptosis, such as ARF and p73.62 Loss of Rb releases E2F1 from its inhibitory interaction with Rb, allowing it to activate proapoptotic genes. In general, this mechanism is dependent on the p53 pathway.63 Second, loss of Rb derepresses apoptotic genes that are under basal inhibition by Rb–E2F complexes.64 This mechanism is largely independent of p53 and does not require E2F transactivation.65,66 Using a dominant-negative E2F mutant (dnE2F) that contains the DNA binding domain but not the transactivation domain, we recently identified one of these basally inhibited proapoptotic genes and identified a potentially new Rb-mediated apoptotic pathway.67 The dnE2F mutant caused a rapid onset of apoptosis through derepression of the protein phosphatase PTP1B, leading to the dephosphorylation and inactivation of the antiapoptotic focal adhesion kinase (FAK), and activation of caspase-8 and the extrinsic death pathway. Hence, loss of Rb sets in motion a multi-pronged apoptotic response through p53-dependent and -independent mechanisms, to eliminate abnormal cells.
Reconciling the Antiproliferative and Antiapoptotic Functions of Rb
Because Rb inhibits both cell division and apoptosis, how do cells inactivate Rb to allow normal cell division without triggering apoptosis? We provided a potential explanation for this paradox by showing that the cell cycle and apoptotic functions of Rb are regulated by distinct mechanisms. The CDK sites in the C terminus are phosphorylated every cell cycle and regulate cell cycle arrest and differentiation. In contrast, phosphorylation of Ser567 is energetically unfavorable and does not appear to occur during normal cell division, but only under abnormal conditions such as excessive mitogenic stimulation or cellular stress. Further, phosphorylation of Ser567 leads not simply to release of E2Fs but also to irreversible Rb degradation, which drives up the intracellular levels of free E2Fs and induces an apoptotic response as described above.
Our current hypothesis is that the energetically unfavorable phosphorylation of Ser567 does not occur in normal cycling cells but provides a buffer to allow cells to partially phosphorylate and inactivate Rb during cell division without inadvertently triggering apoptosis. Further, Ser567 may serve as an apoptotic checkpoint for eliminating abnormal cells that become Rb-deficient. This hypothesis would reconcile the antiproliferative and antiapoptotic properties of Rb and would place Rb at the center of a major cell fate checkpoint (Fig. 4). This model makes several assumptions that previously would have been controversial but now have experimental support. First, the model implies that Rb is only partially inactivated in normal proliferating cells. Indeed, phosphorylation of Rb by CDK4, which is sufficient to allow cell division, results in a partially phosphorylated form of Rb that retains the ability to bind E2Fs and to repress transcription.68,69 Second, the model suggests that Rb does not become completely dissociated from E2Fs every cell cycle, but rather that some Rb-E2F complex persist throughout the cell cycle. Consistent with this idea, the Farnham laboratory has shown using chromatin immunoprecipitation and CpG microarray experiments that Rb continues to colocalize with E2Fs at promoters throughout the cell cycle.70,71
Figure 4.
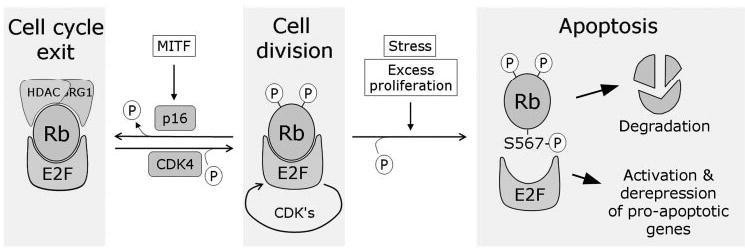
The Rb cell fate checkpoint. Rb regulates a major checkpoint in the cell fate decision between proliferation, differentiation, and apoptosis. Partial phosphorylation of Rb allows cell division without triggering an apoptotic response. Hypophosphorylation of Rb allows it to interact with chromatin remodeling proteins such as HDAC and BRG1, which induces widespread repression of specific genes that leads to cell cycle exit, such as occurs in differentiation and senescence. Ultimate phosphorylation of Rb at Ser567, which may occur only during abnormal hyperproliferation or stress conditions, disrupts the Rb protein, leading to its degradation. This also releases free E2F1 to induce the expression of proapoptotic genes.
The Role of Rb in Development and Differentiation
Surprisingly, Rb is not needed for the cell division cycle to function. Cells lacking Rb progress normally through the cell cycle, but they are unable to exit the cycle normally during senescence or differentiation.72 Indeed, tumors develop in mouse retina lacking Rb/p107 not because of a proliferation defect, but because of an inability of retinal progenitor cells to exit the cell cycle and terminally differentiate.73 Similarly, we showed that the Rb pathway is essential for melanocyte differentiation.74 In these studies, the melanocyte differentiation factor MITF was found to induce expression of the p16Ink4a tumor suppressor, causing an accumulation of hypophosphorylated (active) Rb and cell cycle arrest. This MITF-INK4A-Rb pathway was required for efficient cell cycle exit and melanocyte differentiation in cultured cells and in vivo (Fig. 5). Thus, the tumor suppressor function of Rb appears to be due, at least in part, to its ability to induce permanent cell cycle exit in association with senescence and differentiation.75 However, it remains unclear whether Rb is needed simply to enforce cell cycle exit or whether it actively participates in the differentiation program. In muscle cell differentiation, Rb appears to cooperate with the muscle-specific transcription factor MyoD to activate genes involved in myocyte differentiation, but cell-cycle arrest is not a prerequisite for differentiation.76,77 Whether this is also the case for melanocyte differentiation is yet to be determined.
Figure 5.
