

Abstract
During embryogenesis, a complex interplay between extracellular matrix (ECM) molecules, regulatory molecules, and growth factors mediates morphogenetic processes involved in palatogenesis. Transforming growth factor-β (TGF-β), retinoic acid (RA), and γ-aminobutyric acid (GABA)ergic signaling systems are also potentially involved. Using [3H]glucosamine and [35S]methionine incorporation, anion exchange chromatography, semiquantitative radioactive RT-PCR, and a TGF-β binding assay, we aimed to verify the presence of phenotypic differences between primary cultures of secondary palate (SP) fibroblasts from 2-year-old subjects with familial nonsyndromic cleft lip and/or palate (CLP-SP fibroblasts) and age-matched normal SP (N-SP) fibroblasts. The effects of RA—which, at pharmacologic doses, induces cleft palate in newborns of many species—were also studied. We found an altered ECM production in CLP-SP fibroblasts that synthesized and secreted more glycosaminoglycans (GAGs) and fibronectin (FN) compared with N-SP cells. In CLP-SP cells, TGF-β3 mRNA expression and TGF-β receptor number were higher and RA receptor-α (RARA) gene expression was increased. Moreover, we demonstrated for the first time that GABA receptor (GABRB3) mRNA expression was upregulated in human CLP-SP fibroblasts. In N-SP and CLP-SP fibroblasts, RA decreased GAG and FN secretion and increased TGF-β3 mRNA expression but reduced the number of TGF-β receptors. TGF-β receptor type I mRNA expression was decreased, TGF-β receptor type II was increased, and TGF-β receptor type III was not affected. RA treatment increased RARA gene expression in both cell populations but upregulated GABRB3 mRNA expression only in N-SP cells. These results show that CLP-SP fibroblasts compared with N-SP fibroblasts exhibit an abnormal phenotype in vitro and respond differently to RA treatment, and suggest that altered crosstalk between RA, GABAergic, and TGF-β signaling systems could be involved in human cleft palate fibroblast phenotype.
INTRODUCTION
The normal development of the upper jaw and of the palate starts at about the sixth week of intra-uterine life and requires growth and fusion of the medial nasal processes and maxillary processes to form the lip, while the fusion of the palatal shelves to form the secondary palate occurs later (tenth week).
Craniofacial malformations and in particular orofacial clefting are the most common birth defects that occur in humans. Clefts of the lip, with or without cleft palate (CLP), and those that involve the palate only (CPO), are due to a failure in fusion of the facial processes and/or palatal shelves, and constitute 2 forms of oral-facial clefts considered separate birth defects involving many (but not all) of the same genetic and environmental causes (1). CLP and CPO can be subdivided into syndromic (such as chromosomal, Mendelian, teratogen-based, and uncategorized syndromes) and nonsyndromic forms (about 70% of cases). The nonsyndromic CLP arises when nasal processes and/or palatal shelves fail to fuse because genetic abnormalities and/or a perturbed environment alter extracellular matrix (ECM) composition (2) and affect cell patterning, migration, proliferation (3), and differentiation (4).
ECM, a complex and highly organized mesh of molecules including, for example, GAGs, proteoglycans (PGs), collagens, and fibronectin (FN), constitutes the tissue microenvironment where developmental processes are orchestrated (5). ECM components bind, store, and release endogenous growth factors, which influence ECM synthesis and remodeling (6) thus controlling timescale and placement of their activity (7). FN, together with HA, could act as a scaffold on which normal mesenchyme formation occurs and palatal shelves develop (8). HA, the main GAG that is produced before and during palatal shelf reorientation (9), can bind much water to provide the force for palatal shelf elevation. In fact, the amount of GAG and GAG hydration have been implicated as generating the palatal shelf-elevating force in mammals (10), and sulphated GAGs are believed to contribute to shelf volume (11).
Hence, normal orofacial configuration is the end product of highly regulated interplay between ECM molecules and cells from the epithelium and mesenchyme which produce growth factors such as transforming growth factor-β (TGF-β) family members (TGF-β1, -β2, and -β3). All 3 mammalian TGF-β isoforms are expressed during palatal development, and exact timing and spatial expression are required. TGF-β3 appears to play a pivotal role, because TGF-β3 gene mutations and/or deficiencies give rise to cleft palate in humans (12) and mice (13–15). The TGF-β3 signaling system includes 3 major types of specific cell surface receptor proteins: TGFBR1, TGFBR2, and TGFBR3. TGF-β binding to the cell surface receptor complex leads to phosphorylation and activation of the intracellular mediators of TGF-β signaling, the Smad proteins, which act as transcriptional activators of target genes (16).
The retinoic acid (RA) signaling system may also be involved in nonsyndromic oral clefts, and interestingly, functional interactions and bidirectional cross-talk between the RA and TGF-β signaling pathways had been demonstrated in different settings (17–20). Signaling of RA, a vitamin A metabolite, is critical for morphogenesis (21) and is required by multiple tissues during development. Excessive or deficient RA levels can lead to severe abnormalities in morphological development (teratogenesis) (22). High doses adversely affect craniofacial development (23–25), and cleft palate and craniosynostosis are associated with prenatal exposure (26). Alterations in mesenchyme development and effects on ECM production have been demonstrated: in mice exposed to pharmacological doses of RA, palate mesenchyme hydration was delayed, HA deposition and ECM glycoproteins were reduced, palatal shelf elevation was markedly delayed, and the palatal shelf did not make contact, resulting in a cleft palate formation (27).
RA binds to retinoic acid receptors (RARs), which are proteins belonging to the superfamily of nuclear receptors. The RAR family consists of RAR α (RARA), β, and γ isoforms. In a mouse model, a RARA protein receptor mutant is reported to induce a cleft palate in transgenic offspring (28). Despite evidence from a recent family-based investigation (29), opinions diverge as to whether RARA should be considered as a candidate gene in CLP (30–33).
