

Abstract
Background/Aims: In vivo autofluorescence endoscopic imaging and spectroscopy have been used to detect and differentiate benign (hyperplastic) and preneoplastic (adenomatous) colonic lesions. This fluorescence is composed of contributions from the epithelium, lamina propria, and submucosa. Because epithelial autofluorescence in normal and diseased tissues is poorly understood, this was the focus of the present study.
Methods: Whole colonic crypts were isolated, and short term primary cultures of epithelial cells were established from biopsies of normal, hyperplastic, and adenomatous colon. Autofluorescence (488 nm excitation) was examined by confocal fluorescence microscopy. Fluorescently labelled organelle probes and transmission electron microscopy were used to identify subcellular sources of fluorescence.
Results: Mitochondria and lysosomes were identified as the main intracellular fluorescent components in all cell types. Normal and hyperplastic epithelial cells were weakly autofluorescent and had similar numbers of mitochondria and lysosomes, whereas adenomatous (dysplastic) epithelial cells showed much higher autofluorescence, and numerous highly autofluorescent lysosomal (lipofuscin) granules.
Conclusions: Short term primary cell cultures from endoscopic biopsies provide a novel model to understand differences in colonic tissue autofluorescence at the glandular (crypt) and cellular levels. The differences between normal, hyperplastic, and adenomatous epithelial cells are attributed in part to differences in the intrinsic numbers of mitochondria and lysosomes. This suggests that the detection of colonic epithelial fluorescence alone, if possible, may be sufficient to differentiate benign (hyperplastic) from preneoplastic and neoplastic (adenomatous) colonic intramucosal lesions during in vivo fluorescence endoscopy. Furthermore, highly orange/red autofluorescent intracellular granules found only in dysplastic epithelial cells may serve as a potential biomarker.
Keywords: autofluorescence, colonic dysplasia, primary cell culture, gastrointestinal, confocal fluorescence microscopy, microfluidics
Colon cancer continues to be associated with high mortality rates, primarily because of the failure to detect neoplastic lesions at an early stage.1 Fluorescence endoscopic imaging or point spectroscopy, using ultraviolet or blue light excitation, which detect endogenous (auto)fluorescence emission in the green and red regions of the spectrum, have been used as an adjunct to white light endoscopy to differentiate between hyperplastic and adenomatous colon polyps.2–5 In general, dysplastic lesions have enhanced red fluorescence compared with either normal colon or hyperplastic polyps, and initial clinical studies of autofluorescence endoscopy have shown increased sensitivity and specificity for the detection of colonic adenomas when used adjunctively with white light endoscopy.6–9
“Fluorescence endoscopic imaging or point spectroscopy have been used as an adjunct to white light endoscopy to differentiate between hyperplastic and adenomatous colon polyps”
Several groups have investigated the changes in tissue between normal and abnormal colonic tissues that underlie autofluorescence endoscopy/spectroscopy (RS DaCosta. Mechanisms of fluorescence endoscopy of the human colon. MSc Thesis, Department of Medical Biophysics, University of Toronto, Ontario, Canada, 2000).2,10,11 Possible morphological contributions to the increased red fluorescence relative to green fluorescence in adenomas are mucosal thickening and microvascularity. These contribute to reduce the excitation light intensity reaching the submucosa and, hence, its green collagen fluorescence, and also preferentially attenuate the shorter wavelengths in the fluorescence emission. However, because autofluorescence imaging can differentiate between hyperplastic and adenomatous polyps of the same general size and shape, and also normal mucosa from flat adenomas,6 these gross morphological changes alone cannot provide a complete explanation. Additional microscopic—that is, cellular—factors must also play a role by changing the intrinsic fluorescence of the tissue layers.
Several groups have used fluorescence microscopy to study the fluorescence characteristics of colon tissue, and have exclusively used full thickness sections from biopsies.10–13 In the submucosa, collagen and elastin are the main fluorophores in normal colon and in all stages of preneoplasia and tumour development. However, the mucosal layer shows complex changes in the spectral and spatial distribution of fluorescence between normal, hyperplastic, and adenomatous tissues, as follows.
In the normal lamina propria, thin stands of loosely arranged collagen and elastin show weak green fluorescence, whereas circulating macrophages have bright yellow/orange/red fluorescence.5,14 The increased lamina propria collagen in hyperplastic polyps produces intense green fluorescence. This is not seen in adenomatous mucosa, but the lamina propria does contain numerous highly fluorescent macrophages (RS DaCosta. MSc Thesis, 2000).11
In general, the macroscopic autofluorescence features of colonic tissue are well characterised. In contrast, the autofluorescence characteristics of colonic epithelial cells have not been investigated in detail. Because preneoplastic lesions usually develop intramucosally, and are thought to be of epithelial origin,1 understanding the contribution of epithelial cells to autofluorescence in vivo is important.