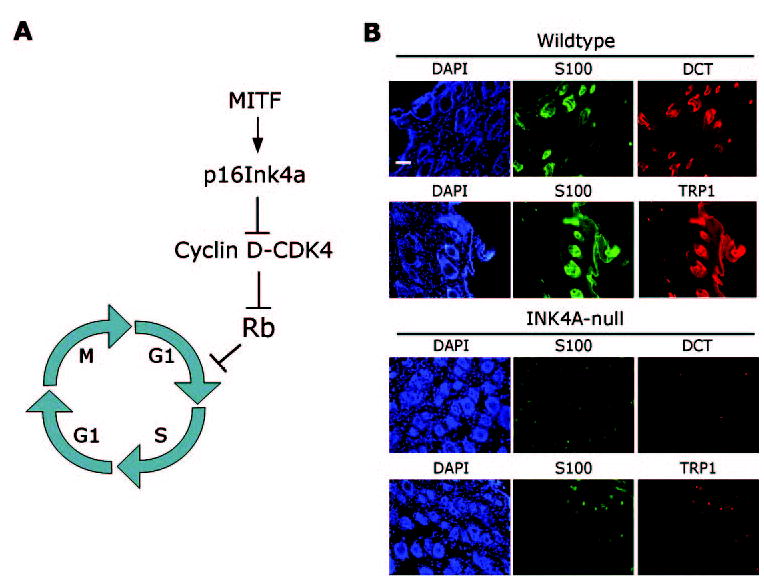
The role of Rb in melanocyte differentiation. (A) The melanocyte differentiation factor MITF transcriptionally activates the p16Ink4a tumor suppressor, which activates Rb by inhibiting CDK4. Active Rb then enforces cell cycle exit at the G1-S boundary, which is required for efficient melanocyte differentiation. (B) Melanocyte differentiation in vivo is hindered when p16Ink4a is not present to activate Rb. Low-power photomicrographs show immunofluorescence analysis of the melanocyte markers S100, DCT, and TRP1 in cutaneous hair follicles of 3-week-old INK4A-wild-type mice and INK4A-null mice. Note that the expression of S100 (green), TRP1 (red), and DCT (red) is greatly diminished in INK4A-null mice. DAPI (blue) indicates cell nuclei. Scale bar, 100 μm.
Of interest, cultured cells that are growth inhibited due to MITF expression occasionally escape this inhibition, spontaneously re-enter the cell cycle and grow into colonies.74 These escape clones usually exhibit methylation and inactivation of the INK4A promoter, resulting in phosphorylation of Rb. Thus, restraining Rb appears to be a key step in melanoma formation by allowing melanocytes to re-enter the cell cycle inappropriately. It is noteworthy that Rb itself is not the target of these escape mutations, suggesting that there may be a selective pressure to partially inactivate Rb through methylation of INK4A to allow cell cycle re-entry, rather than the complete inactivation of Rb, which may promote apoptosis (as discussed earlier). Consistent with this idea, we found that a more complete inactivation of Rb by RNA inhibition or overexpression of cyclins D and E leads to melanocyte apoptosis (Harbour JW, unpublished data, 2005). Further, acute inactivation of Rb in vivo by conditional mutagenesis in the skin results in melanocyte apoptosis and severe depigmentation,78 indicating that Rb has a cell-autonomous role in melanocyte survival.
Thus, our current view is that Rb is dispensable for regulation of the normal cell cycle, but it is essential for coordinating orderly cell cycle exit associated with differentiation and senescence. Further, Rb may act as a buffer against inappropriate apoptosis during normal cell division but can unleash a potent apoptotic signal under abnormal hyperproliferative or stress conditions. Next, we will explore how uveal melanoma has provided further insights into the role of Rb in melanocyte biology and oncogenesis.
The RB Pathway in Uveal Melanoma
Uveal melanoma is the most common eye cancer and the second most common form of melanoma.79,80 Unlike retinoblastoma, uveal melanoma has been extremely recalcitrant to classic molecular genetic analysis. This cancer is rarely hereditary, which has hampered linkage analysis for susceptibility genes.81 Further, none of the major tumor suppressor genes, including RB, are mutated in uveal melanomas with any significant frequency, as evidenced by the normal expression of the Rb protein in most of these tumors.82,83 In parallel studies of the p53 pathway, we found no evidence for p53 mutations but frequent disruption of the p53 pathway, such as overexpression of the p53 inhibitor HDM2.83–86 Likewise for Rb, we found that the Rb pathway is commonly inhibited in uveal melanomas by inappropriate phosphorylation of Rb. In primary uveal melanomas, we showed that Rb is phosphorylated at the C-terminal CDK sites that regulate the repressor activity of Rb.82 Since these sites are known to be phosphorylated by CDK4, this phosphorylation could result from upregulation of cyclin D (which activates CDK4) or inactivation of INK4A (which encodes the CDK4 inhibitor p16Ink4a). Indeed, about two thirds of uveal melanomas express high levels of cyclin D1,83,87 and about one third of these tumors exhibit INK4A promoter methylation.88 Further, many uveal melanomas display constitutive activation of the MAPK pathway,89 which activates cyclin D1 expression.90
Of interest, while Rb is phosphorylated in uveal melanomas at the C-terminal sites that regulate cell proliferation, Ser567is rarely phosphorylated in these tumors (Harbour JW, unpublished data, 2004). This observation is consistent with the very low rate of spontaneous apoptosis observed pathologically in primary uveal melanomas, compared with the high rate of apoptosis in Rb-null retinoblastomas. Hence, uveal melanomas (and many other solid tumors) do not completely abolish Rb activity, but rather, they appear to cause partial inactivation of Rb, to allow cell division without inducing the excessive apoptosis that can accompany complete Rb loss. This phenomenon may explain, at least in part, the extreme resistance to therapy-induced apoptosis in these tumors.91
A New Model of Uveal Melanoma Genetics
The Genetics of Uveal Melanoma Metastasis
Up to half of patients with uveal melanoma die of metastatic disease, even when the eye tumor is successfully treated.92 Do the defects in the Rb pathway explain this propensity for metastasis in uveal melanoma? This notion seems unlikely for several reasons. First, Rb pathway abnormalities are found in virtually all uveal melanomas, whether or not they are associated with metastasis (Harbour JW, unpublished data, 2003). Second, for the reasons discussed earlier, disruption of the Rb pathway is probably an early event in melanoma initiation, being required for melanocytes to re-enter the cell cycle before acquiring additional mutations.74,93 A few recurring chromosomal alterations are associated with increased risk of metastasis, including loss of chromosome 3 and gain of 8q, but no specific cancer genes have been linked to these loci.94 This lack of understanding of the metastatic phenotype using traditional methods led us to explore global changes in gene expression.