A third signaling system constituted by γ-aminobutyric acid (GABA), the major inhibitory neurotransmitter in the mammalian central nervous system, and its biosynthesizing enzymes and receptors (the GABAergic system) is also involved in normal palate development. Targeted mutagenesis of the GABA type A receptor β3 (GABRB3) subunit induces cleft palate in the mouse (34). Mice lacking the subunit die soon after birth, most probably because of feeding problems associated with cleft palate (35). The significant relation between GABRB3 and CLP which emerged in a recent study suggests that GABRB3 is probably involved in human CLP malformation (29).
Because in vitro biochemical data on the effects of RA on ECM composition in primary cultures of CLP-SP fibroblasts are not available, we evaluated its effects on N-SP and CLP-SP fibroblasts and assessed proliferation and GAG and FN synthesis.
Expression of the TGF-β3 gene and the 3 TGF-β receptor genes was also investigated, together with the count of TGF-β receptors. Finally, in an attempt to clarify in vitro phenotype of fibroblasts from infants affected by nonsyndromic cleft lip and/or palate, we evaluated RARA and GABRB3 expression because they are 2 of the genes that could be involved in CLP.
MATERIALS AND METHODS
Cell Cultures
Fibroblasts were obtained from the oral flap edge of hard secondary palate of four 2-year old subjects, 2 boys and 2 girls, with familial nonsyndromic cleft lip and palate during corrective surgery for the malformation (CLP-SP fibroblasts). The 4 patients had CLP, with the involvement of the lip and the primary and secondary palate, as a unique disorder and a positive familial history: the first case (male) was unilateral (R) CLP, with the maternal grandfather reported to carry the malformation; the second case (male) was unilateral (L) CLP, with paternal grandmother; the third case (female) was unilateral (L) CLP, with paternal great-grandfather; the fourth case (female) was bilateral CLP, with a cousin.
Control normal palatal (N-SP) fibroblasts were obtained from the same anatomical site of 4 age-matched normal subjects hospitalized for palate trauma.
Human tissues were obtained with a protocol approved by our institutions. Informed consent was obtained from all parents after the nature of the study had been fully explained.
Human tissues were dissociated and derived fibroblast primary cultures were grown separately in Falcon flasks containing Dulbecco modified Eagle’s minimum essential medium (DMEM) supplemented with 20% fetal calf serum (Invitrogen, Paisley, UK), essential amino acids, vitamins, and antibiotics (Sigma, St. Louis, MO, USA). All tests were performed between passages 6 and 8. For the assays, subconfluent cells (1 × 106 cell/mL) were incubated in DMEM alone with or without 10 μM all-trans retinoic acid (RA) (Sigma) for 48 h according to the following experimental protocols.
Cell Number and Viability
To determine cell number in cultures, untreated or RA-treated human N-SP and CLP-SP fibroblasts were harvested with PBS, sedimented by centrifugation at 720 g, and resuspended in 1 mL medium. Trypan blue was added to the cell suspension to obtain a final concentration of 2 mg/mL. Cells were incubated for 5 min at room temperature, and viable and non-viable cells were counted using the Burker’s camera. Cell growth was expressed by number of cells/well, and cell viability was expressed as the percentage of the counted viable cells.
Total and Individual Glycosaminoglycan Synthesis
N-SP and CLP-SP fibroblasts, after being maintained for 48 h in serum-free DMEM with and without 10 μM RA, were labeled in the last 24 h with 5 μCi/mL [3H]glucosamine hydrochloride (specific activity 29 Ci/mmol; Amersham Biosciences, Little Chalfont, UK). At the end of incubation, media was recovered separately and processed (36). Aliquots of 3H-labeled GAG from media were applied to a DE-52 cellulose anion exchange column. Individual GAGs were identified by their enzymatic susceptibility. Testicular hyaluronate lyase (beef; Miles Italiana, Milano, Italy), streptomyces hyaluronate lyase (Streptomyces hyalurolyticus) (Seikagaku Kogyo Co., Tokyo, Japan), and chondroitin AC-II lyase (Arthrobacter aurescens) (Seikagaku Kogyo) digestions were performed. Standard GAGs (Sigma) were then added and precipitated with 3 volumes of 5% potassium acetate in ethanol. Radioactivity was measured in both supernatants (digested GAGs) and pellet. Results are expressed as cpm/106 cells.
Fibronectin Synthesis and Secretion
To analyze fibronectin synthesis, N-SP and CLP-SP fibroblasts were cultured and treated as described above. For metabolic labeling, fibroblasts were incubated with 20 μCi/mL [35S]methionine (specific activity > 1000 Ci/mmol; Amersham Biosciences) during the last 3 h of incubation. After biosynthetic labeling, fibronectin was isolated from equal aliquots of media by selective and quantitative binding to gelatin-Sepharose resin (Pharmacia, Piscataway, NJ, USA) pre-equilibrated in TBS in individual minicolumns (37). The fibronectin was eluted with 2×SDS-PAGE sample buffer and analyzed by electrophoresis on 6% SDS-polyacrylamide slab gels and fluorography. The fluorographs were analyzed for quantification via densitometric scanning using QuantiScan software (Biosoft, Cambridge, UK) running on a Pentium 4 desktop PC.
RNA Extraction and cDNA Synthesis
Total RNA was extracted from N-SP and CLP-SP fibroblasts incubated for 48 h in DMEM with and without 10 μM RA using TRIzol reagent (Invitrogen) following the manufacturer’s instructions. The RNA was treated immediately with DNAse I (Invitrogen) to eliminate genomic DNA contamination, and the integrity of the treated RNA was examined by detection of ribosomal RNA bands (28S and 18S) in ethidium bromide–stained agarose gels. The RNA was quantified by reading the optical density at 260 nm. One microgram of total RNA was subjected to reverse transcription (RT) in a final volume of 50 μL.
PCR Reaction and cDNA Analysis
Semiquantitative radioactive PCR was used to determine the level of expression of the following specific genes: TGF-β3, TGFBRI, TGFBR2, TGFBR3, RARA, GABRB3. β-Actin was used as an internal control. Primers were designed to the relevant sequences from GenBank and were synthesized by Invitrogen.
Polymerization reactions were performed in a PCR thermocycler (Hybaid, Ashford, UK) in a 50-μL reaction volume containing 2 μL cDNA from RT reaction, 100 μM cold dNTPs, 5 units Platinum Taq DNA polymerase (Invitrogen), and 2.5 μCi (3000 Ci/mmol) [32P]dCTP (Amersham Biosciences). The PCR reaction started with the activation of the Platinum Taq enzyme for 2 min at 94 °C (hot-start PCR). For more accurate quantitative results, the PCR conditions for each pair of primers were optimized for number of cycles and annealing so that the amplifications of all genes were within a linear range. The oligonucleotide primers and conditions used for PCR are indicated in Table 1. At the end of the reaction, 10 μL loading buffer was added to the 50-μL PCR mix, and a 10-μL aliquot was run in a 6% polyacrylamide gel in a Tris-boric acid-EDTA system. Gels were dried and exposed to an InstantImager apparatus (Packard Instrument, Meriden, CT, USA). Radioactivity incorporated by the PCR products was quantified using the Imager software (Packard), and the quantification for each specific gene was expressed as a ratio with the β-actin used as internal control and amplified in parallel PCRs. The values were then transformed in a percentage, assuming as 100 the untreated level for N-SP and CLP-SP cells. All experimental measurements were run in quadruplicate.
Table 1.
Sequences of oligonucleotide primers and PCR conditions
| Gene name | Size, bp | 5′ Oligonucleotide | 3′ Oligonucleotide | GenBank accession no. | Annealing temperature, °C | Cyclesa, n |
|---|---|---|---|---|---|---|
| β-Actin | 351 | (522)–CACACTGTGCCCATC TACGAGG-(543) | (872)-AGTTTCGTGGATGCC ACAGGA-(852) | X00351 | 60 | 23 |
| TGF-β3 | 298 | (262)–GCACTTGCAAAGGGC TCTG-(280) | (559)-GGCATAGTATTCCGA | NM_0003239 | 58 | 28 |
| TGFBRI | 277 | (780)–CCTCTAGAGAAGAAC GTTCGT-(800) | (1056)-GGCTTTCCTTGGGTA CCAACAA-(1035) | NM_004612 | 58 | 26 |
| TGFBR2 | 387 | (1092)–GGTCGCTTTGCTGAG GTCTA-(2011) | (1478)-CTTGAGGTCCCTGTG CACGAT-(1458) | NM_003242 | 58 | 23 |
| TGFBR3 | 363 | (1621)-CCTGTCATTCCCAGC ATACAACT-(1643) | (1983)-ATCACCTGACTCCAG ATCTTCATA-(1960) | XM_001924 | 58 | 25 |
| RARA | 298 | (491)-GCATCATCAACAAGG TGACC-(510) | (788)-CTGAACTTGTCCCAG AGGTCA-(768) | NM_000964 | 56 | 22 |
| GABRB3 | 299 | (243)-CCTTTGTGAAGGAGA CGGT-(261) | (541)-CACTCCATGCACAAA TGAC-(523) | NM_000814 | 54 | 22 |
The number of cycles was adjusted to be in the exponential phase of the amplification of each product.
TGF-β1 Binding Assay
N-SP and CLP-SP human fibroblasts were maintained for 48 h in serum-free DMEM with and without 10 μM RA. TGF-β receptor binding was performed according to Massaguè (38). Briefly, the cells were incubated for 2 h at 37 °C in serum-free DMEM with 0.15% gelatin before the start of each experiment. Cells were then washed twice with cold PBS and 200 μL cold DMEM containing 25 mM Hepes, pH 7.5, plus 0.15% gelatin and increasing concentrations of [125I]TGF-β (0.1 to 10 ng/mL) (Amersham Biosciences) were added to each well. The cells were incubated for 2 h at 4 °C on an orbital shaker. The binding medium was discarded, and the cells were washed twice with ice-cold PBS and twice with DMEM/gelatin. To determine low-affinity binding of TGF-β, the cells were incubated twice for 5 min with cold PBS, pH 7.5, containing 2 M NaCl, and the cell extract was counted in a γ counter (Packard). Control experiments were conducted using cells exhaustively treated with heparinase to determine the amount of the reduced low-affinity receptor–bound TGF-β. Nonspecific binding was estimated in the presence of 100-fold excess of unlabeled recombinant human TGF-β and subtracted from all data. High-affinity bound TGF-β was determined by extraction in 20 mM sodium acetate buffer, pH 4.0, containing 2 M NaCl. All experimental measurements were run in quadruplicate. Receptor binding was normalized with respect to cell number, which was determined using hemocytometer, and analyzed with Scatchard methods.
Statistical Analysis
Results reported in figures are the mean ± SD of 4 separate experiments for each of the 4 patients; each experiment was performed in quadruplicate. Statistical analysis was performed by ANOVA followed by the Sheffé F test. The SDS-PAGE results were analyzed by paired Student t test.
RESULTS
Cell Growth and Viability
RA treatment did not significantly modify cell number in N-SP and CLP-SP fibroblasts (Figure 1). Cell viability was not significantly affected by RA treatment (data not shown).
Figure 1.

Effects of exposure in vitro of N–SP and CLP–SP fibroblasts to RA on cell number. The cells were maintained in DMEM alone for 48 h with or without RA. The results represents the cell number. The values were the mean ± SD of 4 independent experiments for each of the 4 patients; each experiment was performed in quadruplicate. Data were analyzed by ANOVA. CLP–SP fibroblasts vs. N–SP fibroblasts: §not significant; treated vs. untreated cells: NS, not significant.
GAG Synthesis and RA Effects
Data on GAG secretion, as assessed by [3H]glucosamine incorporation, are reported in Figure 2. CLP-SP fibroblasts secreted more total GAG (+65%, F test significant at 99%) than N-SP fibroblasts. In N-SP fibroblasts, RA decreased GAG secretion by about 73% (F test significant at 99%), and in CLP-SP fibroblasts, by about 43% (F test significant at 99%).
Figure 2.

Incorporation of [3H]glucosamine into total and individual GAG classes secreted by N–SP (A) and CLP–SP (B) fibroblasts treated (black bars) or not (white bars) with RA. The results are shown as mean ± SD of 4 separate experiments for each of the 4 patients; each experiment was performed in quadruplicate. HA indicates hyaluronan; HS, heparan sulphate; CS, chondroitin–4 and –6 sulphate; DS, dermatan sulphate. The statistical analysis was performed with ANOVA. CLP–SP fibroblasts vs. N–SP fibroblasts: §F test significant at 99%; treated N–SP and CLP–SP fibroblasts vs. individual GAG classes of untreated group: *F test significant at 99%, **F test significant at 95%.
Analysis of newly secreted GAGs performed by anion exchange chromatography revealed 4 classes of GAG: hyaluronic acid (HA), heparan sulphate (HS), chondroitin sulphate (CS), and dermatan sulphate (DS) (Figure 2).
In the media from both cell populations, HA was the main GAG class, and HA/sulphated GAG ratio was shifted in favor of sulphated GAG. In both cell populations, RA treatment decreased levels of all secreted GAG classes, and HA/sulphated GAG ratio was slightly shifted in favor of sulphated GAG.
Fibronectin Secretion and RA Effects
Densitometric analysis of fibronectin on fluorographs showed that CLP-SP fibroblasts produced higher levels of secreted fibronectin (+67%, P < 0.001) than N-SP cells (Figure 3). RA downregulated secreted fibronectin in both N-SP (−24%, P < 0.01) and CLP-SP (−21%, P < 0.01) fibroblasts (Figure 3).
Figure 3.
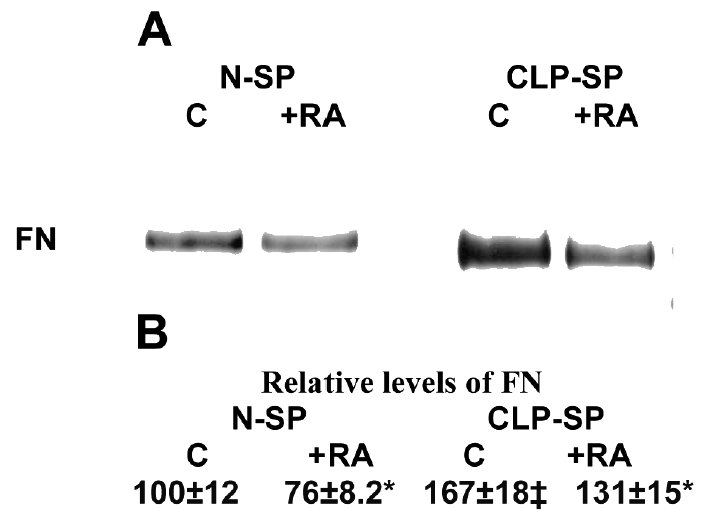
Fibronectin (FN) synthesized by N–SP and CLP–SP fibroblasts. (A) Film autoradiogram of the gels. (B) The fluorographs shown in A were analyzed by computerized scanning densitometry. The values obtained assume as 100 the untreated level for N–SP cells. Similar results were seen in 4 separate experiments for each of the 4 patients; each experiment was performed in quadruplicate. The results are analyzed by paired Student t test. Untreated CLP–SP vs. untreated N–SP fibroblasts: ‡P < 0.001; treated vs untreated cells: *P < 0.01.
Expression of Specific Genes in N-SP and CLP-SP Fibroblasts Cultured with or without RA
We used a semiquantitative radioactive RT-PCR method to measure changes induced by the different culture conditions (without or with RA) on expression of TGF-β3, TGFBR1, TGFBR2, TGFBR3, RARA, and GABRB3 genes. Quantitation of amplimers is expressed as a ratio to the control β-actin transcripts amplified in parallel PCR reactions.
Our results show low levels of TGF-β3 transcription in N-SP cells and a modest but significantly higher level in CLP-SP cells (Figure 4 and Table 2). RA treatment upregulated TGF-β3 mRNA in both cell populations (+142% in N-SP and +59% in CLP-SP cells). In both cell populations, TGFBR1 mRNA was downregulated, TGFBR2 was upregulated, and TGFBR3 was not affected by RA (Figure 4 and Table 2).
Figure 4.

Representative samples of the semiquantitative radioactive RT–PCR used to quantitate the mRNA levels of different specific genes. β–Actin was used as internal control in all PCRs. C and RA represent untreated and 10 μM RA-treated fibroblasts, respectively. The amplification products were electrophoresed on 6% polyacrylamide gels. Gels were dried and exposed for electronic autoradiography. Values of semiquantitative analysis are reported in Table 2. Similar results were seen in 4 independent experiments for each of the 4 patients; each experiment was performed in quadruplicate.
Table 2.
Semiquantitative analysis of mRNA for TGF–β3, TGFBR1, TGFBR2, TGFBR3, RARA, and GABRB3 in N–SP and CLP–SP fibroblasts treated or not with RA.
| N–SP fibroblasts
|
CLP–SP fibroblasts
|
|||
|---|---|---|---|---|
| Control | RA | Control | RA | |
| TGF–β3 | 100 ± 13 | 242 ± 27d | 143 ± 17b | 228 ± 25d |
| TGFBR1 | 100 ± 13 | 65 ± 7d | 103 ± 11c | 57 ± 7d |
| TGFBR2 | 100 ± 11 | 267 ± 28d | 111 ± 11c | 285 ± 30d |
| TGFBR3 | 100 ± 12 | 95 ± 11NS | 107 ± 13c | 123 ± 14NS |
| RARA | 100 ± 14 | 170 ± 19d | 315 ± 35a | 510 ± 54d |
| GABRB3 | 100 ± 11 | 307 ± 35d | 540 ± 59a | 590 ± 68NS |
| β–Actin | 100 ± 11 | 95 ± 10NS | 100 ± 12c | 91 ± 11NS |
The values indicate mRNA levels corrected for β–actin mRNA levels and expressed as the percentage of untreated N–SP and CLP–SP fibroblasts. All values are mean ± SD of 4 separate experiments for each of the 4 patients; each experiment was performed in quadruplicate. The results were analyzed by ANOVA. CLP–SP control fibroblasts vs. N-SP control fibroblasts:
F test significant at 99%;
F test significant at 95%;
not significant; treated vs. untreated control cells:
F test significant at 99%; NS, not significant.
In CLP-SP fibroblasts, more RARA mRNA was expressed than in N-SP cells. RA increased the levels of RARA expression in both cell populations (+70% in N-SP and +62% in CLP-SP cells; F test significant at 99%) (Figure 4 and Table 2).
In addition, we found an elevated level of GABRB3 transcription in CLP-SP fibroblasts compared with the faint levels in N-SP cells (about 5-fold more than N-SP cells). Treatment with RA markedly increased (by about 3-fold) GABRB3 expression in N-SP fibroblasts but did not influence its expression in CLP-SP cells (Figure 4 and Table 2).
Quantitative Analysis of TGF-β Receptors by Binding Assay in N-SP and CLP-SP Fibroblasts Cultured with or without RA
A Scatchard analysis of data showed that a single class of high-affinity binding sites was responsible for binding TGF-β to N-SP and CLP-SP cells, whether treated with RA or not (Figure 5). Binding isotherms with different concentrations of [125I]TGF-β1 and native TGF-β1 indicated TGF-β1 saturation at 37 °C was achieved (0.20–0.30 nM TGF-β1) in controls and treated cells. Analysis of data revealed that the receptor numbers were significantly higher (106697 ± 1686 per cell) on CLP-SP fibroblasts than on N-SP fibroblasts (43739 ± 674 per cell).
Figure 5.

Scatchard analysis of [125I]TGF–β1 binding to N–SP and CLP–SP fibroblasts treated or not with RA. The cells were preincubated at 37 °C for 2 h in serum–free medium containing 0.1% BSA, 25 mM Hepes, pH 7.4, then incubated at 4 °C for 3 h in serum–free medium containing TGF–β1. Cell monolayers were than washed with ice–cold Hanks buffered saline containing 0.1% BSA, 1% Triton X–100, 10% glycerol, and 20 mM Hepes, pH 7.4. Specifically bound (B) and free (F) ligands were measured as described in Materials and Methods. C and RA represent untreated and 10 μM RA-treated fibroblasts, respectively. Data are from a representative determination. Similar results were seen in 4 independent experiments for each of the 4 patients; each experiment was performed in quadruplicate.
RA treatment reduced the number of TGF-β receptors per cell by 60% and 29% in N-SP and CLP-SP fibroblasts, respectively.
DISCUSSION
In previous in vitro studies on human CLP-SP fibroblasts, we showed marked phenotype alterations in GAG and collagen production as well as abnormal growth factor expression and aberrant cross-talk between TGF-β3 and IL-6 (39–41).
In the present study, we found abnormal ECM synthesis in CLP-SP fibroblasts, which secreted more FN and total and individual GAGs. Unlike N-SP cells which secreted HS as the main sulphated GAG, CLP-SP fibroblasts secreted CS. They responded differently to RA treatment, which reduced FN to the same extent (−24% vs. −21%) in both cell populations but reduced GAG secretion less in CLP-SP fibroblasts (−73% vs. −43%). Although the RA effects we found were not due to alterations in cell number, because RA does not affect cell growth in N-SP and CLP-SP primary human fibroblasts, our results suggest that RA disrupts normal ECM stoichiometry by changing the balanced expression of GAG and FN, thus altering normal palate development. As normal palate development requires proper composition and regulation of ECM turnover, it is worth bearing in mind that RA is an important regulator of cell proliferation, differentiation, and ECM production. Indeed, RA might impair the cell–ECM interaction, thus impeding a cell’s ability to migrate through that matrix (42).
As reported by others, RA treatment on monolayer cultures of fibroblasts from other anatomic sites such as human skin fibroblasts decreases the incorporation of [3H]glucosamine into HA by 35% and stimulates the incorporation of [3H]glucosamine into heparan sulfate and chondroitin sulfates by 58% and 72%, respectively (43). The different behavior of CLP-SP fibroblasts compared with N-SP and fibroblasts from other anatomic sites strengthens the hypothesis that they carry an abnormal phenotype.
Among the several genes that regulate ECM production in palate mesenchyme, TGF-β, particularly TGF-β3, affects palate development. Our results demonstrate a low level of TGF-β3 transcription in N-SP cells and a modest but significantly higher level in CLP-SP cells. The low level of transcription could explain our failure to detect TGF-β3 mRNA in a previous work using a less sensitive technique (40). Nevertheless, the same study had demonstrated upregulated TGF-β3 protein secretion in CLP-SP cells, and adding TGF-β3 increased accumulation of ECM molecules. In the present study, the TGF-β receptor number was about 2.4-fold higher in CLP-SP cells, but analysis of TGF-β receptor expression showed that TGFBR1, TGFBR2, and TGFBR3 were similarly expressed in CLP-SP and N-SP fibroblasts. Lack of correspondence between receptor numbers and steady-state levels for mRNA expression of receptor types could be due to differences in posttranscriptional and/or posttranslational regulation.
As TGF-β receptors bind all TGF-β family members that induce ECM molecule expression in mesenchymal cells (6,44), the concomitant enhanced receptor number and increased TGF-β3 expression in CLP-SP cells could lead to abnormally high TGF-β system activity, thus accounting for the high ECM production by CLP-SP cells. Interestingly, excessive TGF-β signaling has been implicated in a newly described human phenotype that is associated with widespread perturbations in development leading to diseases such as cleft palate, craniosynostosis, arterial aneurysms, congenital heart disease, and mental retardation (45). Our results cast new light on the interaction between the TGF-β signaling system and cell phenotype because they suggest that an excess of, rather than a deficit in, TGF-β3 pathway activity in CLP-SP fibroblasts could underlie CLP defects.
Clinical and experimental studies show that RA plays an essential role in maintenance of growth and differentiation in adult tissues (46) and in normal embryonic development (21). In the murine embryonic palate mesenchymal cell model, RA increased TGF-β3 mRNA expression and protein (47,48). Moreover, functional interactions between TGF-β and retinoic systems had been found in different settings (17,19,20). In the present study, RA treatment upregulated TGF-β3 mRNA and decreased TGF-β receptor number less in CLP-SP fibroblasts than in N-SP cells. When we analyzed the transcription of single receptor types, we found that RA treatment of either CLP-SP fibroblasts or N-SP cells down-regulated TGFBR1, upregulated TGFBR2, and did not affect TGFBR3 gene expression. Although a previous study (49) reported RA decreases mRNA expression of TGF-β receptors, the significance of our differing results with each receptor is impossible to establish within the scope of the present study. In the final analysis, RA treatment of both CLP-SP and N-SP fibroblasts appears to modulate TGF-β3 system activity, which could account for the altered ECM composition (less GAG and FN production) that we found in RA-treated cells.
One of the genes that has been linked to CLP pathogenesis, even though its direct involvement has not been demonstrated, is the retinoic acid receptor (RARA). RARA mRNA was expressed about 3-fold more in CLP-SP cells, suggesting it is related to the abnormal CLP phenotype, at least in our 4 Italian families. RA treatment increased RARA expression in N-SP and CLP-SP fibroblasts, confirming results in mouse embryos treated with teratogenic doses of RA (50). Should the upregulated RARA mRNA expression that we found both in untreated CLP-SP cells and in all RA-treated cells be paralleled by enhanced receptor protein synthesis, it could influence the RA signaling system and alter the cross-talk between the RA and TGF-β signaling systems (18), perturbing, for example, TGF-β signaling. We could hypothesize also that variations in endogenous levels of RA would have wide-ranging effects on the expression of genes involved in ECM production and consequently in normal palate development by impairing cell–ECM interaction as well as cell migration through the matrix.
Finally, the present study detected, for the first time, activated GABA receptor expression in CLP-SP cells, at a much higher level of GABRB3 transcription than in the very faint traces found in N-SP cells. This in vitro finding supports the hypothesis that impairment of any GABA function (in our patients, an excess) leads to cleft palate in vivo (51,52) and the involvement of GABRB3 in human CLP malformation (29). Treatment with RA markedly increased GABRB3 expression level in N-SP fibroblasts but had no effect on CLP-SP cells, implying that GABRB3 transcription was already at a maximum. Whether the GABRB3 transcription that we found in CLP-SP cells has been induced by a within-cell unregulated retinoic signaling system remains to be determined in further studies. Recent studies performed in neurons put new insights into interactions between GABAergic and RA signaling systems (53), and our results suggest that interactions between the 2 systems could be involved in the development of N-SP and CLP-SP fibroblast phenotype.
Taken together, our data extend previous findings that CLP-SP fibroblasts retain an abnormal phenotype in vitro, which the present paper has defined in terms of ECM production, TGF-β system, RARA, and GABRB3 expression and different response to RA. The results contribute to a better understanding of the interactions between RA and TGF-β signaling pathways and support the hypothesis that altered cross-talk between TGF-β and RA signaling systems plays a role in eliciting the CLP phenotype in humans.
ACKNOWLEDGMENTS
The authors thank Dr. Geraldine Anne Boyd for her help in English language.
Footnotes
Online address: http://www.molmed.org
REFERENCES
- 1.Carinci F, et al. Recent developments in orofacial cleft genetics. J Craniofac Surg. 2003;14:130–43. doi: 10.1097/00001665-200303000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Brinkley LL, Morris-Wiman J. The role of extracellular matrices in palatal shelf closure. Curr Top Dev Biol. 1984;19:17–36. doi: 10.1016/s0070-2153(08)60393-2. [DOI] [PubMed] [Google Scholar]
- 3.Kochhar DM. Studies of vitamin A-induced teratogenesis: effects on embryonic mesenchyme and epithelium, and on incorporation of H3-thymidine. Teratology. 1968;1:299–310. doi: 10.1002/tera.1420010308. [DOI] [PubMed] [Google Scholar]
- 4.Young DL, Schneider RA, Hu D, Helms JA. Genetic and teratogenic approaches to craniofacial development. Crit Rev Oral Biol Med. 2000;11:304–17. doi: 10.1177/10454411000110030201. [DOI] [PubMed] [Google Scholar]
- 5.Brinkley LL, Morris-Wiman J. Effects of chlorcyclizine-induced glycosaminoglycan alterations on patterns of hyaluronate distribution during morphogenesis of the mouse secondary palate. Development. 1987;100:637–40. doi: 10.1242/dev.100.4.637. [DOI] [PubMed] [Google Scholar]
- 6.Eickelberg O, et al. Extracellular matrix deposition by primary human lung fibroblasts in response to TGF-beta1 and TGF-beta3. Am J Physiol Lung Cell Mol Physiol. 1999;276:L814–24. doi: 10.1152/ajplung.1999.276.5.L814. [DOI] [PubMed] [Google Scholar]
- 7.Taipale J, Keski-Oja J. Growth factors in the extracellular matrix. FASEB J. 1997;11:51–9. doi: 10.1096/fasebj.11.1.9034166. [DOI] [PubMed] [Google Scholar]
- 8.Morris-Wiman J, Brinkley L. An extracellular matrix infrastructure provides support for murine secondary palatal shelf remodelling. Anat Rec. 1992;234:575–86. doi: 10.1002/ar.1092340413. [DOI] [PubMed] [Google Scholar]
- 9.Pratt RM, Goggins JF, Wilk AL, King CT. Acid mucopolysaccharide synthesis in the secondary palate of the developing rat at the time of rotation and fusion. Dev Biol. 1973;32:230–7. doi: 10.1016/0012-1606(73)90237-6. [DOI] [PubMed] [Google Scholar]
- 10.Singh GD, Moxham BJ, Langley MS, Waddington RJ, Embery G. Changes in the composition of glycosaminoglycans during normal palatogenesis in the rat. Arch Oral Biol. 1994;39:401–7. doi: 10.1016/0003-9969(94)90170-8. [DOI] [PubMed] [Google Scholar]
- 11.Young AV, Hehn BM, Cheng KM, Shah RM. A comparative study on the effects of 5-fluorouracil on glycosaminoglycan synthesis during palate development in quail and hamster. Histol Histopathol. 1994;9:515–23. [PubMed] [Google Scholar]
- 12.Lidral AC, et al. Association of MSX1 and TGFB3 with nonsyndromic clefting in humans. Am J Hum Genet. 1998;63:557–68. doi: 10.1086/301956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Proetzel G, et al. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11:409–14. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11:415–21. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- 15.Brunet CL, Sharpe PM, Ferguson MW. Inhibition of TGF-beta 3 (but not TGF-beta 1 or TGF-beta 2) activity prevents normal mouse embryonic palate fusion. Int J Dev Biol. 1995;39:345–55. [PubMed] [Google Scholar]
- 16.Massaguè J, Hata A, Liu F. TGFbeta signaling through the Smad pathway. Trends Cell Biol. 1997;7:187–92. doi: 10.1016/S0962-8924(97)01036-2. [DOI] [PubMed] [Google Scholar]
- 17.Roberts AB, Sporn MB. Mechanistic interrelationships between two superfamilies: the steroid/retinoid receptors and transforming growth factor-beta. Cancer Surv. 1992;14:205–20. [PubMed] [Google Scholar]
- 18.Nugent P, Greene RM. Interactions between the transforming growth factor beta (TGF-β) and retinoic acid signal transduction pathways in murine embryonic palatal cells. Differentiation. 1994;58:149–55. doi: 10.1046/j.1432-0436.1995.5820149.x. [DOI] [PubMed] [Google Scholar]
- 19.Pendaries V, Verrecchia F, Michel S, Mauviel A. Retinoic acid receptors interfere with the TGF-beta/Smad signaling pathway in a ligand-specific manner. Oncogene. 2003;22:8212–20. doi: 10.1038/sj.onc.1206913. [DOI] [PubMed] [Google Scholar]
- 20.Bartholin L, Powers SE, Melhuish TA, Lasse S, Weinstein M, Wotton D. TGIF inhibits retinoid signaling. Mol Cell Biol. 2006;26:990–1001. doi: 10.1128/MCB.26.3.990-1001.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawakami Y, Raya A, Raya RM, Rodriguez-Esteban C, Belmonte JC. Retinoic acid signaling links left-right asymmetric patterning and bilaterally symmetric somitogenesis in the zebrafish embryo. Nature. 2005;435:165–71. doi: 10.1038/nature03512. [DOI] [PubMed] [Google Scholar]
- 22.Morriss-Kay G. Retinoic acid and craniofacial development: molecules and morphogenesis. Bioessays. 1993;15:9–15. doi: 10.1002/bies.950150103. [DOI] [PubMed] [Google Scholar]
- 23.Helms JA, Kim CH, Hu D, Minkoff R, Thaller C, Eichele G. Sonic hedgehog participates in craniofacial morphogenesis and is downregulated by teratogenic doses of retinoic acid. Dev Biol. 1997;187:25–35. doi: 10.1006/dbio.1997.8589. [DOI] [PubMed] [Google Scholar]
- 24.Cuervo R, Valencia C, Chandraratna RA, Covarrubias L. Programmed cell death is required for palate shelf fusion and is regulated by retinoic acid. Dev Biol. 2002;245:145–56. doi: 10.1006/dbio.2002.0620. [DOI] [PubMed] [Google Scholar]
- 25.Song HM, Nacamuli RP, Xia W, Bari AS, Shi YY, Fang TD, Longaker MT. High-dose retinoic acid modulates rat calvarial osteoblast biology. J Cell Physiol. 2005;202:255–62. doi: 10.1002/jcp.20115. [DOI] [PubMed] [Google Scholar]
- 26.Cohen MM., Jr Etiopathogenesis of craniosynostosis. Neurosurg Clin N Am. 1991;2:507–13. [PubMed] [Google Scholar]
- 27.Degitz SJ, Francis BM, Foley GL. Mesenchymal changes associated with retinoic acid induced cleft palate in CD-1 mice. J Craniofac Genet Dev Biol. 1998;18:88–99. [PubMed] [Google Scholar]
- 28.Damm K, Heyman RA, Umesono K, Evans RM. Functional inhibition of retinoic acid response by dominant negative retinoic acid receptor mutants. Proc Natl Acad Sci U S A. 1993;90:2989–93. doi: 10.1073/pnas.90.7.2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scapoli L, Martinelli M, Pezzetti F, Carinci F, Bodo M, Tognon M, Carinci P. Linkage disequilibrium between GABRB3 gene and non-syndromic familial cleft lip with or without cleft palate. Hum Genet. 2002;110:15–20. doi: 10.1007/s00439-001-0639-5. [DOI] [PubMed] [Google Scholar]
- 30.Chenevix-Trench G, Jones K, Green AC, Duffy DL, Martin NG. Cleft lip with or without cleft palate: associations with transforming growth factor alpha and retinoic acid receptor loci. Am J Hum Genet. 1992;51:1377–85. [PMC free article] [PubMed] [Google Scholar]
- 31.Shaw D, Ray A, Marazita M, Field L. Further evidence of a relationship between the retinoic acid receptor alpha locus and nonsyndromic cleft lip with or without cleft palate (CL +/− P) Am J Hum Genet. 1993;53:1156–7. [PMC free article] [PubMed] [Google Scholar]
- 32.Stein JD, Hecht JT, Blanton SH. Exclusion of retinoic acid receptor and a cartilage matrix protein in non-syndromic CL(P) families. J Med Genet. 1995;32:78. doi: 10.1136/jmg.32.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moreno LM, et al. Genetic analysis of candidate loci in non-syndromic cleft lip families from Antioquia-Colombia and Ohio. Am J Med Genet A. 2004;125:135–44. doi: 10.1002/ajmg.a.20425. [DOI] [PubMed] [Google Scholar]
- 34.Ding R, Tsunekawa N, Obata K. Cleft palate by picrotoxin or 3-MP and palatal shelf elevation in GABA-deficient mice. Neurotoxicol Teratol. 2004;26:587–92. doi: 10.1016/j.ntt.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 35.Homanics GE, et al. Mice devoid of gamma-aminobutyrate type A receptor beta3 subunit have epilepsy, cleft palate, and hypersensitive behavior. Proc Natl Acad Sci U S A. 1997;94:4143–8. doi: 10.1073/pnas.94.8.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conrad GW, Hamilton C, Haynes E. Differences in glycosaminoglycans synthesized by fibroblast–like cells from chick cornea, heart and skin. J Biol Chem. 1977;252:6861–70. [PubMed] [Google Scholar]
- 37.Bodo M, et al. Apert’s syndrome: differential in vitro production of matrix macromolecules and its regulation by interleukins. Eur J Clin Invest. 1997;27:36–42. doi: 10.1046/j.1365-2362.1997.660618.x. [DOI] [PubMed] [Google Scholar]
- 38.Massaguè J. Identification of receptors for type-beta transforming growth factor. Methods Enzymol. 1987;146:174–95. doi: 10.1016/s0076-6879(87)46020-5. [DOI] [PubMed] [Google Scholar]
- 39.Bosi G, et al. Diphenylhydantoin affects glycosaminoglycans and collagen production by human fibroblasts from cleft palate patients. J Dent Res. 1998;77:1613–21. doi: 10.1177/00220345980770080901. [DOI] [PubMed] [Google Scholar]
- 40.Bodo M, et al. TGFbeta isoforms and decorin gene expression are modified in fibroblasts obtained from non-syndromic cleft lip and palate subjects. J Dent Res. 1999;78:1783–90. doi: 10.1177/00220345990780120401. [DOI] [PubMed] [Google Scholar]
- 41.Baroni T, et al. Cross-talk between interleukin-6 and transforming growth factor-beta3 regulates extracellular matrix production by human fibroblasts from subjects with non-syndromic cleft lip and palate. J Periodontol. 2003;74:1447–53. doi: 10.1902/jop.2003.74.10.1447. [DOI] [PubMed] [Google Scholar]
- 42.Irving DW, Willhite CC, Burk DT. Morphogenesis of isotretinoin-induced microcephaly and micrognathia studied by scanning electron microscopy. Teratology. 1986;34:141–53. doi: 10.1002/tera.1420340203. [DOI] [PubMed] [Google Scholar]
- 43.Edward M. Effects of retinoids on glycosaminoglycan synthesis by human skin fibroblasts grown as monolayers and within contracted collagen lattices. Br J Dermatol. 1995;133:223–30. doi: 10.1111/j.1365-2133.1995.tb02619.x. [DOI] [PubMed] [Google Scholar]
- 44.D’Angelo M, Greene RM. Transforming growth factor-beta modulation of glycosaminoglycan production by mesenchymal cells of the developing murine secondary palate. Dev Biol. 1991;145:374–8. doi: 10.1016/0012-1606(91)90136-q. [DOI] [PubMed] [Google Scholar]
- 45.Loeys BL, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–81. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 46.De Luca LM. Retinoids and their receptors in differentiation, embryogenesis, and neoplasia. FASEB J. 1991;5:2924–33. [PubMed] [Google Scholar]
- 47.Nugent P, Ma L, Greene RM. Differential expression and biological activity of retinoic acid-induced TGFbeta isoforms in embryonic palate mesenchymal cells. J Cell Physiol. 1998;177:36–46. doi: 10.1002/(SICI)1097-4652(199810)177:1<36::AID-JCP4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 48.Degitz SJ, Morris D, Foley GL, Francis BM. Role of TGF-beta in RA-induced cleft palate in CD-1 mice. Teratology. 1998;58:197–204. doi: 10.1002/(SICI)1096-9926(199811)58:5<197::AID-TERA6>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 49.Makhijani NS, Bischoff DS, Yamaguchi DT. Regulation of proliferation and migration in retinoic acid treated C3H10T1/2 cells by TGF-beta isoforms. J Cell Physiol. 2005;202:304–13. doi: 10.1002/jcp.20128. [DOI] [PubMed] [Google Scholar]
- 50.Harnish DC, Jiang H, Soprano KJ, Kochhar DM, Soprano DR. Retinoic acid receptor beta 2 mRNA is elevated by retinoic acid in vivo in susceptible regions of mid-gestation mouse embryos. Dev Dyn. 1992;194:239–46. doi: 10.1002/aja.1001940309. [DOI] [PubMed] [Google Scholar]
- 51.Wee EL, Zimmerman EF. Involvement of GABA in palate morphogenesis and its relation to diazepam teratogenesis in two mouse strains. Teratology. 1983;28:15–22. doi: 10.1002/tera.1420280104. [DOI] [PubMed] [Google Scholar]
- 52.Condie BG, Bain G, Gottlieb DI, Capecchi MR. Cleft palate in mice with a targeted mutation in the gamma-aminobutyric acid-producing enzyme glutamic acid decarboxylase 67. Proc Natl Acad Sci U S A. 1997;94:11451–5. doi: 10.1073/pnas.94.21.11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song XQ, Meng F, Ramsey DJ, Ripps H, Qian H. The GABA rho1 subunit interacts with a cellular retinoic acid binding protein in mammalian retina. Neuroscience. 2005;136:467–75. doi: 10.1016/j.neuroscience.2005.08.018. [DOI] [PubMed] [Google Scholar]