Although fluorescence microscopy and microspectrofluorimetry of snap frozen biopsies are straightforward, they clearly alter the metabolic state—for example, the redox potential—which may change the fluorescence characteristics compared with in vivo tissues. However, there are no known published reports on autofluorescence of living colonic cells. In fact, the phenomenon of cellular autofluorescence has been a source of unwanted background interference when labelling with organelle or cell specific fluorophores in most cellular fluorescence microscopy studies and in flow cytometry.15,16 However, a few groups have explicitly studied the autofluorescence of established cell lines, such as sarcoma tumour cell lines and immortalised, carcinogen transformed human bronchial epithelial cells.17,18 Cellular autofluorescence has been reported in various cell cultures including mammalian cells,19 pancreatic islet cells,20 human skin fibroblasts,21 and human skeletal cells.22 Specific contributing fluorophores include: aromatic amino acid residues found in most proteins (λex = 200–340 nm, λem = 360–370, 455 nm); reduced pyridine nucleotides (NADH, NADPH), which are cofactors in cellular metabolism and are found mainly in mitochondria, but are also present in the cytoplasm (λex = 360 nm, λem = 460 nm)19; and flavins and flavin nucleotides (riboflavin, flavin mononucleotide, and dinucleotide), which are mostly bound to enzymes as coenzymes of flavoproteins and concentrated in the mitochondria (λex = 360 nm, 445–470 nm, λem = 440 nm, 520 nm).23 Endogenous porphyrins and age related lipofuscin have also been shown to be sources of cellular autofluorescence.21,24
Although cellular autofluorescence is known to depend on the metabolic state,25 no direct studies have reported on the autofluorescence differences between normal, benign, and neoplastic living colonic epithelial cells. There is one recent report in which autofluorescence imaging and excitation and emission matrices were performed on short term cultures of human normal cervical tissue explants.26 In this study, transverse tissue sections (200 μm thick) from fresh biopsies, which remained viable for up to 24 hours in culture medium, showed autofluorescence changes with cellular transformation. Andersson et al examined the effect of the biochemical manipulation of cellular metabolism on autofluorescence in tumour cell lines in vitro.27 Colocalisation with organelle specific fluorescent vital dyes enabled specific subcellular structures (mitochondria and lysosomes) to be identified. This study also showed that, unlike mitochondria, lysosomal autofluorescence was not easily modified using exogenous biochemicals. A more recent use of cellular autofluorescence, using human promyelocytic leukaemic HL60 cells exposed to various apoptosis inducing agents, showed a relation between cellular autofluorescence characteristics and apoptosis induced changes in cells.28 Such studies illustrate the potential usefulness of autofluorescence based microscopy studies for the characterisation of living cells.
The colon provides a unique model to study normal epithelial cells at different stages of proliferation and differentiation, because these depend on the position along the crypt.1 Unfortunately, methods to isolate and establish longterm normal colonic epithelial cultures have generally been unsatisfactory, although five month cultures of epithelial cells from resected normal human fetal colon have been achieved.29,30 There are also a few reports of short term normal cultures from adult colon tissues, but none in which the autofluorescence was studied.31–33
In our present study, short term primary cell cultures of normal, hyperplastic, and dysplastic colonic epithelial cells obtained from biopsies were successfully established. Individual whole crypts were first isolated and imaged by confocal fluorescence microscopy (CFM). Further digestion of the extracellular matrix yielded individual epithelial cells that were similarly imaged to characterise the autofluorescence at different stages of transformation.
MATERIALS AND METHODS
Preparation and assays of primary cell cultures and whole crypts
RPMI-1640 medium (Gibco, Burlington, Ontario, Canada) was used to incubate the primary cell cultures (PCC), supplemented with 10% fetal bovine serum (Wisent Inc, Saint Jean Baptiste de Rouville, Quebec, Canada), 2mM L-glutamine (Sigma-Aldrich Corporation Ltd, Oakville, Ontario, Canada), 100 units/ml penicillin G (Novopharm Limited, Scarborough, Ontario, Canada), 100 μg/ml streptomycin (BioShop, Burlington, Ontario, Canada), 1% kannamycin (Sigma-Aldrich Corporation), and 1% nystatin (Sigma-Aldrich Corporation). RPMI-1640 with double these amounts of penicillin, streptomycin, kannamycin, and nystatin was used for transportation of the biopsies. The tissue digestion solution was prepared as follows (all agents from Sigma): 1 mg/ml collagenase, 1.5% bovine serum albumin, 50 μg/ml DNAse, and 0.95mM CaCl2 were added to 500 ml HAMS F-10 Hepes medium. The pH was then adjusted to 7.4 with NaOH (0.1M).
Human colon biopsies (15 normal, nine hyperplastic, and 11 adenomatous polyps) were obtained with jumbo forceps (FB 50K; Olympus, Tokyo, Japan) during routine surveillance white light endoscopy from 18 patients with informed consent, and under ethical approval of the institutional review board of St Michael’s Hospital, Toronto, Canada. For each polyp, a matched biopsy was also collected from nearby normal colon. One part of each biopsy was fixed for five minutes in formalin, paraffin wax embedded, sectioned, and haematoxylin and eosin stained for pathological diagnosis, whereas another part was prepared for transmission electron microscopy (TEM).