Two Distinct Molecular Classes of Uveal Melanoma
If Rb pathway alterations occur early in uveal melanoma pathogenesis, what are the later events that determine whether these tumors will metastasize? Several clinical, pathologic, and cytogenetic features have been shown to predict metastatic disease, including larger tumor size, anterior tumor location, older patient age, epithelioid cell type, extracellular matrix deposition in looping patterns, and monosomy 3.95–99 To gain insights into the molecular mechanisms of uveal melanoma metastasis, we used microarray gene expression profiling to analyze primary, uncultured uveal melanomas.100 We found that primary uveal melanomas cluster naturally into two groups that we provisionally refer to as classes 1 and 2, based on gene-expression signature (Fig. 6). Survival analysis showed that patients with class 1 tumors rarely died of metastasis, whereas those with class 2 tumors had a high rate of metastatic death. Correspondingly, class 2 tumors were more likely to exhibit epithelioid cells, looping extracellular matrix patterns, and monosomy 3.100,101 Similarly, the Lohmann laboratory102 in Germany independently found that uveal melanomas form two molecular classes that correspond to chromosome 3 status. The gene signatures from these two studies were remarkably similar, despite differences in study design and patient population, indicating that these molecular signatures may be generalizable to other populations. Further, these were the first large-scale studies of gene expression using primary, uncultured uveal melanomas, which is important because the use of cultured cells for such studies can introduce unpredictable artifacts resulting from the stress and selection that occur in cell culture.103 Several other studies have yielded findings consistent with these two landmark papers.104–108 Thus, there appears to be a robust and profound molecular difference between low-grade, nonmetastasizing (class 1) tumors and high-grade, metastasizing (class 2) tumors. The distinctive gene signatures of these two tumor classes provide a valuable new tool for understanding the biological processes underlying metastasis. The results of preliminary work in our laboratory suggests that previous prognostic markers for metastasis, such as epithelioid cytology and extracellular matrix patterns, may be phenotypic indicators of a more fundamental underlying difference in the differentiation program that is operative in class 2 tumors.100 How these changes in gene expression lead to metastasis is now the subject of intense investigation.
Figure 6.
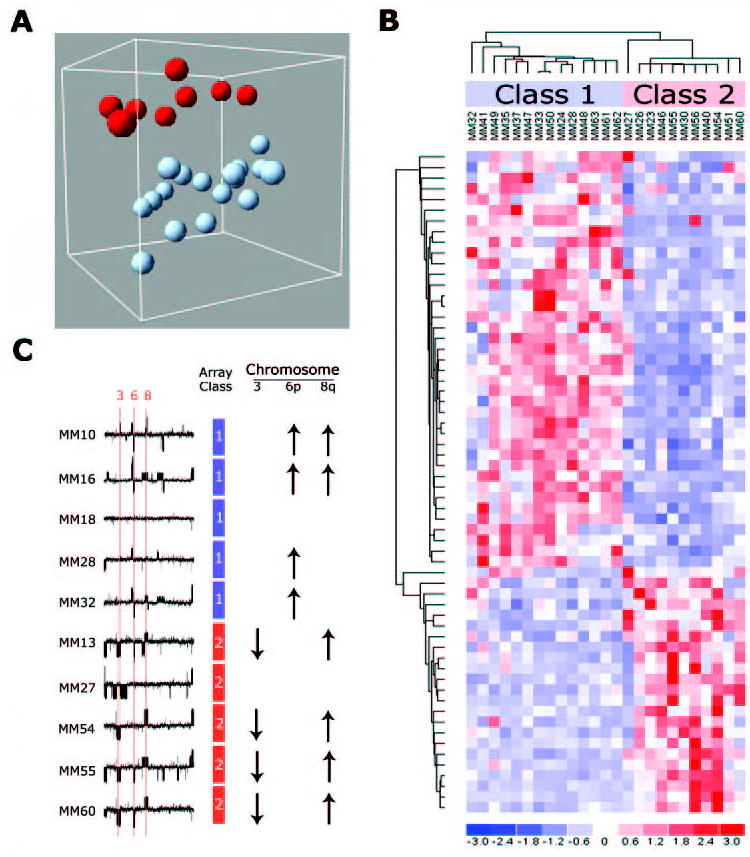
Two molecular classes of uveal melanoma. (A) Unsupervised analysis of primary uveal melanomas using principal component analysis reveals two distinct groups of uveal melanomas based on gene expression patterns. (B) Hierarchical clustering of the top 62 genes that discriminate between class 1 and 2 melanomas shows that the two tumor classes have distinct “molecular signatures.” (C) Microarray-based comparative genomic hybridization experiments show that the class 2 signature is strongly associated with monosomy 3 (down arrows) and inversely associated with gain of chromosome 6p (up arrows). Reprinted, with permission, from Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004;64:7205–7209. © 2004 American Association for Cancer Research.
Implications for the Future
If these recent discoveries in uveal melanoma stand the test of time, they may eventually be viewed similarly to the identification of the RB gene and the profound impact that that event had on our understanding of retinoblastoma. The discovery that uveal melanomas form a binary classification of low- and high-risk tumors, rather than a continual spectrum of tumors of intermediate metastatic risks, will profoundly affect future research directions, the design of clinical trials, and the management of patients. Inherent in this novel classification is a key question: do class 1 tumors evolve into class 2 tumors, or do the two classes develop along distinct pathways? If the former possibility is correct, then treatment of the primary eye tumor has the potential to be curative, and early treatment of small tumors before growth may be recommended. Alternatively, if the latter possibility is correct, then treatment of the primary eye tumor may have no effect on survival, which would have profound implications for how primary uveal melanomas should be managed. Regardless of which of model is more correct, the better we understand how uveal melanomas acquire the ability to metastasize, the more likely we will be to develop new therapeutic strategies to delay or prevent the development of metastasis in high-risk patients.
Conclusions
A cogent argument could be made that the modern era of molecular oncology started with ocular cancer. The discovery of the retinoblastoma gene heralded a revolution in cancer research and ushered in the concept of tumor suppressors. The importance of Rb in developmental, cellular, and cancer biology has stood the test of time and will continue to influence more and more areas of biomedical research, including neuro-protection, stem cell biology, and tissue regeneration.46 Likewise, the impact of uveal melanoma on cancer biology far outweighs its incidence. This cancer provides an important model for studying the pathobiology of tumor progression and metastasis. Though there are still many gaps in our understanding, we can now begin to construct a provisional sequence of major genetic events in uveal melanoma progression (Fig. 7). It is likely that scientific advances in retinoblastoma and uveal melanoma will continue to benefit patients with these potentially lethal cancers, as well as those with a wide array of other maladies.
Figure 7.
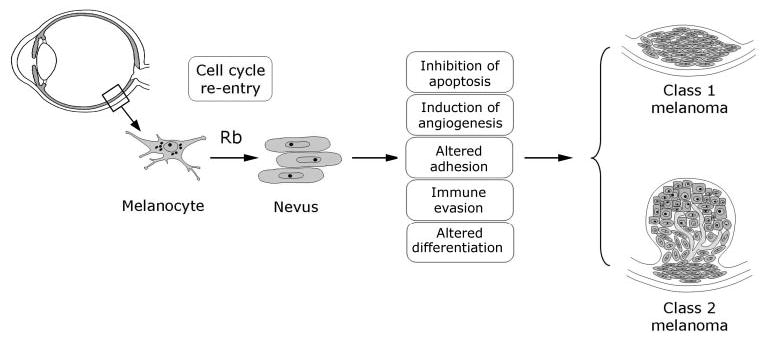
Provisional model of the major genetic events in uveal melanoma. Re-entry of uveal melanocytes into the cell cycle by disruption of the Rb pathway is probably an early event. Limited proliferation may then lead to a population of low-grade neoplastic melanocytes that clinically would be recognized as a nevus. Further mutations that inhibit apoptosis, induce angiogenesis, alter cell-cell and cell–matrix adhesion, alter immunogenicity and dispense with constraints of the differentiated phenotype are probably necessary for the tumor to progress further toward malignancy. Finally, there is a major bifurcation as the melanoma exhibits either a class 1 or 2 molecular profile. Currently, it is not known whether class 1 tumors evolve into class 2 tumors, or whether they progressed along different lines from a very early point in tumor development. Cartoon of a normal eye adapted from image #NEA05 from the Web site of the National Eye Institute, National Institutes of Health.
Acknowledgments
It is an honor for me to receive the Cogan Award, and I am deeply indebted to the many people who have made it possible. First, I thank my wife Tonya, my children, and other loving family members and friends for their patience and support. I especially thank Hugh Browder, whose unwavering friendship has been an anchor through the years. I owe a special thanks to all the people who have worked in my laboratory and contributed to the advances we have made, including Lori Worley, Michael Onken, Rachel Delston, Amy Loercher, Jon Lieman, Justis Ehlers, Yang Sun, Binh Tran, Erica Person, Duan Ma, Ping Zhou, and many others. I thank my teachers and mentors, without whose encouragement and support I would not be where I am today, including (in alphabetical order) Dan Albert, Jim Augsburger, Devron Char, Doug Dean, Stuart Fine, Barrett Haik, Tim Murray, Jerry Shields, Bill Tasman, and Bradley Straatsma. Singular in his impact on my personal and professional life has been the late J. Donald M. Gass, MD, whose brilliance was matched only by his humility and devotion to God and family. I am also blessed by the friendship of many outstanding colleagues from whom I have learned much, including David Abramson, Bertil Damato, Laurence Desjardins, Patrick DePotter, Paul Finger, Robert Folberg, Brenda Gallie, Evan Gragoudas, Hans Grossniklaus, John Hungerford, Martine Jager, Zeynel Karcioglu, Bruce Ksander, Tom Lee, Edoardo Midena, Linn Murphree, Joan M. O’Brien, Jacob Pe’er, Ian Rennie, Stefan Seregard, Carol Shields, Arun Singh, and Matt Wilson. There are many others that I owe a debt of gratitude, and I thank you all.
Footnotes
Supported by National Eye Institute Grants EY00382, EY13169, Career Development and Physician-Scientist Awards from Research to Prevent Blindness, Inc., and grants from the Knights Templar Foundation, Macula Society and Tumori Foundation.
References
- 1.Harbour JW. Retinoblastoma: pathogenesis and diagnosis. In: Char DH, editor. Tumors of the Eye and Orbit. Philadelphia: BC Decker; 2001. pp. 253–265. [Google Scholar]
- 2.Neel JV, Falls HF. The rate of mutation of the gene responsible for retinoblastoma in man. Science. 1951;114:419–422. doi: 10.1126/science.114.2964.419. [DOI] [PubMed] [Google Scholar]
- 3.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friend SH, Bernards R, Rogelj S, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643–646. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- 5.Lee C, Chang JH, Lee HS, Cho Y. Structural basis for the recognition of the E2F transactivation domain by the retinoblastoma tumor suppressor. Genes Dev. 2002;16:3199–3212. doi: 10.1101/gad.1046102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fung YK, Murphree AL, T’Ang A, Qian J, Hinrichs SH, Benedict WF. Structural evidence for the authenticity of the human retinoblastoma gene. Science. 1987;236:1657–1661. doi: 10.1126/science.2885916. [DOI] [PubMed] [Google Scholar]
- 7.Harbour JW. Overview of RB gene mutations in patients with retinoblastoma: implications for clinical genetic screening. Ophthalmology. 1998;105:1442–1447. doi: 10.1016/S0161-6420(98)98025-3. [DOI] [PubMed] [Google Scholar]
- 8.Eng C, Li FP, Abramson DH, et al. Mortality from second tumors among long-term survivors of retinoblastoma. J Natl Cancer Inst. 1993;85:1121–1128. doi: 10.1093/jnci/85.14.1121. [DOI] [PubMed] [Google Scholar]
- 9.Moll AC, Imhof SM, Bouter LM, Tan KE. Second primary tumors in patients with retinoblastoma: a review of the literature. Ophthalmic Genet. 1997;18:27–34. doi: 10.3109/13816819709057880. [DOI] [PubMed] [Google Scholar]
- 10.Harbour JW, Lai SL, Whang-Peng J, Gazdar AF, Minna JD, Kaye FJ. Abnormalities in structure and expression of the human retinoblastoma gene in SCLC. Science. 1988;241:353–357. doi: 10.1126/science.2838909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bookstein R, Rio P, Madreperla SA, et al. Promoter deletion and loss of retinoblastoma gene expression in human prostate carcinoma. Proc Natl Acad Sci USA. 1990;87:7762–7766. doi: 10.1073/pnas.87.19.7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee EY, To H, Shew JY, Bookstein R, Scully P, Lee WH. Inactivation of the retinoblastoma susceptibility gene in human breast cancers. Science. 1988;241:218–221. doi: 10.1126/science.3388033. [DOI] [PubMed] [Google Scholar]
- 13.T’Ang A, Varley JM, Chakraborty S, Murphree AL, Fung YK. Structural rearrangement of the retinoblastoma gene in human breast carcinoma. Science. 1988;242:263–266. doi: 10.1126/science.3175651. [DOI] [PubMed] [Google Scholar]
- 14.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 15.DeCaprio JA, Ludlow JW, Figge J, et al. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell. 1988;54:275–283. doi: 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- 16.Whyte P, Buchkovich KJ, Horowitz JM, et al. Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature. 1988;334:124–129. doi: 10.1038/334124a0. [DOI] [PubMed] [Google Scholar]
- 17.Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 18.Horowitz JM, Park SH, Bogenmann E, et al. Frequent inactivation of the retinoblastoma anti-oncogene is restricted to a subset of human tumor cells. Proc Natl Acad Sci USA. 1990;87:2775–2779. doi: 10.1073/pnas.87.7.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodrich DW, Wang NP, Qian YW, Lee EY, Lee WH. The retinoblastoma gene product regulates progression through the G1 phase of the cell cycle. Cell. 1991;67:293–302. doi: 10.1016/0092-8674(91)90181-w. [DOI] [PubMed] [Google Scholar]
- 20.Kim HY, Cho Y. Structural similarity between the pocket region of retinoblastoma tumour suppressor and the cyclin-box. Nat Struct Biol. 1997;4:390–395. doi: 10.1038/nsb0597-390. [DOI] [PubMed] [Google Scholar]
- 21.Lee JO, Russo AA, Pavletich NP. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature. 1998;391:859–865. doi: 10.1038/36038. [DOI] [PubMed] [Google Scholar]
- 22.Zhang HS, Gavin M, Dahiya A, et al. Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell. 2000;101:79–89. doi: 10.1016/S0092-8674(00)80625-X. [DOI] [PubMed] [Google Scholar]
- 23.Xiao ZX, Chen J, Levine AJ, et al. Interaction between the retinoblastoma protein and the oncoprotein MDM2. Nature. 1995;375:694–698. doi: 10.1038/375694a0. [DOI] [PubMed] [Google Scholar]
- 24.Uchida C, Miwa S, Kitagawa K, et al. Enhanced Mdm2 activity inhibits pRB function via ubiquitin-dependent degradation. EMBO J. 2005;24:160–169. doi: 10.1038/sj.emboj.7600486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. The E2F transcription factor is a cellular target for the RB protein. Cell. 1991;65:1053–1061. doi: 10.1016/0092-8674(91)90557-f. [DOI] [PubMed] [Google Scholar]
- 26.Nevins JR. Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ. 1998;9:585–593. [PubMed] [Google Scholar]
- 27.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 28.Flemington EK, Speck SH, Kaelin WJ. E2F-1-mediated transactivation is inhibited by complex formation with the retinoblastoma susceptibility gene product. Proc Natl Acad Sci USA. 1993;90:6914–6918. doi: 10.1073/pnas.90.15.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Helin K, Harlow E, Fattaey A. Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol Cell Biol. 1993;13:6501–6508. doi: 10.1128/mcb.13.10.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weintraub SJ, Prater CA, Dean DC. Retinoblastoma protein switches the E2F site from positive to negative element. Nature. 1992;358:259–261. doi: 10.1038/358259a0. [DOI] [PubMed] [Google Scholar]
- 31.Harbour JW, Dean DC. Chromatin remodeling and Rb activity. Curr Opin Cell Biol. 2000;12:685–689. doi: 10.1016/s0955-0674(00)00152-6. [DOI] [PubMed] [Google Scholar]
- 32.Frolov MV, Dyson NJ. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J Cell Sci. 2004;117:2173–2181. doi: 10.1242/jcs.01227. [DOI] [PubMed] [Google Scholar]
- 33.Dunaief JL, Strober BE, Guha S, et al. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell. 1994;79:119–130. doi: 10.1016/0092-8674(94)90405-7. [DOI] [PubMed] [Google Scholar]
- 34.Trouche D, Le CC, Muchardt C, Yaniv M, Kouzarides T. RB and hbrm cooperate to repress the activation functions of E2F1. Proc Natl Acad Sci USA. 1997;94:11268–11273. doi: 10.1073/pnas.94.21.11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schumacher A, Magnuson T. Murine Polycomb- and trithorax-group genes regulate homeotic pathways and beyond. Trends Genet. 1997;13:167–170. [PubMed] [Google Scholar]
- 36.Dahiya A, Wong S, Gonzalo S, Gavin M, Dean DC. Linking the Rb and polycomb pathways. Mol Cell. 2001;8:557–569. doi: 10.1016/s1097-2765(01)00346-x. [DOI] [PubMed] [Google Scholar]
- 37.Eden S, Hashimshony T, Keshet I, Cedar H, Thorne AW. DNA methylation models histone acetylation. Nature. 1998;394:842. doi: 10.1038/29680. [DOI] [PubMed] [Google Scholar]
- 38.Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25:338–342. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- 39.Nielsen SJ, Schneider R, Bauer UM, et al. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412:561–565. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- 40.Vandel L, Nicolas E, Vaute O, Ferreira R, Ait-Si-Ali S, Trouche D. Transcriptional repression by the retinoblastoma protein through the recruitment of a histone methyltransferase. Mol Cell Biol. 2001;21:6484–6494. doi: 10.1128/MCB.21.19.6484-6494.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- 42.Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92:463–473. doi: 10.1016/s0092-8674(00)80940-x. [DOI] [PubMed] [Google Scholar]
- 43.Magnaghi JL, Groisman R, Naguibneva I, et al. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- 44.Lai A, Lee JM, Yang WM, et al. RBP1 recruits both histone deacetylase-dependent and -independent repression activities to retinoblastoma family proteins. Mol Cell Biol. 1999;19:6632–6641. doi: 10.1128/mcb.19.10.6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu X, Horvitz HR. lin-35 and lin-53, two genes that antagonize a C. elegans Ras pathway, encode proteins similar to Rb and its binding protein RbAp48. Cell. 1998;95:981–991. doi: 10.1016/s0092-8674(00)81722-5. [DOI] [PubMed] [Google Scholar]
- 46.Panteleeva I, Rouaux C, Larmet Y, Boutillier S, Loeffler JP, Boutillier AL. HDAC-3 participates in the Repression of e2f-dependent gene transcription in primary differentiated neurons. Ann N Y Acad Sci. 2004;1030:656–660. doi: 10.1196/annals.1329.076. [DOI] [PubMed] [Google Scholar]
- 47.Harbour JW, Luo RX, Dei Sante A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859–869. doi: 10.1016/s0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- 48.Chen PL, Scully P, Shew JY, Wang JY, Lee WH. Phosphorylation of the retinoblastoma gene product is modulated during the cell cycle and cellular differentiation. Cell. 1989;58:1193–1198. doi: 10.1016/0092-8674(89)90517-5. [DOI] [PubMed] [Google Scholar]
- 49.Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell. 1992;70:993–1006. doi: 10.1016/0092-8674(92)90249-c. [DOI] [PubMed] [Google Scholar]
- 50.Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 1998;18:753–761. doi: 10.1128/mcb.18.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 52.DeCaprio JA, Ludlow JW, Lynch D, et al. The product of the retinoblastoma susceptibility gene has properties of a cell cycle regulatory element. Cell. 1989;58:1085–1095. doi: 10.1016/0092-8674(89)90507-2. [DOI] [PubMed] [Google Scholar]
- 53.Buchkovich K, Duffy LA, Harlow E. The retinoblastoma protein is phosphorylated during specific phases of the cell cycle. Cell. 1989;58:1097–1105. doi: 10.1016/0092-8674(89)90508-4. [DOI] [PubMed] [Google Scholar]
- 54.Knudsen ES, Wang JY. Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J Biol Chem. 1996;271:8313–8320. doi: 10.1074/jbc.271.14.8313. [DOI] [PubMed] [Google Scholar]
- 55.Knudsen ES, Wang JY. Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol Cell Biol. 1997;17:5771–5783. doi: 10.1128/mcb.17.10.5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mackeigan JP, Murphy LO, Dimitri CA, Blenis J. Graded mitogen-activated protein kinase activity precedes switch-like c-Fos induction in mammalian cells. Mol Cell Biol. 2005;25:4676–4682. doi: 10.1128/MCB.25.11.4676-4682.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee EY, Chang CY, Hu N, et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 58.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 59.Clarke AR, Maandag ER, van Roon M, et al. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–330. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- 60.Morgenbesser SD, Williams BO, Jacks T, Depinho RA. p53-dependent apoptosis produced by Rb-deficiency in the developing mouse lens. Nature. 1994;371:72–74. doi: 10.1038/371072a0. [DOI] [PubMed] [Google Scholar]
- 61.Howes KA, Ransom N, Papermaster DS, Lasudry JG, Albert DM, Windle JJ. Apoptosis or retinoblastoma: alternative fates of photoreceptors expressing the HPV-16 E7 gene in the presence or absence of p53. Genes Dev. 1994;8:1300–1310. doi: 10.1101/gad.8.11.1300. [DOI] [PubMed] [Google Scholar]
- 62.Irwin M, Marin MC, Phillips AC, et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature. 2000;407:645–648. doi: 10.1038/35036614. [DOI] [PubMed] [Google Scholar]
- 63.Qin XQ, Livingston DM, Kaelin WJ, Adams PD. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci USA. 1994;91:10918–10922. doi: 10.1073/pnas.91.23.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Young AP, Longmore GD. Differential regulation of apoptotic genes by Rb in human versus mouse cells. Oncogene. 2004;23:2587–2599. doi: 10.1038/sj.onc.1207330. [DOI] [PubMed] [Google Scholar]
- 65.Hsieh JK, Fredersdorf S, Kouzarides T, Martin K, Lu X. E2F1-induced apoptosis requires DNA binding but not transactivation and is inhibited by the retinoblastoma protein through direct interaction. Genes Dev. 1997;11:1840–1852. doi: 10.1101/gad.11.14.1840. [DOI] [PubMed] [Google Scholar]
- 66.Phillips AC, Bates S, Ryan KM, Helin K, Vousden KH. Induction of DNA synthesis and apoptosis are separable functions of E2F- 1. Genes Dev. 1997;11:1853–1863. doi: 10.1101/gad.11.14.1853. [DOI] [PubMed] [Google Scholar]
- 67.Lieman JH, Worley LA, Harbour JW. Loss of Rb-E2F repression results in caspase-8-mediated apoptosis through inactivation of focal adhesion kinase. J Biol Chem. 2005;280:10484–10490. doi: 10.1074/jbc.M409371200. [DOI] [PubMed] [Google Scholar]
- 68.Ezhevsky SA, Nagahara H, Vocero AA, Gius DR, Wei MC, Dowdy SF. Hypo-phosphorylation of the retinoblastoma protein (pRb) by cyclin D:Cdk4/6 complexes results in active pRb. Proc Natl Acad Sci USA. 1997;94:10699–10704. doi: 10.1073/pnas.94.20.10699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ezhevsky SA, Ho A, Becker-Hapak M, Davis PK, Dowdy SF. Differential regulation of retinoblastoma tumor suppressor protein by G(1) cyclin-dependent kinase complexes in vivo. Mol Cell Biol. 2001;21:4773–4784. doi: 10.1128/MCB.21.14.4773-4784.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wells J, Boyd KE, Fry CJ, Bartley SM, Farnham PJ. Target gene specificity of E2F and pocket protein family members in living cells. Mol Cell Biol. 2000;20:5797–5807. doi: 10.1128/mcb.20.16.5797-5807.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wells J, Yan PS, Cechvala M, Huang T, Farnham PJ. Identification of novel pRb binding sites using CpG microarrays suggests that E2F recruits pRb to specific genomic sites during S phase. Oncogene. 2003;22:1445–1460. doi: 10.1038/sj.onc.1206264. [DOI] [PubMed] [Google Scholar]
- 72.Sage J, Mulligan GJ, Attardi LD, et al. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 2000;14:3037–3050. doi: 10.1101/gad.843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen D, Livnebar I, Vanderluit JL, Slack RS, Agochiya M, Bremner R. Cell-specific effects of RB or RB/p107 loss on retinal development implicate an intrinsically death-resistant cell-of-origin in retinoblastoma. Cancer Cell. 2004;5:539–551. doi: 10.1016/j.ccr.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 74.Loercher AE, Tank EM, Delston RB, Harbour JW. MITF links differentiation with cell cycle arrest in melanocytes by transcriptional activation of INK4A. J Cell Biol. 2005;168:35–40. doi: 10.1083/jcb.200410115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dannenberg JH, van Rossum A, Schuijff L, te Riele H. Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev. 2000;14:3051–3064. doi: 10.1101/gad.847700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gu W, Schneider JW, Condorelli G, Kaushal S, Mahdavi V, Nadal-Ginard B. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell. 1993;72:309–324. doi: 10.1016/0092-8674(93)90110-c. [DOI] [PubMed] [Google Scholar]
- 77.Sellers WR, Novitch BG, Miyake S, et al. Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Genes Dev. 1998;12:95–106. doi: 10.1101/gad.12.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu BD, Becker-Hapak M, Snyder EL, Vooijs M, Denicourt C, Dowdy SF. Distinct and nonoverlapping roles for pRB and cyclin D:cyclin-dependent kinases 4/6 activity in melanocyte survival. Proc Natl Acad Sci USA. 2003;100:14881–14486. doi: 10.1073/pnas.2431391100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harbour JW. Clinical overview of uveal melanoma: introduction to tumors of the eye. In: Albert DM, Polans A, editors. Ocular Oncology. New York: Marcel Dekker; 2003. pp. 1–18. [Google Scholar]
- 80.Singh AD, Topham A. Incidence of uveal melanoma in the United States: 1973–1997. Ophthalmology. 2003;110:956–961. doi: 10.1016/S0161-6420(03)00078-2. [DOI] [PubMed] [Google Scholar]
- 81.Loercher AE, Harbour JW. Molecular genetics of uveal melanoma. Curr Eye Res. 2003;27:69–74. doi: 10.1076/ceyr.27.2.69.15952. [DOI] [PubMed] [Google Scholar]
- 82.Brantley MA, Jr, Harbour JW. Inactivation of retinoblastoma protein in uveal melanoma by phosphorylation of sites in the COOH-terminal region. Cancer Res. 2000;60:4320–4323. [PMC free article] [PubMed] [Google Scholar]
- 83.Brantley MA, Jr, Harbour JW. Deregulation of the Rb and p53 pathways in uveal melanoma. Am J Pathol. 2000;157:1795–1801. doi: 10.1016/s0002-9440(10)64817-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sun Y, Tran BN, Worley LA, Delston RB, Harbour JW. Functional analysis of the p53 pathway in response to ionizing radiation in uveal melanoma. Invest Ophthalmol Vis Sci. 2005;46:1561–1564. doi: 10.1167/iovs.04-1362. [DOI] [PubMed] [Google Scholar]
- 85.Coupland SE, Anastassiou G, Stang A, et al. The prognostic value of cyclin D1, p53, and MDM2 protein expression in uveal melanoma. J Pathol. 2000;191:120–126. doi: 10.1002/(SICI)1096-9896(200006)191:2<120::AID-PATH591>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 86.Harbour JW, Worley L, Ma D, Cohen M. Transducible peptide therapy for uveal melanoma and retinoblastoma. Arch Ophthalmol. 2002;120:1341–1346. doi: 10.1001/archopht.120.10.1341. [DOI] [PubMed] [Google Scholar]
- 87.Coupland SE, Bechrakis N, Schuler A, et al. Expression patterns of cyclin D1 and related proteins regulating G1-S phase transition in uveal melanoma and retinoblastoma. Br J Ophthalmol. 1998;82:961–970. doi: 10.1136/bjo.82.8.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van der Velden PA, Metzelaar-Blok JA, Bergman W, et al. Promoter hypermethylation: a common cause of reduced p16(INK4a) expression in uveal melanoma. Cancer Res. 2001;61:5303–5306. [PubMed] [Google Scholar]
- 89.Zuidervaart W, van Nieuwpoort F, Stark M, et al. Activation of the MAPK pathway is a common event in uveal melanomas although it rarely occurs through mutation of BRAF or RAS. Br J Cancer. 2005;92:2032–2038. doi: 10.1038/sj.bjc.6602598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mulloy R, Salinas S, Philips A, Hipskind RA. Activation of cyclin D1 expression by the ERK5 cascade. Oncogene. 2003;22:5387–5398. doi: 10.1038/sj.onc.1206839. [DOI] [PubMed] [Google Scholar]
- 91.Shackney SE, Shankey TV. Cell cycle models for molecular biology and molecular oncology: exploring new dimensions. Cytometry. 1999;35:97–116. doi: 10.1002/(sici)1097-0320(19990201)35:2<97::aid-cyto1>3.3.co;2-x. [DOI] [PubMed] [Google Scholar]
- 92.Eskelin S, Pyrhonen S, Summanen P, Hahka-Kemppinen M, Kivela T. Tumor doubling times in metastatic malignant melanoma of the uvea: tumor progression before and after treatment. Ophthalmology. 2000;107:1443–1449. doi: 10.1016/s0161-6420(00)00182-2. [DOI] [PubMed] [Google Scholar]
- 93.Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 94.Tschentscher F, Prescher G, Zeschnigk M, Horsthemke B, Lohmann DR. Identification of chromosomes 3, 6, and 8 aberrations in uveal melanoma by microsatellite analysis in comparison to comparative genomic hybridization. Cancer Genet Cytogenet. 2000;122:13–17. doi: 10.1016/s0165-4608(00)00266-1. [DOI] [PubMed] [Google Scholar]
- 95.Mooy CM, De Jong PT. Prognostic parameters in uveal melanoma: a review. Surv Ophthalmol. 1996;41:215–228. doi: 10.1016/s0039-6257(96)80024-5. [DOI] [PubMed] [Google Scholar]
- 96.Makitie T, Summanen P, Tarkkanen A, Kivela T. Microvascular loops and networks as prognostic indicators in choroidal and ciliary body melanomas. J Natl Cancer Inst. 1999;91:359–367. doi: 10.1093/jnci/91.4.359. [DOI] [PubMed] [Google Scholar]
- 97.Augsburger JJ, Gamel JW. Clinical prognostic factors in patients with posterior uveal malignant melanoma. Cancer. 1990;66:1596–1600. doi: 10.1002/1097-0142(19901001)66:7<1596::aid-cncr2820660726>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 98.Seddon JM, Polivogianis L, Hsieh CC, Albert DM, Gamel JW, Gragoudas ES. Death from uveal melanoma: number of epithelioid cells and inverse SD of nucleolar area as prognostic factors. Arch Ophthalmol. 1987;105:801–806. doi: 10.1001/archopht.1987.01060060087039. [DOI] [PubMed] [Google Scholar]
- 99.Folberg R, Pe’er J, Gruman LM, et al. The morphologic characteristics of tumor blood vessels as a marker of tumor progression in primary human uveal melanoma: a matched case-control study. Hum Pathol. 1992;23:1298–1305. doi: 10.1016/0046-8177(92)90299-i. [DOI] [PubMed] [Google Scholar]
- 100.Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004;64:7205–7209. doi: 10.1158/0008-5472.CAN-04-1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Onken MD, Lin AY, Worley LA, Folberg R, Harbour JW. Association between microarray gene expression signature and extravascular matrix patterns in primary uveal melanomas. Am J Ophthalmol 2005. 2005;140:748–749. doi: 10.1016/j.ajo.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 102.Tschentscher F, Husing J, Holter T, et al. Tumor classification based on gene expression profiling shows that uveal melanomas with and without monosomy 3 represent two distinct entities. Cancer Res. 2003;63:2578–2584. [PubMed] [Google Scholar]
- 103.Halliwell B. Oxidative stress in cell culture: an under-appreciated problem? FEBS Lett. 2003;540:3–6. doi: 10.1016/s0014-5793(03)00235-7. [DOI] [PubMed] [Google Scholar]
- 104.Ehlers JP, Harbour JW. NBS1 expression as a prognostic marker in uveal melanoma. Clin Cancer Res. 2005;11:1849–1853. doi: 10.1158/1078-0432.CCR-04-2054. [DOI] [PubMed] [Google Scholar]
- 105.Ehlers JP, Worley LA, Onken MD, Harbour JW. DDEF1 is located in an amplified region of chromosome 8q and is overexpressed in uveal melanoma. Clin Cancer Res. 2005;11:3609–3613. doi: 10.1158/1078-0432.CCR-04-1941. [DOI] [PubMed] [Google Scholar]
- 106.Smith SL, Damato BE, Scholes AG, Nunn J, Field JK, Heighway J. Decreased endothelin receptor B expression in large primary uveal melanomas is associated with early clinical metastasis and short survival. Br J Cancer. 2002;87:1308–1313. doi: 10.1038/sj.bjc.6600620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zuidervaart W, van der Velden PA, Hurks MH, et al. Gene expression profiling identifies tumour markers potentially playing a role in uveal melanoma development. Br J Cancer. 2003;89:1914–1919. doi: 10.1038/sj.bjc.6601374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.van der Velden PA, Zuidervaart W, Hurks MH, et al. Expression profiling reveals that methylation of TIMP3 is involved in uveal melanoma development. Int J Cancer. 2003;106:472–479. doi: 10.1002/ijc.11262. [DOI] [PubMed] [Google Scholar]