

Abstract
The soluble epoxide hydrolase (sEH) plays a significant role in the biosynthesis of inflammation mediators as well as xenobiotic transformations. Herein, we report the discovery of substituted ureas and carbamates as potent inhibitors of sEH. Some of these selective, competitive tight-binding inhibitors with nanomolar Ki values interacted stoichiometrically with the homogenous recombinant murine and human sEHs. These inhibitors enhance cytotoxicity of trans-stilbene oxide, which is active as the epoxide, but reduce cytotoxicity of leukotoxin, which is activated by epoxide hydrolase to its toxic diol. They also reduce toxicity of leukotoxin in vivo in mice and prevent symptoms suggestive of acute respiratory distress syndrome. These potent inhibitors may be valuable tools for testing hypotheses of involvement of diol and epoxide lipids in chemical mediation in vitro or in vivo systems.
Epoxide hydrolases (EH; E.C.3.3.2.3) catalyze the hydrolysis of epoxides or arene oxides to their corresponding diols by the addition of water (1). In mammals, the hepatic microsomal and soluble epoxide hydrolase forms are known to complement each other in detoxifying a wide array of mutagenic, toxic, and carcinogenic, xenobiotic epoxides (2, 3). Soluble EH (sEH) is also involved in the metabolism of arachidonic (4) and linoleic (5) acid epoxides. Arachidonate epoxides and diols are elevated in association with pregnancy-induced hypertension and modulate vascular permeability in the heart and kidneys (6). Diols derived from epoxy-linoleate (leukotoxin) perturb membrane permeability and calcium homeostasis (5), resulting in inflammation modulated by nitric oxide synthase and endothelin-1 (7, 8). Micromolar concentrations of leukotoxin reported in association with inflammation and hypoxia (9) depress mitochondrial respiration in vitro (10) and cause mammalian cardiopulmonary toxicity in vivo (7, 11, 12). Leukotoxin toxicity presents symptoms suggestive of multiple organ failure and acute respiratory distress syndrome (9). In both cellular and organismal models, leukotoxin-mediated toxicity depends on epoxide hydrolysis (5).
The bioactivity of these epoxide hydrolysis products and their association with inflammation suggest that inhibition of vicinal-dihydroxylipid biosynthesis may have therapeutic value, making sEH a promising pharmacological target. Previously described selective sEH inhibitors, substituted chalcone oxides (as compound 1 in Table 1), and phenylglycidols (13, 14) are epoxides that are hydrolyzed slowly by the target enzyme. Inhibition stems from an electronically stabilized covalent intermediate that results in low turnover and transient inhibition (15). Moreover, these compounds are relatively unstable, particularly in the presence of glutathione (13), making them of limited use in vivo. We describe herein the discovery of new potent and stable inhibitors of soluble EH and their application to both in vitro and in vivo models.
Table 1.
Inhibition of MsEH and HsEH by several pharmacophores.
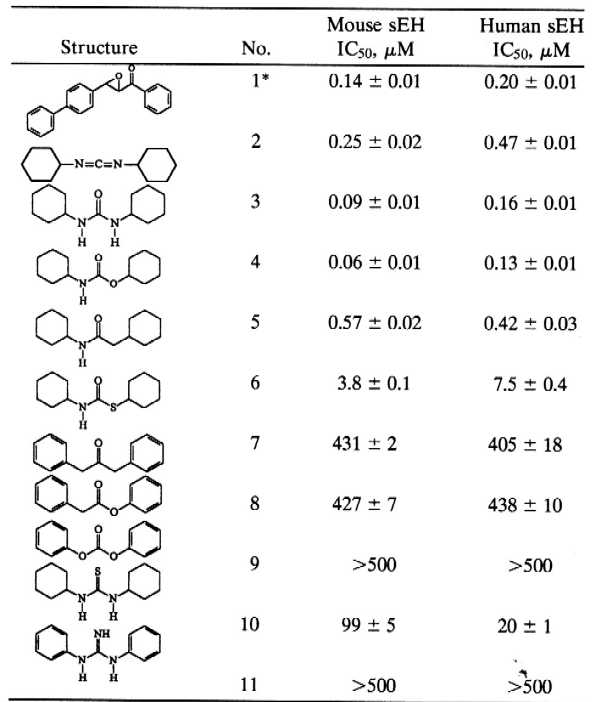 |
Enzymes (0.12 μM MsEH or 0.24 μM HsEH) were incubated with inhibitors for 5 min in sodium phosphate buffer (pH 7.4) at 30°C before substrate introduction ([S] = 40 μM). IC90-to-IC50 ratios between 5 and 10 were found.
Results are means ± SD of three separate experiments.
From Morisseau et al. (15).
MATERIALS AND METHODS
Synthesis.
Compounds 2, 3, 7–9, and 11–18 were obtained from Aldrich. Compounds 10 and 19– 21 were synthesized by the condensation of the appropriate isocyanate or isothiocyanate and amine, whereas compounds 4 and 6 were synthesized by the condensation of the appropriate isocyanate and alcohol or thiol. Reaction products were purified by recrystallization and characterized structurally by 1H- and/or 13C-NMR, infrared, and mass spectroscopy. As an example, synthesis of compound 20 is described below.
N-Cyclohexyl-N′-(3-phenylpropyl)-Urea 20.
To a stirred solution of 0.406 g (3.00 mmol) of 3-phenyl-1-propylamine in 20 ml of hexane was added 0.40 ml (0.39 g, 3.1 mmol) of cyclohexylisocyanate over 5 min. After 3 days at ambient temperature, a white solid was obtained, which, after thorough washing with cold hexane, provided 0.773 g (99%) of the title compound, mp 103.5–104.0°C. IR (KBr): 3,331 (s, NH), 1,621 (vs, C⩵O), 1,581 (s, amide II) cm−1. 1H-NMR (CDCl3): δ 7.2 (m, 2 H, 2 ArH), 7.1 (m, 3 H, 3 ArH), 5.05 (s, 1 H, NH), 4.9 (m, 1 H, NH), 3.5 (m, 1 H, CH), 3.17 (dd, J = 6.5 Hz, 6.1 Hz, 2 H, CH2N), 2.62 (t, J = 7.4 Hz, 2 H, ArCH2), 1.8 (m, 4 H, C—CH2—C plus 2 H of cyclohexyl), 1.6 (m, 3 H, 3 H of cyclohexyl), 1.3 (m, 2 H, 2 H of cyclohexyl), 1.1 (m, 3 H, 3 H of cyclohexyl) ppm. 13C-NMR (CDCl3): δ 158.1 (C⩵O), 141.7 (ArC-1), 128.3 (ArC-2, 3, 5, 6), 125.8 (ArC-4), 48.9 (CH-1), 40.0 (CH2N), 33.9 (CH2-2, 6), 33.2 (ArCH2), 32.0 (C—CH2—C), 25.6 (CH2-4), 24.9 (CH2-3, 5) ppm. FAB-MS m/z (relative intensity): 521 (1, 2 M + H+), 262 (22, M + H+ + 1), 261 (100, M + H+). High-resolution MS m/z: 261.1964 (Th. 261.1967).
Enzyme Preparation.
Recombinant mouse sEH (MsEH) and human sEH (HsEH) were produced in a baculovirus expression system (16, 17) and purified by affinity chromatography (18). The preparations were at least 97% pure as judged by SDS/PAGE and scanning densitometry. No detectable esterase or glutathione transferase activity, which can interfere with this sEH assay, was observed (19). Protein concentration was quantified by using the Pierce BCA assay using BSA as the calibrating standard.
IC50 Assay Conditions.
IC50 and IC90 values were determined as described by using racemic 4-nitrophenyl-trans-2,3-epoxy-3-phenylpropyl carbonate as substrate (19). Enzymes (0.12 μM MsEH or 0.24 μM HsEH) were incubated with inhibitors for 5 min in sodium phosphate buffer, pH 7.4, at 30°C before substrate introduction ([S] = 40 μM). Activity was assessed by measuring the appearance of the 4-nitrophenolate anion at 405 nm at 30°C during 1 min (Spectramax 200; Molecular Devices). Assays were performed in triplicate. By definition, IC50 and IC90 are concentrations of inhibitor, which reduce enzyme activity by 50 and 90%, respectively. IC50 and IC90 were determined by regression of at least five datum points with a minimum of two points in the linear region of the curve on either side of the IC50 or IC90. The curve was generated from at least three separate runs, each in triplicate, to obtain the SD in Tables 1 and 2.
Table 2.
Inhibition of MsEH and HsEH ureas
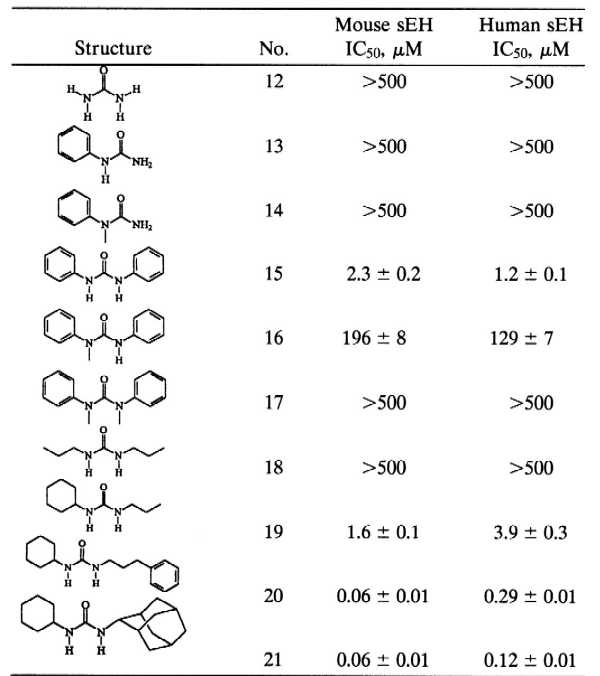 |
Enzymes (0.12 μM MsEH or 0.24 μM HsEH) were incubated with inhibitors for 5 min in sodium phosphate buffer (pH 7.4) at 30°C before substrate introduction ([S] = 40 μM).
Results are means ± SD of three separate experiments.
Kinetic Assay Conditions.
Dissociation constants were determined by following the method described by Dixon (20) for competitive tight-binding inhibitors, using [3H]-1,3-diphenyl-trans-propene oxide (t-DPPO) as substrate (21). Inhibitors at concentrations between 0 and 100 nM for 3 and 0 and 10 nM for 20 were incubated in triplicate for 5 min in sodium phosphate buffer, pH 7.4, at 30°C with the enzyme (2 nM MsEH or 3 nM HsEH). Substrate (2.5 ≤ [S]final ≤ 50 μM) then was added. Velocity was measured as described (21). For each substrate concentration, the plots of the velocity as a function of the inhibitor concentration (Fig. 1) allow the determination of an apparent inhibition constant (Kiapp) (20). The plot of these Kiapp values as a function of the substrate concentration (Fig. 1 Inset) allows the determination of Ki when [S] = 0. The results are mean ± SD of three separate determinations of Ki.
Figure 1.

Determination of the Ki of compound 3 with the MsEH (2 nM) by using [3H]-1,3-diphenyl-trans-propene oxide as substrate (21). For each substrate concentration (2.5–50.0 μM), the velocity is plotted as a function of the inhibitor concentration (0–100 nM), allowing the determination of an apparent inhibition constant (Kiapp) (20). Kiapp values are plotted as a function of the substrate concentration (Inset). For [S] = 0, a Ki value of 26 nM is found. Similar plots were obtained with the human enzyme and 3 (Ki = 30 nM) and the murine enzyme and 20 (Ki = 3 nM).
RESULTS
Mechanistic studies of EHs have demonstrated the presence of an important aspartate residue at the catalytic site (2, 22). Initial attempts at sEH inhibition focused on carboxylate-modifying agents. Dicyclohexylcarbodiimide 2, classically used in peptide synthesis (23), gave the best results (Table 1). However, a kinetic study showed that in the buffer solution used, this compound was hydrolyzed rapidly to the corresponding urea 3 (N,N′-dicyclohexylurea; DCU), which is, in fact, the active inhibitor. To optimize the pharmacophore for inhibition, several structural analogs were tested (Table 1). Compared with the urea 3, the replacement of a single nitrogen by an oxygen, to give the carbamate 4, slightly increased inhibitor potency. Analogous replacement with a methylene, yielding the amide 5, or with a sulfur, yielding the thiocarbamate 6, resulted in 5- and 50-fold-less potent inhibitors, respectively. Moreover, structures without a nitrogen atom on either side of the carbonyl function, such as compounds 7–9, were poor sEH inhibitors. Finally, replacing the oxygen of the urea function with a sulfur, compound 10 compared with 3, or with an imino group, compound 11 compared with 15, resulted in far less active inhibitors.
We then explored the effect of N-substitution on the urea pharmacophore on sEH inhibition potency (Table 2). From the nonsubstituted to the tetra-substituted ureas (12–17), inhibition was obtained only for the 1,3-disubstituted 15 or the 1,1,3-trisubstituted 16 ureas, with the former being 100-fold more potent than the latter. These results indicated that at least one hydrogen on one nitrogen of the urea function is necessary for the inhibition potency and agreed with the results observed for compounds 4, 5, and 6 compared with 3. Comparison of inhibition obtained with compounds 18 and 19 to compound 3 indicated that the best inhibition is obtained when two relatively big groups are present on each side of the urea function. Moreover, two cyclohexyl groups 3, which are more hydrophobic and not planar, gave better inhibition potency than two phenyl groups 15. Better inhibition potency is obtained when one of these two groups is larger, as illustrated by compounds 20 and 21. Over the series of compounds tested, the relative inhibitor potencies between the recombinant murine and human enzymes were similar. Thus, the mouse enzyme is a reasonable model for the development of human sEH inhibitors.
Compared with phenylchalcone oxide 1, the most potent sEH inhibitor reported previously (13, 15), the best new compounds (3, 4, 20, and 21) have similar or better IC50 values after 5 min of incubation. After 30 min, a 5-fold increase in the IC50 of 1 is observed (15), whereas those of compounds 3, 4, 20, and 21 are unchanged, indicating that these compounds are not transformed by the sEH as is 1 (15). Moreover, an additive effect is observed for the inhibition of the HsEH by compound 1 when 25 nM of compound 20 is added to the enzyme solution (Fig. 2). Such a result suggested that the chalcone oxides and ureas act at the same site on the enzyme. The chalcone oxides have been shown to act at the catalytic site of the EH (13, 15). Fast-gel filtration (Bio-Spin column P-30; Bio-Rad) of an aqueous solution of MsEH (3.3 μM) and DCU 3 (100 μM) permitted the recovery of more than 90% of the activity, indicating a rapid dissociation of the inhibitor from the enzyme. IC50 values obtained for compounds 4, 20 and 4, 21 for the MsEH and the HsEH, respectively, are equal to half of the enzyme present, indicating a tight inhibition of the enzyme by these compounds. To test this hypothesis, the dissociation constant of DCU 3 and 20 [N-cyclohexyl-N′-(3-phenyl)propylurea; CPU] was determined (Fig. 1). Ki values of 22 ± 3 and 30 ± 2 nM were obtained for the DCU with MsEH and HsEH, respectively, whereas a Ki of 3.1 ± 0.2 nM was obtained for MsEH in the presence of CPU. These values are between 1- and 10-fold the enzyme concentrations, supporting a tight association between these inhibitors and the enzymes and that they acted at stoichiometric concentrations. For the MsEH, these inhibitors have dissociation constants 20- to 150-fold better than compound 1 (Kd = 430 nM) (15) and 100- to 1,000-fold better than the Km values of the best-known endogenous and surrogate substrates (2.5 < Km < 11 μM) (4, 21). Lineweaver–Burk analyses of some weaker inhibitors in this series support the hypothesis that they act in a purely competitive fashion.
Figure 2.

The inhibition effect of phenyl chalcone oxide 1 (from 1 to 3 × 104 nM) was assayed on HsEH (240 nM) as described (15) in the absence (●) or presence (⋄) of 25 nM PCU 20. The open bar indicates the inhibitory effect of 25 nM CPU 20. Results are means ± SDs of three separate experiments. An additive effect is observed.
Before testing the effect of ureas on epoxide metabolism, the effects of DCU (100 μM) on activities of carboxylesterases, glutathione transferases, EHs, four P450s, and a CoA-oxidase from rat liver were evaluated. Significant inhibition is observed only for cytosolic and peroxisomal EH activities, indicating selectivity of this compound for sEH. The effect of DCU 3 on the metabolism of two epoxides was examined in vitro with cultured cells (Fig. 3). The toxicity of trans-stilbene oxide (Fig. 3a) is reduced strongly by the expression of the MsEH, resulting in increased cell viability. In the presence of 60 μM DCU, MsEH protection is lost and toxicity similar to that obtained with cells expressing β-galactosidase (Lac Z) is observed. For the 1:1 mixture of leukotoxin/isoleukotoxin methyl esters (MeLTX/ILTX) (Fig. 3b), the expression of MsEH results in an increased toxicity as observed previously (5). Addition of DCU protects cells expressing MsEH from the toxicity of leukotoxin.
Figure 3.

Viability of cultured cells of Spodoptera frugiperda (Sf-21) expressing the murine sEH (MsEH) in the absence (●) or presence (▴) of compound 3 at 60 μM. Cells expressing β-galactosidase (Lac Z; ♦) were used as control. (a) Effect of trans-stilbene oxide from 0 to 1 mM. sEH detoxifies trans-stilbene oxide (Inset) to its diol, and the cytotoxicity of stilbene oxide is enhanced by the sEH inhibitor DCU. (b) Effect of leukotoxin/isoleukotoxin methyl ester from 0 to 1 mM. sEH activates leukotoxin to its toxic diol, and the cytotoxicity of this compound is reduced dramatically by the sEH inhibitor DCU. Results are means of three separate experiments; bars = SD. Experiments were performed as described previously (32).
A similar protection from leukotoxin toxicity also is observed in vivo. Male Swiss–Webster mice (25–30 g, four per group; Simonsen Laboratories, Gilroy, CA) were pretreated i.p. with DCU 3 suspended in corn oil (400 mg/kg, 7 ml/kg) or corn oil as control. After 30 min of pretreatment, the mice were exposed to MeLTX/ILTX in ethanol (700 mg/kg, 1.0 ml/kg) or a 1:1 mixture of leukotoxin/isoleukotoxin diols in ethanol (350 mg/kg, 1.0 ml/kg) by tail-vein injection. Mice died shortly (less than 1 hr) after exposure to MeLTX/ILTX from respiratory distress. Death follows accelerated breathing and gasping and is associated with a bloody nasal discharge. Pretreatment with DCU 3 either lengthens the life of the mice at least 4-fold or blocks death induced by leukotoxin/isoleukotoxin entirely. However, as expected, DCU did not demonstrate a protective effect on leukotoxin diol-induced toxicity (5). The results indicated that ureas can influence sEH activity in vivo and may prove valuable for the treatment of diseases such as acute respiratory distress syndrome, where sEH activation of epoxy lipids can lead to tissue damage (unpublished data).
DISCUSSION
We investigated the effect of several pharmacophores on the inhibition of sEH. The results obtained clearly show that compounds containing a carbamide function, especially ureas and carbamates, represent a new class of potent sEH inhibitors. Moreover, the inhibition depends on the presence of at least one hydrogen on the nitrogen(s) of the active pharmacophore and is enhanced by 1,3-disubstitutions. These two substitutions need to be relatively large and hydrophobic and do not need to be of the same size. Unlike with previously described sEH inhibitors (13–15), inhibition caused by these compounds does not decrease over time. Moreover, the more potent compounds have nanomolar Ki values, indicating that this new class of sEH inhibitors interacts stoichiometrically with the murine and human sEHs and that they act as competitive tight-binding inhibitors.
The exact mechanism by which the ureas, carbamates, and related compounds inhibit the sEH is not known. However, their high potency suggests that they may mimic one or more of the transition states along the reaction coordinate leading from epoxide to product diol. The best inhibitors appear to have one hydrophobic group on each side of the urea. This finding agrees with the structure–activity relationships found among both substrates (3) and previously described inhibitors (15). Moreover, it suggests that the best inhibitors are able to interact with a hydrophobic pocket on either side of the catalytic site. These hypothetical hydrophobic pockets, indicated by hatch marks in Fig. 4, are not necessarily equivalent (13, 15), with one site able to accommodate a much larger substitutent (R′). In normal catalysis, the epoxide carbon of the substrate is attacked by Asp333 to yield a covalent intermediate termed the hydroxyalkyl enzyme (2, 22). The nucleophilic nature of Asp333 suggests a possible tight association between the urea inhibitor and this residue. However, the pKa values of ureas and carbamates (24) are high enough that the formation of a true base in the catalytic site as is shown at the bottom of Fig. 4 is not likely. Based on the mechanism of the related haloalkane dehalogenase (2) and on kinetic measurements (15), in normal catalysis one or more amino acids are likely involved in polarizing the epoxide to facilitate the nucleophilic Asp333 attack. The carbonyl of the inhibitor may be polarized by this group (marked P for proton donor; Fig. 4), forcing the compound into a form approaching the charged base in Fig. 4a. Alternatively, in a less conventional way, the electron doublet of the inhibitor nitrogen could interact with the hydrogen carried by P (Fig. 4b) and also could result in a form approaching the charged base drawn at the bottom of Fig. 4b. These products could form two or more hydrogen bonds and a partial salt bridge with the enzyme and, thus, mimic a hypothetical transition state between the enzyme and its substrate.
Figure 4.

Hypothetical mechanism of sEH inhibition by ureas or carbamates. Asp333 has been established as the catalytic nucleophile, and P-H is a hypothetical proton donor.
Numerous pesticides have pharmacophores similar to the new inhibitors described herein (25), and, thus, they may present a risk by inhibiting the sEH. However, from all the agrochemical compounds we tested, only Siduron, a phenylurea herbicide, is a very potent inhibitor, with IC50 values of 0.16 and 0.36 μM for MsEH and HsEH, respectively. This compound is metabolized poorly by plants and has a soil half-life of 4–6 months (25); however, only a small quantity of this compound is used annually (26), indicating a low potential risk. Pesticide compounds tested were of analytic grade (purity >99%); however, technical material used in the pesticide formulations often contains trace contaminants. For example, technical-grade Diuron has been reported to contain up to 3% of N,N′-di-(3,4-dichloro)-phenylurea (27). The sEH inhibition potency of this contaminant (IC50 = 1 μM for MsEH) is approximately 100-fold greater than that of the pure Diuron [the Diuron used here contains 0.05% of the contaminant as determined by HPLC (27)]. One percent contamination of the Diuron, a percentage commonly found in technical grade Diuron (27), resulted in a 2-fold increase in the apparent inhibitory potency of the Diuron. This result highlights potential effects from trace contaminants in technical mixtures.
The more powerful inhibitors described herein were able to block sEH activity in vitro and in vivo in mice. Ureas, carbamates, and related compounds represent powerful tools to explore the toxicological and pharmacological roles of sEH. These compounds are potential leads for the development of new therapeutic drugs for the treatment of epoxy-lipid-induced symptoms in conditions such as acute respiratory distress syndrome, which affects more than 150,000 individuals per year in the United States with >50% mortality (28). Similar pharmacophores likely will yield inhibitors of other epoxide hydrolases of importance in insects, plants, and mammals (2, 29, 30). As a cautionary note, however, high-level exposure to such therapeutic drugs or other inhibitors could alter both our normal inflammatory regulation and ability to transform xenobiotics. Additionally, if complimentary epoxide-detoxification pathways are compromised, an increased health risk may result (31).
Acknowledgments
We thank A. D. Jones (Pennsylvania State University) for performing the mass spectra analysis and J. W. Newman for helpful discussions. This work was supported in part by National Institute on Environmental Health Sciences (NIEHS) Grant R01-ES02710, NIEHS Superfund Basic Research Program ES04699, NIEHS Center for Environmental Health Sciences Grant 1P30-ES05707, and University of California at Davis Environmental Protection Agency Center for Ecological Health Research Grant CR819658.
ABBREVIATIONS
- EH
epoxide hydrolase
- sEH
soluble EH
- MsEH
mouse sEH
- HsEH
human sEH
- DCU
N,N′-dicyclohexylurea
References
- 1.Oesch F. Xenobiotica. 1972;3:305–340. doi: 10.3109/00498257309151525. [DOI] [PubMed] [Google Scholar]
- 2.Hammock B D, Grant D, Storms D. In: Comprehensive Toxicology. Sipes I, McQueen C, Gandolfi A, editors. Oxford: Pergamon; 1997. pp. 283–305. [Google Scholar]
- 3.Wixtrom R N, Hammock B D. In: Biochemical Pharmacology and Toxicology. Zakim D, Vessey D A, editors. New York: Wiley; 1985. pp. 1–93. [Google Scholar]
- 4.Zeldin D C, Wei S, Falck J R, Hammock B D, Snapper J R, Capdevila J H. Arch Biochem Biophys. 1995;316:443–451. doi: 10.1006/abbi.1995.1059. [DOI] [PubMed] [Google Scholar]
- 5.Moghaddam M F, Grant D F, Cheek J M, Greene J F, Williamson K C, Hammock B D. Nat Med. 1997;3:562–566. doi: 10.1038/nm0597-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oltman C L, Weintraub N L, Vanrollins M, Dellsperger K C. Circ Res. 1998;83:932–939. doi: 10.1161/01.res.83.9.932. [DOI] [PubMed] [Google Scholar]
- 7.Ishizaki T, Shigemori K, Nakai T, Miyabo S, Ozawa T, Chang S W, Voelkel N F. Am J Physiol. 1995;269:L65–L70. doi: 10.1152/ajplung.1995.269.1.L65. [DOI] [PubMed] [Google Scholar]
- 8.Ishizaki T, Shigemori K, Nakai T, Miyabo S, Hayakawa M, Ozawa T, Voelkel N F, Chang S W. J Appl Physiol. 1995;79:1106–1111. doi: 10.1152/jappl.1995.79.4.1106. [DOI] [PubMed] [Google Scholar]
- 9.Dudda A, Spiteller G, Kobelt F. Chem Phys Lipids. 1996;82:39–51. doi: 10.1016/0009-3084(96)02557-1. [DOI] [PubMed] [Google Scholar]
- 10.Sakai T, Ishizaki T, Ohnishi T, Sasaki F, Ameshima S, Nakai T, Miyabo S, Matsukawa S, Hayakawa M, Ozawa T. Am J Physiol. 1995;269:L326–L331. doi: 10.1152/ajplung.1995.269.3.L326. [DOI] [PubMed] [Google Scholar]
- 11.Fukushima A, Hayakawa M, Sugiyama S, Ajioka M, Ito T, Satake T, Ozawa T. Cardiovasc Res. 1988;22:213–218. doi: 10.1093/cvr/22.3.213. [DOI] [PubMed] [Google Scholar]
- 12.Ishizaki T, Takahashi H, Ozawa T, Chang S W, Voelkel N F. Am J Physiol. 1995;268:L123–L128. doi: 10.1152/ajplung.1995.268.1.L123. [DOI] [PubMed] [Google Scholar]
- 13.Mullin C A, Hammock B D. Arch Biochem Biophys. 1982;216:423–439. doi: 10.1016/0003-9861(82)90231-4. [DOI] [PubMed] [Google Scholar]
- 14.Dietze E C, Kuwano E, Casas J, Hammock B D. Biochem Pharmacol. 1991;42:1163–1175. doi: 10.1016/0006-2952(91)90250-9. [DOI] [PubMed] [Google Scholar]
- 15.Morisseau C, Du G, Newman J W, Hammock B D. Arch Biochem Biophys. 1998;356:214–228. doi: 10.1006/abbi.1998.0756. [DOI] [PubMed] [Google Scholar]
- 16.Grant D E, Storms D H, Hammock B D. J Biol Chem. 1993;268:17628–17633. [PubMed] [Google Scholar]
- 17.Beetham J K, Tian T, Hammock B D. Arch Biochem Biophys. 1993;305:197–201. doi: 10.1006/abbi.1993.1411. [DOI] [PubMed] [Google Scholar]
- 18.Wixtrom R N, Silva M H, Hammock B D. Anal Biochem. 1988;169:71–80. doi: 10.1016/0003-2697(88)90256-4. [DOI] [PubMed] [Google Scholar]
- 19.Dietze E C, Kuwano E, Hammock B D. Anal Biochem. 1994;216:176–187. doi: 10.1006/abio.1994.1023. [DOI] [PubMed] [Google Scholar]
- 20.Dixon M. Biochem J. 1972;129:197–202. doi: 10.1042/bj1290197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borhan B, Mebrahtu T, Nazarian S, Kurth M J, Hammock B D. Anal Biochem. 1995;231:188–200. doi: 10.1006/abio.1995.1520. [DOI] [PubMed] [Google Scholar]
- 22.Armstrong R N. Drug Metab Rev. 1999;31:71–86. doi: 10.1081/dmr-100101908. [DOI] [PubMed] [Google Scholar]
- 23.Bodanszky M, Bodanszky A. The Practice of Peptide Synthesis. 2nd Ed. Berlin: Springer; 1994. [Google Scholar]
- 24.Strichartz G R, Sanchez V, Arthur G R, Chafetz R, Martin D. Anesth Analg. 1990;71:58–70. doi: 10.1213/00000539-199008000-00008. [DOI] [PubMed] [Google Scholar]
- 25.Tomlin C, editor. British Crop Protection Council and Royal Society of Chemistry. The Pesticide Manual. 10th Ed. Bath, U.K.: Bath Press; 1994. [Google Scholar]
- 26.United States Geographical Survey. National Water Quality Assessment and Pesticide National Synthesis Project. Reston, VA: U.S. Geographical Survey National Center; 1997. [Google Scholar]
- 27.Blein J P, Ducruet J M, Gauvrit C. Weed Res. 1979;19:117–121. [Google Scholar]
- 28.Demling R H. Annu Rev Med. 1995;46:193–202. doi: 10.1146/annurev.med.46.1.193. [DOI] [PubMed] [Google Scholar]
- 29.Debernard S, Morisseau C, Severson T F, Feng L, Wojtasek H, Prestwich G D, Hammock B D. Insect Biochem Mol Biol. 1998;28:409–419. doi: 10.1016/s0965-1748(98)00014-9. [DOI] [PubMed] [Google Scholar]
- 30.Hammock B D. Chem Eng News. 1998;76:28. [Google Scholar]
- 31.Seidegård J, Ekström G. Environ Health Perspect. 1997;105:791–799. doi: 10.1289/ehp.105-1470052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grant D F, Greene J F, Pinot F, Borhan B, Moghaddam M F, Hammock B D, McCutchen B, Ohkawa H, Luo G, Guenthner T M. Biochem Pharmacol. 1996;51:503–515. doi: 10.1016/0006-2952(95)02227-9. [DOI] [PubMed] [Google Scholar]