Biopsies were kept in transport medium for less than two hours at room temperature until processing. The procedure for establishing the primary cell cultures was based on that of Csoka, with minor modifications to maximise cell viability and yield (K Csoka. The fluorimetric microculture cytotoxicity assay (FMCA) in assessment of the clinical activity of cytotoxic drugs, with special reference to ovarian carcinoma. PhD Thesis, Division of Clinical Pharmacology, University Hospital, Uppsala University, Sweden, 1997). Briefly, the biopsies were thoroughly minced with a scalpel and washed several times in N-acetylcysteine (Sigma) to remove excess mucin. The homogenised mixture was digested for ∼10–20 minutes at 37°C in 5% CO2/95% air at 90% relative humidity and shaken vigorously several times. The suspension was then centrifuged for five minutes at 112 ×g. The supernatant was discarded and the pellet resuspended in 2 ml supplemented RPMI-1640 media. A Percoll gradient (Pharmacia, Uppsala, Sweden) was prepared, consisting of 2.5 ml each of 25%, 15%, and 10% Percoll in phosphate buffered saline, layered in a centrifuge tube. The cell suspension was pipetted on to the gradient and centrifuged at 30 ×g for five minutes. The isolated tumour cells settled to the bottom, whereas red blood cells remained in the 10–15% layer. The supernatant was discarded and the cell pellet resuspended in supplemented RPMI-1640 medium. The viability of the isolated cells was confirmed by trypan blue exclusion assay and the number of cells/ml was determined in a haemocytometer (Fisher Scientific Company, Ottawa, Ontario, Canada). The isolated epithelial cells were seeded in poly-L-lysine coated four well chambered coverglasses for inverted microscopy (Nalge Nunc International, Rochester, New York, USA) and maintained at 37°C in 5% CO2/95% air at 90% relative humidity. Cells were imaged by CFM within approximately one hour of seeding.
One biopsy of each tissue type was also used to isolate whole crypts, for which collagenase digestion was used for only two to five minutes. The solution containing the partially digested tissue was transferred to four well chambered coverglasses for confocal imaging. To confirm that the isolated cells were of epithelial origin, a subpopulation from each sample was fixed in 10% buffered formalin phosphate for 10 minutes, permeabilised in 0.1% Triton X-100 in 0.1% sodium citrate (two minutes at 4°C), and stained for vimentin with antivimentin 3B4 (American Research Products, Belmont, Massachusetts, USA)34 and cytokeratin with antikeratin AE1/AE3 (Zymed Laboratories, San Francisco, California, USA).35,36 Fluorescein conjugated human monoclonal antibodies against epithelial specific antigen (ESA; Novocastra, Mississauga, Canada) were used to identify the live epithelial cells by incubating separately with the conjugated antibody solution for 30–60 minutes at 37°C in 5% CO2/95% air at 90% relative humidity.
Confocal fluorescence microscopy
Light transmission and CFM were performed on the isolated whole crypts and epithelial cells at room temperature using an inverted confocal fluorescence microscope (model LSM510; Karl Zeiss, Jena, Germany). Cellular autofluorescence was excited with 488 nm from an argon ion laser, with the fluorescence emission detected at > 505 nm, 505–550 nm, and > 585 nm. The confocal pinhole diameter was 160 μm and 10× Fluar and 63× C-Apochromat water immersion objectives (Nikon Corporation, Tokyo, Japan) were used for both transmitted light and fluorescence imaging. All fluorescence images were made with the same settings for each cell type studied and for whole crypts. False colour fluorescent images and transmitted light images (512 × 512 8 bit), were stored and manipulated using the resident Zeiss LSM Image Browser software. This allowed fluorescence intensity line profiles and intensity histograms to be performed, thereby providing mean (SD) autofluorescence intensity values for green (505–550 nm) and red (> 585 nm) emission channels of each image.
CFM images of isolated crypts (normal (n = 15), hyperplastic (n = 18), dysplastic crypts (n = 8)) and individual epithelial cells (normal (n = 45), hyperplastic (n = 35), dysplastic (n = 55)) were collected with a scan time of approximately one second. Background subtraction of fluorescence images was applied by obtaining a confocal fluorescence image of a cell free area of the four well chamber where there were no cells. In addition, images were flat field corrected by dividing by a confocal fluorescence micrograph of a uniform fluorescence standard.
The viability of the isolated cells was checked during and after CFM imaging by incubating in 0.1% trypan blue (Fluka, Oakville, Ontario, Canada) in phosphate buffered saline for one to three minutes and examining by light microscopy (×10 magnification). Cells completely excluding trypan blue were considered viable.
Mitochondrial and lysosomal specific fluorescent probes
Rhodamine 123 (Molecular Probes, Eugene, Oregon, USA) was used to identify mitochondria under CFM. Approximately 4 × 103 each of HT-29 (ATCC, Manasses, Virginia, USA) and RIF-8A (donated by Dr G Singh, Hamilton Regional Cancer Center, Hamilton, Ontario, Canada) were seeded separately in four well chambered coverglasses and incubated for 24 hours at 37°C, in 5% CO2/95% air and 90% relative humidity. The samples were exposed to rhodamine 123 at a concentration of 3μM for 30 minutes before imaging. Using 488 nm excitation, cellular autofluorescence (collected with a > 500 nm fluorescence filter) was differentiated from rhodamine 123 fluorescence with peak fluorescence emission at ∼ 530 nm (detected using a band pass 550 nm (±25) filter). Furthermore, the addition of rhodamine 123 resulted in a significant increase in fluorescence intensity of the cellular organelles, compared with the original autofluorescence signal, indicating colocalisation of the mitochondria and the autofluorescent organelles.
Lysosomes were identified with LysoTracker green DND-26 (Molecular Probes) at a concentration of 75nM for 15 minutes with the same excitation and emission filters. Despite the spectral overlap, the autofluorescence and tracer probe fluorescence could be distinguished.
It should be noted that taking images of the same cell under autofluorescence and then after addition of rhodamine 123 (or Lysotracker green) would have been ideal. However the primary cultured colon epithelial cells were not adherent to the coverslip (even with poly-L-lysine coated coverslips), so that washing of the unattached “labelled” cells, after addition of the labelling agent (rhodamine 123 or Lysotracker green), resulted in movement of the original cell. Thus, imaging of the original cell from which the autofluorescence images was first collected was not possible. This was also true for the uncoupling experiment. HT29 cells, which were adherent, were imaged by autofluorescence and then rhodamine 123 or Lysotracker green was added to them. Images of the cellular autofluorescence and the fluorescence from rhodamine 123 or Lysotracker green labelling was possible from the same cell.
Mitochondrial uncoupling
The mitochondrial toxin carbonyl cyanide m-chlorophenylhydrazone (CCCP; Sigma), which uncouples oxidative phosphorylation, was added to the cells for 10 minutes at 2μM. Autofluorescence imaging was performed before and after this exposure.
Transmission electron microscopy
TEM was used to assess the ultrastructural integrity and features of the isolated epithelial cells. Briefly, normal and dysplastic biopsies were fixed in 2% gluteraldehyde (Fisher Scientific Company, Fair Lawn, New Jersey, USA) plus 0.1M sodium cacodylate buffer (Fisher Scientific Company) for two hours and transferred to 2% gluteraldehyde for ∼ 12 hours, following which they were washed in sodium cacodylate buffer and postfixed in 1% osmium tetroxide. The samples were dehydrated in graded ethanol, infiltrated, and imbedded in Epon 812 resin (Fisher Scientific Company). Ultrathin sections (∼ 70 nm) were cut with a diamond knife, and imaged on a TEM (JEOL-1200EX; JEOL Ltd, Tokyo, Japan), operating at 80 kV.
RESULTS AND DISCUSSION
Typically, the PCC procedure yielded ∼ 1–2 × 106 epithelial cells from each biopsy. The presence of non-epithelial cells was confirmed at the initial stage of disaggregation by haematoxylin and eosin staining. Following the PCC protocol there was little contamination of the epithelial cell population (< 5% fibroblasts), based on cytokeratin and vimentin staining. Subsequent fibroblast overgrowth was also not a problem, because the imaging was performed within two to 10 hours after seeding. The epithelial origin of the cells was further confirmed by CFM imaging with fluorescein conjugated antihuman ESA antibodies. The epithelial specificity of ESA was checked in a co-culture of human HT-29 and RIF-8A cells, in which only HT-29 cells stained positively for ESA.
The viability of the isolated epithelial cells, demonstrated by trypan blue exclusion, varied considerably between PCC samples. Those from normal colon were viable for approximately one to two days, and thereafter become rounded and non-adherent, whereas those from hyperplastic and adenomatous polyps were viable for about five to seven days and remained more adherent to the substrate.
Autofluorescence of isolated normal epithelial cells
Figure 1A shows an example of cellular autofluorescence of isolated normal epithelial cells, elongated and lying side by side, superimposed on a transmitted light micrograph, whereas fig 1B shows only the autofluorescence of the same cells. The non-fluorescent nuclei appear as round spaces located at the basal end of the cells, whereas non-fluorescent mucin vacuoles were located apically. Autofluorescence appeared to be granular (∼ 0.5–1 μm diameter) and was distributed in the cytoplasm. There was little variation in the autofluorescence intensity or spectral features between these granules in the normal cells. Normal epithelial cells were characterised by higher average green (505–550 nm) than red (> 585 nm) autofluorescence (mean (SD) normal epithelial cell autofluorescencegreen count, 233 782 (16 364); mean (SD) normal epithelial cell autofluorescencered count, 152 796 (11 307)). These observations were consistent for all normal colonic epithelial cells studied.
Figure 1.

(A) Confocal autofluorescence micrograph of primary cultured normal colonic epithelial cells superimposed on a corresponding transmitted white light image. (B) autofluorescence only of the same cells. Note the green fluorescent cell in the bottom left hand corner, which was epithelial specific antigen (ESA) negative and, therefore, not of epithelial origin. (C) Fluorescein conjugated anti-ESA staining (false coloured grey). (D) Transmitted light and (E) autofluorescence of primary cultured dysplastic epithelial cells.
Figure 1C shows a grey scale image of the same cells stained with fluorescein conjugated anti-ESA, confirming their epithelial origin. ESA staining also confirmed the epithelial phenotype of all hyperplastic and dysplastic epithelial cells.
Autofluorescence of isolated dysplastic epithelial cells
Figure 1D shows a transmitted light image of primary cultured dysplastic epithelial cells isolated from a tubulovillous adenoma, whereas fig 1E shows the corresponding autofluorescence. Intensity histograms showed that, compared with normal epithelial cells, dysplastic cells had lower average green autofluorescence (505–550 nm) (mean (SD) normal epithelial cell autofluorescencegreen count, 233 782 (16 364); mean (SD) dysplastic epithelial cell autofluorescencegreen count, 115 903 (9272)), whereas the average red autofluorescence (>585 nm) was increased (mean (SD) normal epithelial cell autofluorescencered count, 152 796 (11 307); mean (SD) dysplastic epithelial cell autofluorescencered count, 219 186 (16 220)). This observation was consistent for all the dysplastic cells studied. There was also significantly higher red (> 585 nm) (approximately eight to 10 times normal) relative to green autofluorescence (505–550 nm), which was most pronounced in the apical region of the dysplastic cells. The nuclei of dysplastic cells were always non-fluorescent but, against the background cytoplasmic autofluorescence, were seen to be larger than those of normal epithelial cells, a common hallmark of dysplasia (increased nuclear to cytoplasmic ratio).1 Furthermore, a reduction in mucous vacuoles was seen in most of the dysplastic cells studied, which is also another feature of dysplastic transformation in the colon.1 In general, two different sizes of round fluorescent granules were seen, tightly packed in the apical region: ∼ 0.5–1 μm granules similar to those found in normal epithelial cells, and fewer numbers of ∼ 2–3 μm granules present only in the dysplastic cells. These granules were seen in all the dysplastic cells studied by CFM.
Rhodamine 123 staining
Figure 2A shows a magnified view of the distribution of autofluorescent granules in normal epithelial cells. This image was contrast (×11) and brightness (×11) enhanced in both the green and red fluorescent channels because the autofluorescence is intrinsically weak. The non-fluorescent nuclei are apparent, as are the non-fluorescent mucous vacuoles towards the apical end. Figure 2B shows normal epithelial cells stained with rhodamine 123, and demonstrates the dense perinuclear distribution of the mitochondria, and reduced density near the vacuoles at the apical ends. This distribution generally correlated well with the autofluorescence patterns, indicating that mitochondria were, in part (see LysoTracker staining), responsible for the (weak) autofluorescence of the normal cells.
Figure 2.

Confocal fluorescence micrographs. (A, C) Autofluorescence; (B, D) rhodamine 123 fluorescence. (A, B) Normal epithelial cells; (C, D) dysplastic epithelial cells; (A) and (C) have been brightness and contrast enhanced for display, whereas (B) and (D) are pseudo coloured blue.
Figure 2C shows an example of autofluorescence from a single dysplastic epithelial cell. The source of the thin band of bright red fluorescence at the apical region of the cell is unknown, but was seen in 50–70% of these cells. We hypothesise that this is caused by endogenous bacteria containing porphyrins that may not have been removed during the cell isolation procedure.37 The non-fluorescent nucleus is seen close to the base. Many of the large and highly yellow/orange/red fluorescent granules are visible in the cytoplasm, together with homogeneously distributed small, weakly green fluorescing granules. Figure 2D shows an example of a rhodamine 123 stained dysplastic epithelial cell, and demonstrates the colocalisation of the mitochondria and the green fluorescent granules.
LysoTracker staining
Figure 3A shows the weakly positive staining of lysosomes in normal epithelial cells using LysoTracker green. This staining reveals a diffuse distribution of lysosomes in the cytoplasm, which may also contribute to the autofluorescence pattern seen in the cytoplasm of these cells.
Figure 3.

Confocal fluorescence micrographs showing LysoTracker lysosomal staining of (A) normal epithelial cells, (B) isolated dysplastic crypts, and (C) disaggregated dysplastic epithelial cells (pseudo coloured red).
Figure 3B and C shows isolated dysplastic crypts and individual dysplastic epithelial cells, respectively, stained with LysoTracker green. The apical portions of the crypts are heavily stained by LysoTracker green. The individual cells show several lysosomes in the cytoplasm. Compared with normal epithelial cells, dysplastic crypts and cells were found to contain large numbers of individual lysosomes and lysosomal aggregates.
Mitochondrial uncoupling
The mitochondrial uncoupler CCCP was used to provide an additional check on the mitochondrial source of the intracellular fluorescent granules. CCCP uncouples the electron transport chain, resulting in an accumulation of fluorescent FAD. As illustrated in fig 4, after 10 minutes of exposure, CCCP increased the autofluorescence intensity of all the cytoplasmic granules. Comparison with the known FAD fluorescence emission (500–580 nm) characteristics at 488 nm excitation38 indicated that mitochondrial FAD is partially responsible for the green (505–550 nm) autofluorescence seen in the cytoplasm of normal and dysplastic epithelia.
Figure 4.

Autofluorescence micrograph of normal epithelial cells, (A) before and (B) after the addition of m-chlorophenylhydrazone.
Autofluorescence of isolated whole crypts
Figure 5A shows the red and green autofluorescence of normal crypts. The signal was relatively weak, but images of reasonable signal to noise ratio could be obtained by increasing the diameter of the confocal pinhole. The top third of the crypt was more fluorescent (for both green and red) than the middle, whereas the crypt base had the highest intensity. The band of nuclei located basally within the epithelial cells was non-fluorescent. A few brightly autofluorescent cells were scattered near the basal side along the length of the crypts and, particularly, near the base (magnified in fig 5C). The use of the term “basal” refers to any region closest to the basement membrane, whereas the term “base” refers to the bottom end of isolated crypts. Schmorl staining (fig 5D) was used to confirm that these were endocrine cells, which are known to reside at the base of normal crypts.1 This feature was seen in all isolated normal crypts examined (n = 35).
Figure 5.
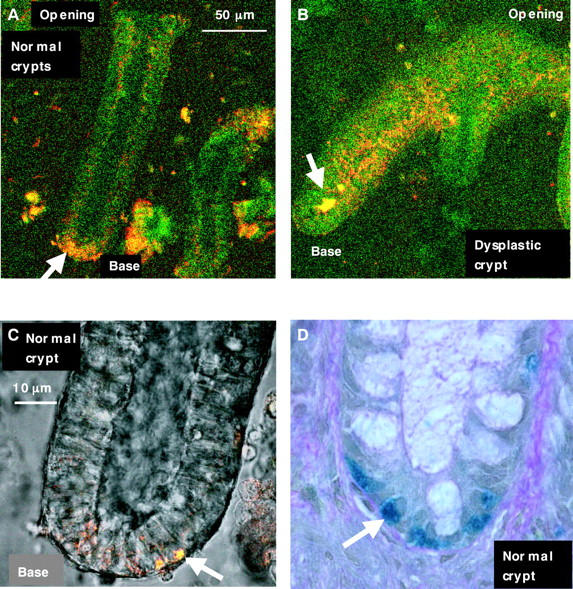
Autofluorescence micrograph of isolated living (A) normal colonic crypt and (B) dysplastic crypt. Note the intensely fluorescent granules at the base of the normal crypt (arrow). (C) A magnified view of these granules is shown overlapped on to the transmission image. (D) A normal crypt base stained with Schmorl indicating endocrine cells (arrow).
Figure 5B shows the autofluorescence micrograph of a dysplastic crypt. In general, the use of collagenase to isolate dysplastic crypts had limited success, partly because of their fragility during processing, as also found by Araki and Ogata.39 However, some fluorescence images were captured before the crypts disintegrated. Most isolated dysplastic crypts were visualised as elongated fan-like structures, with several protuberances and a few short branchings. Some heterogeneity was seen in the autofluorescence emission characteristics along the height of single dysplastic crypts, such that some segments of the dysplastic crypts were more fluorescent than others. The reason for this is unknown. A large number of small bright red fluorescent granules were also found in the apical region along the length of the crypts. Bright yellow fluorescent structures (arrow in fig 5B) were seen within the lumen of some dysplastic crypts but were unidentifiable by CFM alone. It is thought that these are autofluorescent luminal contents (such as faecal bacteria) remaining after isolation of some crypts, although this needs to be studied further.
Figure 6 shows the autofluorescence micrograph of an isolated hyperplastic crypt. In general, the green and red autofluorescence intensities of these were higher along the basal side and length compared with normal crypts. This finding was inconsistent with previous ex vivo observations using CFM to characterise the autofluorescence of frozen tissue sections of hyperplastic mucosa, in which the autofluorescence of these crypts was weak, similar to normal crypts. The reason for this difference is not clear. The development of hyperplastic polyps is accompanied by an increase in the collagen content of the basal membrane and the surrounding subepithelial collagen sheath,40 but this is unlikely to be the explanation for this increase in fluorescence because the collagenase used to isolate the crypts separates the epithelial cells from the basement membranes. It may be caused by an increase in cellular metabolic activity, associated with hyperproliferation, a known characteristic of hyperplastic polyps.41,42 The observations of cellular autofluorescence in isolated living normal and dysplastic colonic epithelial cells were consistent with those previously noted in ex vivo tissue studies using CFM to characterise the autofluorescence of frozen tissue sections (RS DaCosta. MSc Thesis, 2000).5 For example, similar to ex vivo tissues, cultured normal epithelial cells showed weak green autofluorescence, whereas cultured dysplastic epithelial cells contained highly orange/red fluorescent intracellular “granules” (RS DaCosta. MSc Thesis, 2000).
Figure 6.

Autofluorescent micrograph of an isolated hyperplastic crypt.
TEM of normal and dysplastic epithelial cells
The distribution of the autofluorescent granules seen in normal epithelial cells was compared with TEM of normal colon mucosa (fig 7A). The size and distribution of these granules were consistent with those of mitochondria and lysosomes. In particular, the fluorescent granules in the basal region corresponded to the location of mitochondria in the TEM images. A similar autofluorescence pattern was seen in hyperplastic epithelial cells (data not shown). Moderately electron dense granules were seen in the same apical locations of dysplastic epithelial cells by TEM (fig 7B, C). Based upon previous studies of autofluorescence and TEM, these structures are probably lipofuscin, a lipid peroxidation byproduct.43–46 The TEM results were consistent for all normal and dysplastic samples studied.
Figure 7.

Transmission electron microscopy micrographs of (A) a normal colonic crypt (scale bar, 2 μm) and (B) a dysplastic crypt (scale bar, 5 μm). The inset (C) shows a magnified view of the pigmented structures in the apical region of the dysplastic crypt, identified as “lipofuscin-like” material (scale bar, 1 μm).
CONCLUSIONS
We have described a novel biological model that allows the autofluorescence characterisation of isolated “living” colonic normal and dysplastic crypts and epithelial cells, which more accurately reflects autofluorescence measured in vivo. All normal epithelial cells had weak autofluorescence, originating from mitochondria (for example, FAD autofluorescence) and diffusely distributed lysosomes. Epithelial cells from hyperplastic polyps were similar in autofluorescence intensity to normal cells. Dysplastic epithelial cells had an overall increased red autofluorescence intensity compared with normal and hyperplastic cells. A significant increase in orange/red fluorescence (λem > 585 nm) was seen in the apical regions of the dysplastic cells relative to green fluorescence (λem = 505–550 nm), as a result of the presence of large numbers of highly autofluorescent granules, which were shown to be lysosomes. The green fluorescence in dysplastic cells is attributed to mitochondria (FAD autofluorescence).
Take home messages.
We used short term primary cell cultures from endoscopic biopsies to investigate autofluorescence at the glandular (crypt) and cellular levels
The differences between normal, hyperplastic, and adenomatous epithelial cells were attributable in part to differences in the intrinsic numbers of mitochondria and lysosomes
The detection of colonic epithelial fluorescence alone might be sufficient to differentiate benign (hyperplastic) from preneoplastic and neoplastic (adenomatous) colonic intramucosal lesions during in vivo fluorescence endoscopy
Importantly, highly orange/red autofluorescent intracellular granules found only in dysplastic epithelial cells may serve as a potential biomarker
Normal colonic crypts were weakly fluorescent except for several bright yellow/orange endocrine cells concentrated at the crypt base and scattered sparsely along the crypt height. Dysplastic crypts showed increased red fluorescence generally along the central core of the crypt, corresponding to the presence of red fluorescent granules in the apical regions of the epithelium (lysosomes). The brightly fluorescent endocrine cells were seen only for some dysplastic crypts, and generally not at the crypt base. Hence, as normal colonic crypts become dysplastic, changes in mitochondrial and lysosomal composition, and the presence of endocrine cells, contribute to changes in autofluorescence intensity and spectral features.
“Dysplastic epithelial cells had an overall increased red autofluorescence intensity compared with normal and hyperplastic cells”
We have characterised autofluorescence at the single cell level, although the isolation and CFM imaging of individual cultured epithelial cells were technically challenging and time consuming. However, recent technological advances in microfluidic devices may offer a means to perform high throughput, live cell sorting without the need for cell specific fluorescent labelling dyes, thereby reducing the sample preparation time.47 Such microfluidic devices may have applications in sorting normal and cancerous cells based on their autofluorescence features (cytopathology), and may exploit autofluorescence as a means of studying the effects of various pharmacological agents at the single cell level.
In vivo fluorescence techniques (imaging or point spectroscopy) collect fluorescence from the full colon thickness. However, our results suggest that measuring epithelial or mucosal autofluorescence alone may be sufficient, and potentially more accurate, for the detection of preneoplastic lesions. Hence, future modification of fluorescence endoscopy devices may incorporate some means (such as confocal detection)48,49 to sample only the superficial mucosal surface where, for example, the presence of bright autofluorescent “lysosomal granules” may serve as a useful “dysplasia specific” biomarker.
Acknowledgments
This work was supported by Photonics Research Ontario and the Ontario Research and Development Challenge Fund in partnership with Xillix Technologies, British Columbia and Ontario, Canada. Ultrasectioning and TEM imaging of all tissue samples studied were performed at the Department of Pathology, Electron Microscopy Division, McMaster University (Hamilton, Ontario, Canada). The authors wish to thank Mr Y M Heng for assistance with TEM processing and imaging.
Abbreviations
CCCP, m-chlorophenylhydrazone
CFM, confocal fluorescence microscopy
ESA, epithelial specific antigen
PCC, primary cell culture
TEM, transmission electron microscopy
REFERENCES
- 1.Lev R. Pathology. In: Adenomatous polyps of the colon: pathological and clinical features. New York: Springer Verlag, 1990:33–6.
- 2.Richards-Kortum R, Rava RP, Fitzmaurice M, et al. Spectroscopic diagnosis of colonic dysplasia. Photochem Photobiol 1991;53:777–86. [DOI] [PubMed] [Google Scholar]
- 3.DuVall A, Wilson BC, Marcon N. Tissue autofluorescence. In: Annals of gastrointestinal endoscopy. 10th ed. London: Rapid Science Publishers, 1997:25–30.
- 4.Haringsma J, Tytgat GN. Fluorescence and autofluorescence. Baillieres Best Pract Res Clin Gastroenterol 1999;13:1–10. [DOI] [PubMed] [Google Scholar]
- 5.DaCosta RS, Wilson BC, Marcon NE. Photodiagnostic techniques for the endoscopic detection of premalignant gastrointestinal lesions. Digestion and Endoscopy 2003;15:153–73. [Google Scholar]
- 6.Haringsma J, Tytgat GN, Yano H, et al. Autofluorescence endoscopy: feasibility of detection of GI neoplasms unapparent to white light endoscopy with an evolving technology. Gastrointest Endosc 2001;53:642–50. [DOI] [PubMed] [Google Scholar]
- 7.Marcon NE, Wilson BC. The value of fluorescence techniques in gastrointestinal endoscopy—better than the endoscopist’s eye? II: the North American experience, Endoscopy 1998;30:419–21. [DOI] [PubMed] [Google Scholar]
- 8.Cothren RM, Richards-Kortum R, Sivak MV Jr, et al. Gastrointestinal tissue diagnosis by laser-induced fluorescence spectroscopy at endoscopy. Gastrointest Endosc 1990;36:105–11. [DOI] [PubMed] [Google Scholar]
- 9.Cothren RM, Sivak MV, Van Dame J, et al. Detection of dysplasia at colonoscopy using laser-induced fluorescence: a blinded study. Gastrointest Endosc 1996;44:168–76. [DOI] [PubMed] [Google Scholar]
- 10.Schomacker KT, Frisoli JK, Compton CC, et al. Ultraviolet laser-induced fluorescence of colonic tissue: basic biology and diagnostic potential. Lasers Surg Med 1992;12:63–78. [DOI] [PubMed] [Google Scholar]
- 11.Zonios GI, Cothren RM, Arendt JT, et al. Morphological model of human colon tissue fluorescence. IEEE Trans Biomed Eng 1996;43:113–22. [DOI] [PubMed] [Google Scholar]
- 12.Bottiroli G, Croce AC, Locatelli D, et al. Natural fluorescence of normal and neoplastic human colon: a comprehensive “ex vivo” study. Lasers Surg Med 1995;16:48–60. [DOI] [PubMed] [Google Scholar]
- 13.DaCosta RS, Lilge L, Kost J, et al. Confocal fluorescence microscopy/macroscopy and microspectrofluorimetry analysis of human colorectal tissues. Journal of Analytical Morphology: Cell Vision 1997;4:24–9. [Google Scholar]
- 14.DaCosta RS, Wilson BC, Marcon NE. Light-induced fluorescence endoscopy of the gastrointestinal tract. Gastrointest Endosc Clin N Am 2000;10:37–69. [PubMed] [Google Scholar]
- 15.Mosiman VL, Patterson BK, Canterero L, et al. Reducing cellular autofluorescence in flow cytometry: an in situ method. Cytometry 1997;30:151–6. [PubMed] [Google Scholar]
- 16.Staughton TJ, McGillicuddy CJ, Weinberg PD. Techniques for reducing the interfering effects of autofluorescence in fluorescence microscopy: improved detection of sulphorhodamine B-labelled albumin in arterial tissue. J Microsc 2001;201:70–6. [DOI] [PubMed] [Google Scholar]
- 17.Croce AC, Spano A, Locatelli D, et al. Dependence of fibroblast autofluorescence properties on normal and transformed conditions. Role of the metabolic activity. Photochem Photobiol 1999;69:364–74. [DOI] [PubMed] [Google Scholar]
- 18.Pitts JD, Sloboda RD, Dragnev KH, et al. Autofluorescence characteristics of immortalized and carcinogen-transformed human bronchial epithelial cells. J Biomed Opt 2001;6:31–40. [DOI] [PubMed] [Google Scholar]
- 19.Aubin JE. Autofluorescence of viable cultured mammalian cells. J Histochem Cytochem 1979;27:36–43. [DOI] [PubMed] [Google Scholar]
- 20.Bennett BD, Jetton TL, Ying G, et al. Quantitative subcellular imaging of glucose metabolism within intact pancreatic islets. J Biol Chem 1996;271:3647–51. [DOI] [PubMed] [Google Scholar]
- 21.Jongkind JF, Verkerk A, Visser WJ, et al. Isolation of autofluorescent ‘aged’ human fibroblasts by flow sorting. Exp Cell Res 1982;138:409–17. [DOI] [PubMed] [Google Scholar]
- 22.Perriott LM, Kono T, Whitesell RR, et al. Glucose uptake and metabolism by cultured human skeletal muscle cells: rate-limiting steps. Am J Physiol Endocrinol Metab 2001;281:E72–80. [DOI] [PubMed] [Google Scholar]
- 23.Benson RC, Meyer RA, Zaruba ME, et al. Cellular autofluorescence—is it due to flavins? J Histochem Cytochem 1979;27:44–8. [DOI] [PubMed] [Google Scholar]
- 24.Loh CS, Vernon D, MacRobert AJ, et al. Endogenous porphyrin distribution induced by 5-aminolaevulinic acid in the tissue layers of the gastrointestinal tract. J Photochem Photobiol B 1993;20:47–54. [DOI] [PubMed] [Google Scholar]
- 25.Chance B, Schoener B, Oshino R, et al. Oxidation–reduction ratio studies of mitochondria in freeze-trapped samples. NADH and flavoprotein fluorescence signals. J Biol Chem 1979;254:4764–71. [PubMed] [Google Scholar]
- 26.Drezek R, Brookner C, Pavlova I, et al. Autofluorescence microscopy of fresh cervical-tissue sections reveals alterations in tissue biochemistry with dysplasia. Photochem Photobiol 2001;73:636–41. [DOI] [PubMed] [Google Scholar]
- 27.Andersson H, Baechi T, Hoechli M, et al. