

Abstract
Aims: To evaluate the usefulness of denaturing high performance liquid chromatography (DHPLC) as a high throughput tool in: (1) DNA mutation detection in familial hypertrophic cardiomyopathy (FHC), and (2) single nucleotide polymorphism (SNP) discovery and validation in sporadic motor neurone disease (MND).
Methods: The coding sequence and intron–exon boundaries of the cardiac β myosin heavy chain gene (MYH7) were screened by DHPLC for mutation identification in 150 unrelated patients diagnosed with FHC. One hundred and forty patients with sporadic MND were genotyped for the A67T SNP in the poliovirus receptor gene. All DHPLC positive signals were confirmed by conventional methods.
Results: Mutation screening of MYH7 covered 10 kb with a total of 5700 amplicons, and more than 6750 DHPLC injections were completed within 35 days. The causative mutation was identified in 14% of FHC cases, including seven novel missense mutations (L227V, E328G, K351E, V411I, M435T, E894G, and E927K). Genotyping of the A67T SNP was performed at two different temperatures both in MND cases and 280 controls. This coding SNP was found more frequently in MND cases (13.6%) than in controls (6.8%). Furthermore, 19 and two SNPs were identified in MYH7 and the poliovirus receptor gene, respectively, during DHPLC screening.
Conclusions: DHPLC is a high throughput, sensitive, specific, and robust platform for the detection of DNA variants, such as disease causing mutations or SNPs. It enables rapid and accurate screening of large genomic regions.
Keywords: DNA variant analysis, denaturing high performance liquid chromatography, familial hypertrophic cardiomyopathy, motor neurone disease, high throughput
The ability to screen for DNA sequence variants is fundamental to understanding conventional Mendelian disorders and individual susceptibility to common complex diseases. Numerous DNA screening techniques have been developed.1 Denaturing gradient gel electrophoresis and chemical cleavage have high detection sensitivity, but they are formatted for manual use, involve toxic chemicals, and are technically challenging. Single stranded conformation polymorphism and heteroduplex analyses are simple and inexpensive, although they are labour intensive and are associated with low sensitivity (∼ 80%). Immobilised DNA hybridisation arrays and enzymatic mismatch cleavage are promising techniques, but produce high numbers of false positive signals and are expensive.1,2 In comparison, DHPLC can screen for DNA variants in a high throughput mode and with a superior detection rate (> 95%).1,3–5
“The ability to screen for DNA sequence variants is fundamental to understanding conventional Mendelian disorders and individual susceptibility to common complex diseases”
Despite the discovery of hundreds of causative genes in rare genetic conditions through positional cloning (http://archive.uwcm.ac.uk/uwcm/mg/docs/hahaha.html), detection of underlying sequence changes in candidate or known target genes remains a challenge, both in research and diagnostic settings. To illustrate this, we have chosen to investigate familial hypertrophic cardiomyopathy (FHC), an autosomal dominant disorder (OMIM 192600) caused by at least nine genes encoding sarcomere proteins (http://www.angis.org.au/Databases/Heart/). The cardiac β myosin heavy chain gene (MYH7; OMIM 160760) is often involved in FHC, with 108 mutations.6 However, most mutations are rare and family specific, and are widely distributed over the 40 exons of the gene.
Another area of immense effort involves the mapping of susceptibility genes for diseases that have complex gene–gene and gene–environment interactions.7 The candidate disease related genes are usually present within large genomic intervals. Linkage disequilibrium mapping is one approach to narrowing this interval, but requires high density DNA markers, such as single nucleotide polymorphisms (SNPs).7 This makes a robust experimental SNP discovery method highly desirable. Although in silico mining of SNPs from publicly available databases is inexpensive, this method has a low sensitivity of 27%.8 The usefulness of DHPLC in SNP discovery and validation is illustrated by the neurodegenerative disorder sporadic motor neurone disease (MND).9 Although 5–10% of MND cases are familial, more than 90% of cases are sporadic, and genetic contributing factors to these cases remain unknown. Viruses such as polio can be one environmental factor involved in sporadic MND (for review see Talbot9), and a key gene influencing the infectivity of polio would be that encoding the poliovirus receptor (PVR).
The purpose of our study was to evaluate the application of DHPLC as a high throughput tool in FHC mutation detection, and its applicability to SNP analysis in a sporadic MND association study.
METHODS
Study subjects
FHC
Probands came from 150 unrelated Australian families including six patients with known MYH7 mutations (who acted as positive controls).10,11 The putative disease causing mutations in MYH7 were defined as rare missense changes with a population frequency of < 0.5%. Further functional and structural evaluation is beyond the scope of our present study. Controls (n = 100) were matched for ethnicity. Identified synonymous and non-synonymous coding SNPs with a population frequency > 1% might also represent cryptic exonic splicing signals, but this cannot be excluded.
MND
SNP genotyping was performed in 140 white patients with sporadic MND. The diagnostic criteria have been described previously.12,13 The control group for MND comprised 280 unrelated subjects with no history of neurological disease. MND controls were matched with the patients with regard to ethnicity, age, and sex, and there was a control to case ratio of 2 : 1. Approval was obtained from the University of Sydney and the Royal Prince Alfred Hospital human research ethics committees.
DNA analysis
DNA was isolated from peripheral nucleated blood cells using a phenol chloroform method.14 MYH7 primers for the coding regions were designed using Oligo 6 (Molecular Biology Insights, Cascade, Colorado, USA) and Navigator software (v1.5) (Transgenomic, Omaha, Nebraska, USA) based on the GenBank sequence (accession X52889) or adopted from the literature (table 1). Polymerase chain reaction (PCR) details are available on request. Primers to amplify the 465 bp amplicon from PVR exon 2 were designed using the same software based on reference sequence (accession AC068948).13
Table 1.
MYH7 primers, amplicon sizes, and DHPLC analysis conditions
| Exon | Forward primer 5′→3′ | Reverse primer 5′→3′ | Amplicon size (bp) | Temp (°C) | % Buffer B* |
| 3 | CCT CTT GAC TCT TGA GCA TGG | GTA CCC CTC TCT GTC ACC CA | 392 | 61.9 | 58.9–68.9 |
| 4 | TCC CTG TGA GAT CCT GGT TC | CAA GGA TGT TGG GAC GAG TT | 398 | 62.5 | 59.0–69.0 |
| 5 | AAC TCG TCC CAA CAT CCT TG | AGG TTA GGA GCT GCA CAG GA | 443 | 61.5 | 59.8–69.8 |
| 6 | ACC TTT CCC CCC CAC CCT CT | GGG CTG GAG GCT GGG ATC A | 114 | 62.0 | 43.0–53.0 |
| 7 | GGT CTC CAG TAG TAT TGT TCA | CAA GAA GGA GGC AGG TGA GAG | 229 | 61.8 | 54.2–64.2 |
| 8 | AGG GAG AAG AGC TCT CAC CTG | AAG TCC CAA GGC CAA GGT CAG | 167 | 63.9 | 50.7–60.7 |
| 9 | CCT TGG GAC TTG GAC TGG TG | AAA GAG GAG AAA AAC AGA GGG AGG | 252 | 59.7 | 55.1–65.1 |
| 10 | CCT TCT TCT CCC CAC CTG TTC | GTC TCA GTC GGT GGC TCT GAC | 150 | 58.4 | 49.4–59.4 |
| 11 | GCG AGC AGC CTC CAT GAG | CCC CTC ACT GCC AAT CCT C | 216 | 62.0 | 53.6–63.6 |
| 12 | CAT CAT ACT TCT TTT TTG GGG TCC | GCC CTC CAT GAC TTG ACA GC | 232 | 64.0, 61.0 | 50.8–60.8 |
| 13 | TTA CAG GCA TGA ACC ACA CAC C | GTG AAC TTG AAA ACT CTC ATC CC | 267 | 62.1 | 55.7–65.7 |
| 14 | AGA TGA TAA TGG GTG GGC AG | GAA ATA GCT GTT GAA TGT GGG AG | 328 | 61.5 | 57.5–67.5 |
| 15 | ACT CAC ACC CAC TTT CTG ACT | GAG GGG CTG CTA TTT TGT CTA | 293 | 61.5 | 56.6–66.5 |
| 16 | TTG ACC ATA GAG CAG AAT CCA TGT | GAC ATT GAA GTG GTG GGG TGT AG | 475 | 61.0 | 60.2–70.2 |
| 17 | TTC TTT TGT TGA CTC TCC TTC | GAT GGG GAG CCA AGT TGG CTG | 134 | 61.7 | 48.0–58.0 |
| 18 | TCT CTA TTG CAC TTT TTG GCC | TGG GTT GGC CTG AGT TTG TGG | 140 | 60.6 | 48.6–58.6 |
| 19 | CAG TCC AGT TCT CAC AGA CTC C | GGC TCC CCC TGT TCT ATG AG | 206 | 62.0 | 53.1–63.1 |
| 20 | GGA TCT GCA GGT GAC CCT GAA T | ACA ACA GGA AAA GCA TCA GAG G | 283 | 59.6 | 56.2–66.2 |
| 21 | ACT CCC CTC ATC CCA GCT CCA | CTT TTT TTC CTG ACA CTG CCC | 342 | 61.2 | 57.8–67.8 |
| 22 | AGG CTC AGC ACT CCT TTC AAT | AAG GCA GAG CAG GGT GGA AGA | 335 | 61.3 | 59.7–69.7 |
| 23 | CCC TCC TAT TTG AGT GAT GTG C | GGT CAG TAT GGT CTG AGA GTC C | 404 | 61.0, 59.1 | 59.1–69.1 |
| 24 | CCA CAG ATG GCA CCA AGC TG | TAT CTA GGC CCC ACA ACT CTC A | 271 | 61.3 | 55.8–65.8 |
| 25 | TGG GCC TGG GCT CCT TCT C | GGC AGC AGG GAG GGG ACA | 197 | 62.9 | 52.6–62.6 |
| 26† | CGC CCG CCG CCG CCC TAC CTC ACC ATC CCT CCT TCC | AGC CCA GGG ACT CAG CAT C | 160 | 63.4 | 52.7–62.7 |
| 27 | GCG TGG GTC TGA GCC TTG TGT | GGG GAG GTG GGA GGA GGA AG | 468 | 65.1, 62.2 | 60.1–70.1 |
| 28 | TGC ACC TCT TAC ACC CCT TCA | GGG GAG ACT GTG GTG GGA AC | 242 | 63.1, 61.1 | 54.7–64.7 |
| 29 | GAG GAG GTG GGG ATA GAG AGG A | GTC AGT GTG CTC CTT GCT TGG | 215 | 63.9 | 53.5–63.5 |
| 30 | AGA AAG CTG AAC CCA CCT CCT | CCA GAA GTC AGG CTG CTC AGA | 264 | 66.5, 64 | 55.6–65.6 |
| 31 | CAT CCT CCC CAC CCT CTG C | GCT CTG GCC TCT CAC TGA ACC | 293 | 63.8 | 56.5–66.5 |
| 32 | CTT GGG GGC TGA AGA GTG AG | CCC CTC CCC AGC CTC TTG | 234 | 62.7 | 54.4–64.4 |
| 33 | TCA AAC CGA GTT ACC GTG TTC | TGA GAA CAG GGA CCA AAA GC | 219 | 62.7 | 53.7–63.7 |
| 34 | CCC TGA CTG TCT GCC TGC ATC | GGG GCA GGA GGA ATC TGG TG | 436 | 62.8 | 59.7–69.7 |
| 35 | GCT CAT GCC CAC TCT CCT GAT | TCA GGA ATC AGC AGG GGA GC | 256 | 63.5 | 55.3–65.3 |
| 36 | TCT ACC CAA CCC TCC CCC AAC | GGA TCG GGT CGG TGG AGT G | 180 | 63.4, 61.4 | 48.6–58.6 |
| 37 | CAG ACC ATG TGC CAC CTC TCT | GCA AAC TCT TCA TTC TCC TCA GC | 388 | 64.9, 62.7 | 58.8–68.8 |
| 38 | CAC CCC CTG CCT ACC CTC TGG | GGG AGG TGG GAG CAT GAG GTG | 232 | 63.9 | 54.3–64.3 |
| 39 | CTC ACC TCA TGC TCC CAC CT | TGA CTA GCA AAG CCC AAA AGAG | 192 | 64.1 | 52.3–62.3 |
| 40 | GAT CCT GGC TTT GTT TCC TTT CA | TGG GGC TTT GCT GGC ACC T | 95 | 60.4 | 41.6–51.6 |
The amplicon size includes the corresponding exon and intron–exon boundaries.
Temperature refers to the oven temperature(s) at which DHPLC analysis was performed.
*Range of elution buffer B that was determined by Navigator software based on amplicon sizes; †the underlining highlights the additional GC clamp.
DHPLC, denaturing high performance liquid chromatography.
All amplicons were generated by PCR in a reaction volume of 50 μl using GenAmp 9700 (Applied Biosystems, Foster City, California, USA). After amplification, the amplicons underwent a denaturation step of 95°C for five minutes, followed by ramping to ambient temperature over 45 minutes to encourage heteroduplex formation in samples heterozygous for a mutant allele. One representative sample from each positive profile group was sequenced using the ABI Prism 3700 DNA sequencer (Applied Biosystems). Mutations in other samples with a similar elution profile were confirmed by restriction fragment length polymorphism analysis or amplification refractory mutation system.
DHPLC platform and software
DHPLC analysis was undertaken on the Transgenomic Wave® Nucleic Acid Fragment Analysis System (Model 3500 HT; Transgenomic), controlled by Navigator software (fig 1). The autosampler can hold 192 samples and introduces 5–20 μl of each sample to the DNASep® Cartridge. The DNA fragments were eluted by linear gradient changes of buffers A and B (table 1), with ratios determined by Navigator software based on amplicon size. Eluted DNA fragments were detected by the system’s ultraviolet detector. DNA sequence variant detection depends on heteroduplex formation between wild-type and mutant DNA single strands.15,16 At elevated temperatures, the less thermostable heteroduplexes start to melt at the mismatched region, and as a result the DNA elutes earlier than corresponding homoduplexes. Thus, oven temperature prediction is crucial.
Figure 1.

Diagram of the denaturing high performance liquid chromatography Wave system. Buffers (A, 0.1M triethylammonium acetate (TEAA); B, 0.1M TEAA with 25% acetonitrile; C, 8% acetonitrile for syringe wash; and D, 75% acetonitrile for cartridge cleanup) are pumped into the system individually or in a mixture through a multivalve mechanism. A sample is introduced via the autosampler, which holds 2 × 96 well chilled plates. The sample is eluted from the DNASep® Cartridge by linear gradient changes of buffers A and B (mobile phase) within an oven that has a precisely controlled temperature, and then travels through an ultraviolet (UV) detector. The amount of sample DNA and the elution time from the cartridge are automatically recorded in the computer.
Navigator software integrates several activities including amplicon design, melting profile prediction, large scale analysis, normalisation, and automated mutation calling. A unique feature of the software is the superimposing and normalising of DHPLC profiles to increase the sensitivity, speed, and accuracy of data analysis. Multiple computer stations can work in a networked client/server mode so that data can be shared between collaborating laboratories.
RESULTS
FHC mutation screening in MYH7
Mutation screening of MYH7 covered 10 kb with 38 amplicons (table 1) and was performed on 150 patients. The high throughput HT3500 model allows analysis of one injection in only 2.5 minutes compared with eight minutes for earlier models. One hundred and ninety two injections can easily be completed in an overnight run. Analysis of 5700 amplicons with over 6750 injections can be completed within 35 days with a continuous supply of PCR products. The analysis conditions were predicted using Navigator software (table 1).
Screening of MYH7 revealed 17 disease related mutations (table 2) in 21 families, which accounted for 14% of all families tested. These included seven putative novel mutations (L227V, E328G, K351E, V411I, M435T, E894G, and E927K) that were absent in 200 normal control chromosomes. All six positive controls were verified. In addition, 19 SNPs were identified, including one non-synonymous, 12 synonymous coding SNPs, and six intronic polymorphisms (table 2).
Table 2.
Identified DNA variants in MYH7
| FHC no. | Location of nucleotide change | Amino acid change | Note |
| 644 | Exon 8, 6475 C>G | L227V† | Novel missense |
| 274 | Exon 11, 7550 A>G | E328G† | Novel missense |
| 739 | Exon 12, 8266 A>G | K351E† | Novel missense |
| 125* | Exon 13, 8847 C>T | R403W | Reported missense |
| 113* | Exon 13, 8848 G>A | R403Q | Reported missense |
| 400 | Exon 13, 8871G>A | V411I† | Novel missense |
| 1022 | Exon 14, 9070 T>C | M435T† | Novel missense |
| 59* | Exon 14, 9123 C>T | R453C | Reported missense |
| 1227 | Exon 14, 9124 G>A | R453H | Reported missense |
| 676* | Exon 16, 10457 G>A | V606M | Reported missense |
| 484 | Exon 18, 11281 C>T | R663C | Reported missense |
| 1203 | Exon 18, 11281 C>T | R663C | Reported missense |
| 97* | Exon 19, 12147 C>T | R719W | Reported missense |
| 473 | Exon 19, 12148 G>A | R719Q | Reported missense |
| 1232 | Exon 19, 12148 G>A | R719Q | Reported missense |
| 6 | Exon 21, 12765 G>A | R787H | Reported missense |
| 114 | Exon 23, 13968 A>G | E894G† | Novel missense |
| 280 | Exon 23, 13968 A>G | E894G† | Novel missense |
| 743 | Exon 23, 13968 A>G | E894G† | Novel missense |
| 707* | Exon 23, 14009 C>G | L908V | Reported missense |
| 1275 | Exon 23, 14066 G>A | E927K† | Novel missense |
| 968 | Exon 32, 17153 C>G | S1519C‡ | Novel coding SNP |
| 22 | Exon 3, 4582 C>T | T63T | Reported coding SNP |
| 976 | Exon 7, 6311 A>G | A199A | Reported coding SNP |
| 1053 | Exon 8, 6528 T>C | F244F | Reported coding SNP |
| 895 | Exon 11, 7542 C>T | D325D | Reported coding SNP |
| 1101 | Exon 12, 8310 G>A+8343 C>T | K365K+D376D | Reported coding SNPs |
| 8 | Exon 12, 8310 G>A | K365K | Reported coding SNP |
| 11 | Exon 12, 8310 G>A+8277 C>T | K365K+G354G | Reported coding SNPs |
| 1204 | Exon 12, 8277 C>T+8343 C>T | G354G+D376D | Reported coding SNPs |
| 1146 | Exon 12, 8277 C>T | G354G | Reported coding SNP |
| 1199 | Exon 12, 8343 C>T | D376D | Reported coding SNP |
| 739 | Exon 12, 8266 A>G+8310 G>A | K351E+K365K | Novel and reported coding SNPs |
| 256 | Exon 24, 14438 T>C | I989I | Reported coding SNP |
| 579 | Exon 24, 14438 T>C+14507 C>T | I989I+A1022A | Reported and novel coding SNPs |
| 482 | Exon 25, 15807 G>A | A1061A | Novel coding SNP |
| 451 | Exon 27, 17852 G>A | Q1127Q | Novel coding SNP |
| 1199 | Intron 2, 4360 G>T+ Exon 3, 4582 C>T | Non-coding and T63T | Novel non-coding and reported coding SNPs |
| 256 | Intron 2, 4360 G>T | Non-coding | Novel non-coding SNP |
| 674 | Intron 4, 5217 G>A | Non-coding | Novel non-coding SNP |
| 2079 | Intron 19, 12245 A>G | Non-coding | Novel non-coding SNP |
| 973 | Intron 26, 17833insC | Non-coding | Novel non-coding SNP |
| 1199 | Intron 29, 18910 C>T | Non-coding | Novel non-coding SNP |
| 929 | Intron 38, 23485 G>A | Non-coding | Novel non-coding SNP |
Bold fonts are novel DNA variants based on the literature and dbSNP (Build 121, http://www.ncbi.nlm.nih.gov/SNP). The base numbers are based on the reference sequence (GenBank accession X52889).
*Known MYH7 mutations that have been reported previously10,11; †novel missense mutations were identified in patients with FHC, but absent from 200 normal control chromosomes; ‡this DNA variant was identified in an FHC proband; however, it was also present in 200 normal control chromosomes, with a population frequency of 3%.
FHC, familial hypertrophic cardiomyopathy; SNP, single nucleotide polymorphism.
The M435T, R453C, and R453H changes identified in the exon 14 amplicon had unique elution profiles (fig 2A). Even within the same codon, two point mutations one base pair apart—R453C (M2-9123, C>T) and R453H (M3-9124, G>A)—had distinct profiles. Similar observations were made for R403W–R403Q and R719W–R719Q, which are also one base pair apart. Moreover, the same point mutations had a consistent DHPLC profile—for example, the E894G mutation could be identified in three apparently unrelated families (fig 2B) and the R663C and R719Q changes were the same in two sets of two unrelated families. Combinations of multiple DNA variants altered the elution profiles and different combinations resulted in unique profiles (fig 2C).
Figure 2.
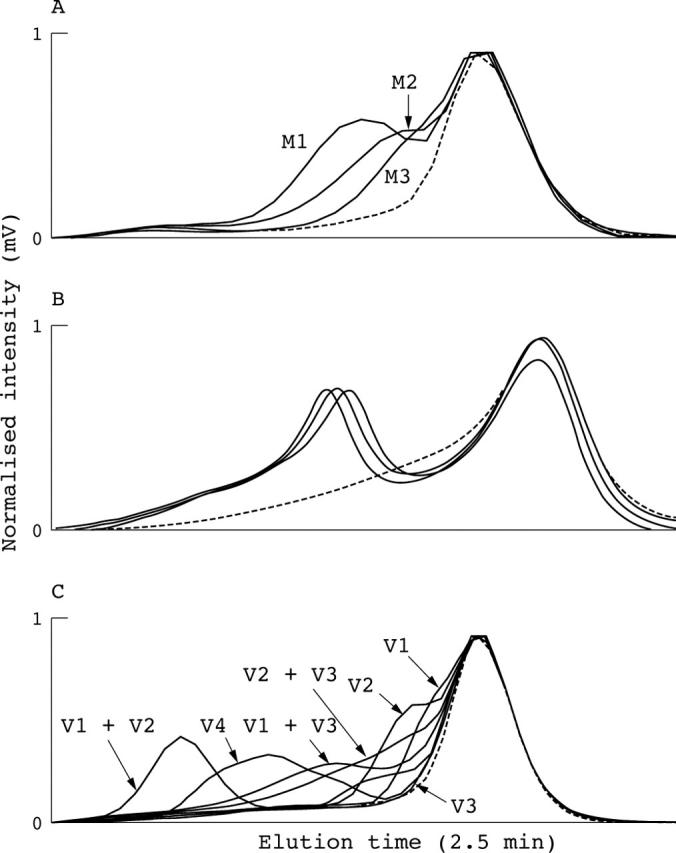
Detection of unknown DNA variants. All denaturing high performance liquid chromatography (DHPLC) profiles are detected at the optimal temperatures and are shown after normalisation by Navigator software. The wild-type control is indicated by the dashed line. (A) Different mutations have unique profiles. The M435T, R453C, and R453H changes were found in the MYH7 exon 14 amplicon (M1 = 9070 T>C, M2 = 9123 C>T, and M3 = 9124 G>A, respectively). M2 and M3 at the same codon of R453 are two point mutations one base pair apart, but they have distinct profiles. (B) The same changes have consistent profiles, especially obvious after normalisation. A missense change (MYH7 exon 23, 13968 A>G, E894G) was found in three different unrelated patients. (C) Combinations of different variants have unique DHPLC profiles. Multiple DNA variants were found in the MYH7 exon 12 amplicon: V1 = 8310 G>A, K365K; V2 = 8277 C>T, G354G; V3 = 8343 C>T, N376N; and V4 = 8266 A>G+8310 G>A; K351E+K365K.
To reduce the number of amplicons analysed by DHPLC, especially when screening for rare mutations, we tested a pooling strategy by mixing multiple DNA samples before PCR amplification. A sample containing a heterozygous polymorphism (MYH7 exon 38, 23485 G>A) was mixed with wild-type sample at a frequency of one variant/diploid up to one variant/40 chromosomes (fig 3). The polymorphism was successfully identified at a range of 2.5–50% chromosomes.
Figure 3.
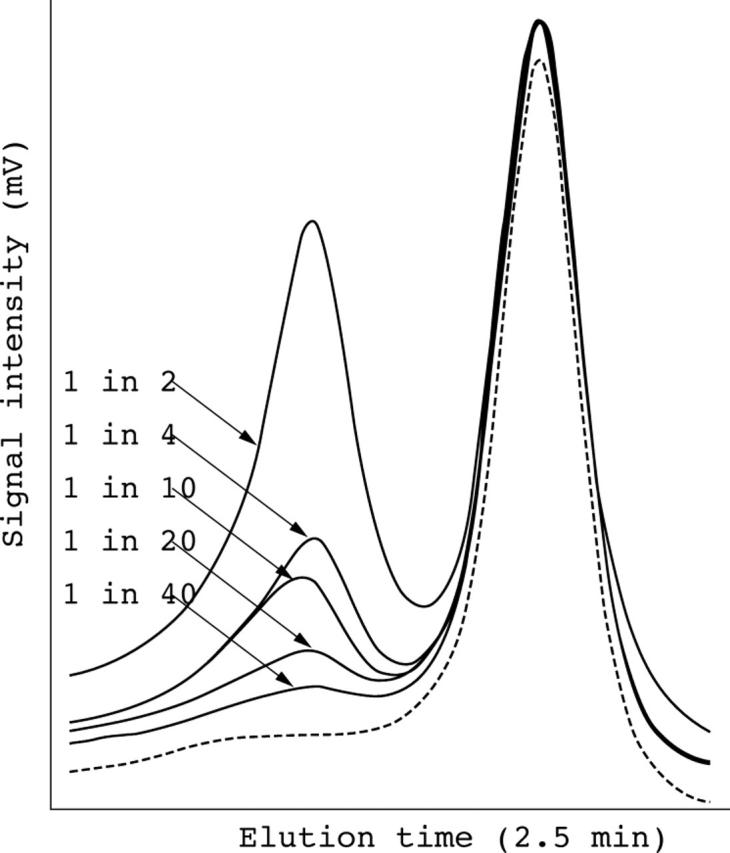
Sensitivity of pooling DNA samples. A heterozygous single nucleotide polymorphism was identified in MYH7 intron 38 (23485 G>A) in a sample. Various amounts of known wild-type sample (10 ng/sample) were mixed with the above heterozygous sample before polymerase chain reaction amplification. Different profiles after normalisation by Navigator software are shown (one variant in 2–40 copies of normal alleles). The homozygous wild-type control is indicated by a dashed line.
SNP discovery, validation, and genotyping in MND
In PVR exon 2, one sample with the A67T change (15952 G>A) was identified via direct sequencing.13 To validate that this coding SNP was polymorphic in our target subjects, DHPLC was used for genotyping. Initial genotyping was performed at 63.4°C based on Navigator software. However, the mutant DNA could not be differentiated at this temperature or even at 65.9°C (fig 4A). The DHPLC profile change was seen when the temperature was increased to 66.4°C, and was optimal at 66.9°C (fig 4B). The optimal temperature was then re-estimated using the δ helicity algorithm (http://www.mutationdiscovery.com/), and was found to be 67°C.
Figure 4.
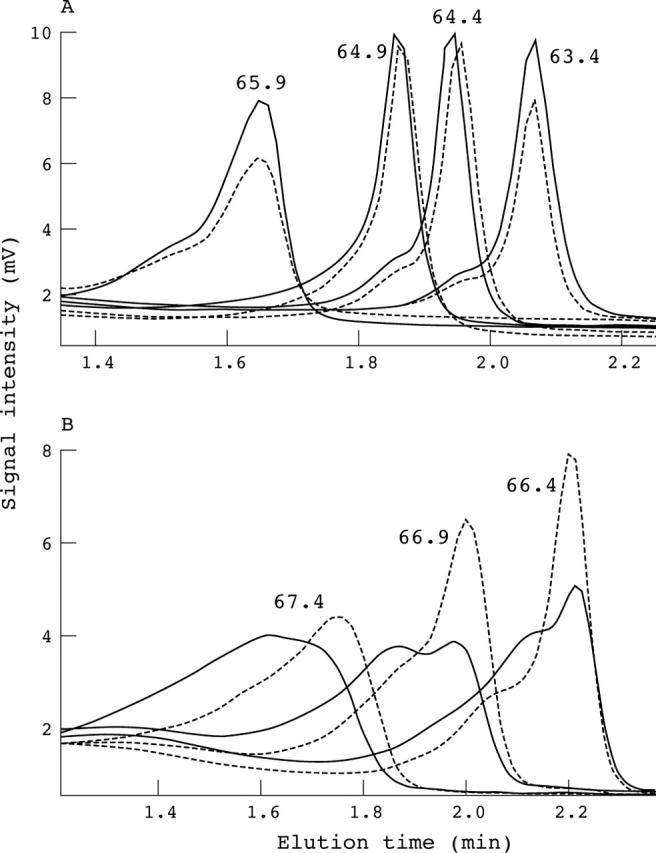
Denaturing high performance liquid chromatography (DHPLC) analysis at different temperatures without normalisation. The wild-type control is indicated by the dashed line. (A) Samples with and without a heterozygous A67T single nucleotide polymorphism in PVR exon 2 run at 63.4, 64.4, 64.9, and 65.9°C. No significant difference was seen between the A67T heterozygote and the wild type. (B) Samples with and without the heterozygous A67T allele run at 66.4, 66.9, and 67.4°C with a timeshift of 0.8 minutes. Significant changes in DHPLC profiles were seen compared with the wild type at these three temperatures, with the most obvious one at 66.9°C.
Subsequently, genotyping was performed at 64.1°C (manually adjusted from 63.4°C) and 66.9°C in 140 MND cases and 280 controls. In total, 840 injections were completed within a week. The frequency of the A67T change in MND cases (13.6%) was significantly higher than in controls (6.8%; p = 0.03; odds ratio, 2.16; 95% confidence interval, 1.10 to 4.22). All genotyping results were confirmed by CfoI restriction enzyme digestion. Two novel coding SNPs (E116E, 16101 G>A and R133R, 16152 G>A) were identified by DHPLC analysis (fig 5). The E116E SNP was present at both temperatures (fig 5A, B), whereas a second SNP (R133R) was seen only at 64.1°C, and not at 66.9°C (fig 5C, D).
Figure 5.

Denaturing high performance liquid chromatography (DHPLC) profiles of DNA variants at two different temperatures without normalisation. A sample with a heterozygous change in PVR exon 2 (E116E, 16101 G>A) shows significant differences in DHPLC profile compared with the wild-type control (dashed line) at both 64.1°C (A) and 66.9°C (B). A second heterozygous change in PVR exon 2, (R133R, 16152 G>A) shows a significant difference at 64.1°C (C) only, and not at 66.9°C (D).
DISCUSSION
Mutation detection and SNP discovery in large genomic areas require automated high throughput technologies. Ideally, there should be minimal modification of PCR components and post PCR steps. Although considered the gold standard, direct DNA sequencing to detect variants has drawbacks, including two post PCR purifications, false positive signals as a result of data derivation from non-variant sequence background, and a low signal to background noise ratio.1 Data analysis can be effort intensive and error prone, especially when screening large numbers of samples.
DHPLC analysis meets most of the above requirements for high throughput detection of DNA variants, and has become the method of choice.1,16 It is considerably cheaper and simpler than sequencing, and enables sequencing to be limited to a few abnormal amplicons identified by DHPLC. In our current study, we demonstrated the high throughput capacity of DHPLC by screening 150 FHC families for disease causing mutations in a large multi-exon gene (MYH7). PVR A67T SNP was also efficiently genotyped in 420 subjects. Twenty one SNPs were identified from analysis of the MYH7 and PVR genes. Our results were consistent with previous evaluations, which concluded that DHPLC is an accurate, reliable, and cost effective method for mutation screening and SNP discovery.1,16–19
Several conditions are important to ensure optimal results in high throughput DHPLC analysis. First, specific and efficient PCR amplification is crucial to avoid false positives because group analysis is essential in high throughput work. It is hard to correct samples manually one by one for their intensity. Normalisation of DHPLC signals below 2 mV is prone to false positives.
Second, to optimise resolution in the rapid analysis option, the elution window and times must be adjusted so that heteroduplex and homoduplex peaks are separated by at least 1.5 minutes from the injection peak and 0.4 minutes from the wash peak (fig 4).
Next, prediction of the melting temperature is essential. Regions that differ by > 10°C in the same amplicon should be avoided. If this is not possible, the addition of a GC clamp may be necessary, as is seen in the MYH7 exon 27 amplicon (table 1), to reduce the number of temperatures required to screen an amplicon. However, its use should be minimised because PCR yields may fall.20 In our current study, temperatures predicted by Navigator software usually allowed the successful detection of most DNA variants, regardless of their number and type. However, the estimated temperature of 63.4°C failed to identify the PVR A67T change (fig 4A). The correct higher temperature was predicted by the δ helicity algorithm, so the use of different algorithms can be helpful in predicting melting temperature.
Finally, to maintain continued efficiency and reproducibility over large sample sizes, known mutation samples with defined profiles should be checked regularly for quality control. The DNASep cartridge showed satisfactory reproducibility, which extended over 6000 injections in our experience. However, the profile for some variants differed slightly between and within columns over time. This reinforces the importance of the positive controls during each run when genotyping.
“The identification of DNA variants with low level mosaicism is an additional feature of denaturing high performance liquid chromatography analysis”
For all samples tested, DHPLC was found to be reproducible and unique for a particular variant (fig 2). With experience, specificity for detecting DNA variants can approach 100% because all positives for MYH7 mutation detection or PVR SNP genotyping were confirmed by direct sequencing or restriction fragment length polymorphism analysis/amplification refractory mutation system analysis. All six FHC families with known MYH7 mutations were identified using the estimated optimal temperatures, suggesting high sensitivity, although this cannot be determined accurately because we sought unknown mutations. Nevertheless, the sensitivity in our study was probably close to 100% because false negatives were not found in PVR genotyping of 420 individuals when compared with the genotypes obtained from restriction fragment length polymorphism analysis. Although the prevalence of MYH7 mutations in our study population (14%) is lower than some earlier estimations, it is not inconsistent with more recently reported mutation frequencies (12–13%) for this gene.21,22
The identification of DNA variants with low level mosaicism is an additional feature of DHPLC analysis.19,23,24 We found that DHPLC could detect as few as 2.5% variants (fig 3) when a sample was pooled with up to 20 normal samples, as reported previously.23 However, care should be taken because the DHPLC positive signal diminished rapidly with increasing numbers of normal chromosomes. This function is superior to direct sequencing and can be used for the detection of somatic mosaicism, loss of heterozygozity in tumour DNA, and heteroplasmy with mitochondrial DNA.1,18,23,25
With the software provided, objective comparisons using DHPLC analysis were more sensitive and less time consuming than subjective assessment using single stranded conformational polymorphism or heteroduplex analysis. In most cases, heterozygous profiles consisted of multiple peaks that were easily distinguishable from the wild-type profile. In others, heterozygotes were only distinguishable from the wild-type by a shoulder in the peak, or by a wider peak (for example, M2 and M3, respectively, in fig 2A). In these circumstances, the ability to overlay and normalise chromatographic profiles with the wild-type profile enhanced identification.
Take home messages.
We found denaturing high performance liquid chromatography (DHPLC) to be a high throughput, automated technique that enabled rapid screening of a large gene (MYH7) in patients with familial hypertrophic cardiomyopathy
The estimation of melting temperature using two different algorithms, in addition to empirical testing with a few additional temperatures, made this technique highly sensitive and specific for DNA variant detection
DHPLC also offered considerable advantages for efficient single nucleotide polymorphism validation, as assessed by analysis of the poliovirus receptor gene in motor neurone disease
DHPLC is distinct from other DNA variant detection techniques because of its purification capacity in conjunction with a fragment collector. DHPLC requires only 8–16 μl of PCR product for screening, so the remaining product from a 50 μl reaction can be used for confirmatory procedures, such as sequencing or cloning. The ability to screen only for heterozygous variants is a limitation of DHPLC analysis. However, this can be overcome by a mixing strategy—a homozygous mutant can be mixed with a “wild-type” sample at a 1 : 1 ratio before denaturing to generate the heteroduplex population.
In summary, we found that DHPLC offers a high throughput, automated technique that enabled rapid screening of a large gene (MYH7) in many patients with FHC. With the estimation of melting temperature using two different algorithms, in addition to empirical testing with a few additional temperatures, we found this technique to be highly sensitive and specific for DNA variant detection. It also offers considerable advantages for efficient SNP validation, as shown in our MND study.
Acknowledgments
This study was partially supported by the National Heart Foundation of Australia.
Abbreviations
DHPLC, denaturing high performance liquid chromatography
FHC, familial hypertrophic cardiomyopathy
MND, motor neurone disease
MYH7, cardiac β-myosin heavy chain gene
PCR, polymerase chain reaction
PVR, poliovirus receptor gene
SNP, single nucleotide polymorphism
REFERENCES
- 1.Taylor CF, Taylor GR. Current and emerging techniques for diagnostic mutation detection: an overview of methods for mutation detection. Methods Mol Med 2004;92:9–44. [DOI] [PubMed] [Google Scholar]
- 2.Wang DG, Fan JB, Siao CJ, et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science 1998;280:1077–82. [DOI] [PubMed] [Google Scholar]
- 3.Gross E, Arnold N, Goette J, et al. A comparison of BRCA1 mutation analysis by direct sequencing, SSCP and DHPLC. Hum Genet 1999;105:72–8. [DOI] [PubMed] [Google Scholar]
- 4.Jones AC, Austin J, Hansen N, et al. Optimal temperature selection for mutation detection by denaturing HPLC and comparison to single-stranded conformation polymorphism and heteroduplex analysis. Clin Chem 1999;45:1133–40. [PubMed] [Google Scholar]
- 5.Klein B, Weirich G, Brauch H. DHPLC-based germline mutation screening in the analysis of the VHL tumor suppressor gene: usefulness and limitations. Hum Genet 2001;108:376–84. [DOI] [PubMed] [Google Scholar]
- 6.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA 2002;287:1308–20. [DOI] [PubMed] [Google Scholar]
- 7.Gibbs RA, Belmont JW, Hardenbol P, et al. The international HapMap project. Nature 2003;426:789–96. [DOI] [PubMed] [Google Scholar]
- 8.Cox D, Boillot C, Canzian F. Data mining: efficiency of using sequence databases for polymorphism discovery. Hum Mutat 2001;17:141–50. [DOI] [PubMed] [Google Scholar]
- 9.Talbot K . Motor neurone disease. Postgrad Med J 2002;78:513–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smart RV, Yu B, Le H, et al. DNA testing in familial hypertrophic cardiomyopathy: clinical and laboratory implications. Clin Genet 1996;50:169–75. [DOI] [PubMed] [Google Scholar]
- 11.Semsarian C, Yu B, Ryce C, et al. Sudden cardiac death in familial hypertrophic cardiomyopathy: are “benign” mutations really benign? Pathology 1997;29:305–8. [DOI] [PubMed] [Google Scholar]
- 12.Yu B, French JA, Jeremy RW, et al. Counselling issues in familial hypertrophic cardiomyopathy. J Med Genet 1998;35:183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sauderson R, Yu B, Trent RJ, et al. Persistent infection polymorphism in the polio virus receptor gene differs in motor neuron disease. Neuroreport 2004;15:383–6. [DOI] [PubMed] [Google Scholar]
- 14.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988;16:1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber CG, Oefner PJ, Bonn GK. High-resolution liquid chromatography of oligonucleotides on nonporous alkylated styrene–divinylbenzene copolymers. Anal Biochem 1993;212:351–8. [DOI] [PubMed] [Google Scholar]
- 16.Xiao W, Oefner PJ. Denaturing high-performance liquid chromatography: a review. Hum Mutat 2001;17:439–74. [DOI] [PubMed] [Google Scholar]
- 17.Kuklin A, Munson K, Gjerde D, et al. Detection of single-nucleotide polymorphisms with the WAVE DNA fragment analysis system. Genet Test 1997;1:201–6. [DOI] [PubMed] [Google Scholar]
- 18.Liu W, Smith DI, Rechtzigel KJ, et al. Denaturing high performance liquid chromatography (DHPLC) used in the detection of germline and somatic mutations. Nucleic Acids Res 1998;26:1396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Donovan MC, Oefner PJ, Roberts SC, et al. Blind analysis of denaturing high-performance liquid chromatography as a tool for mutation detection. Genomics 1998;52:44–9. [DOI] [PubMed] [Google Scholar]
- 20.McDowell DG, Burns NA, Parkes HC. Localised sequence regions possessing high melting temperatures prevent the amplification of a DNA mimic in competitive PCR. Nucleic Acids Res 1998;26:3340–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erdmann J, Daehmlow S, Wischke S, et al. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin Genet 2003;64:339–49. [DOI] [PubMed] [Google Scholar]
- 22.Havndrup O, Bundgaard H, Andersen PS, et al. Outcome of clinical versus genetic family screening in hypertrophic cardiomyopathy with focus on cardiac beta-myosin gene mutations. Cardiovasc Res 2003;57:347–57. [DOI] [PubMed] [Google Scholar]
- 23.Jones AC, Sampson JR, Cheadle JP. Low level mosaicism detectable by DHPLC but not by direct sequencing. Hum Mutat 2001;17:233–4. [DOI] [PubMed] [Google Scholar]
- 24.Pan KF, Liu W, Lu YY, Zhang L, et al. High throughput detection of microsatellite instability by denaturing high-performance liquid chromatography. Hum Mutat 2003;22:388–94. [DOI] [PubMed] [Google Scholar]
- 25.Conley YP, Brockway H, Beatty M, et al. Qualitative and quantitative detection of mitochondrial heteroplasmy in cerebrospinal fluid using denaturing high-performance liquid chromatography. Brain Res Brain Res Protoc 2003;12:99–103. [DOI] [PubMed] [Google Scholar]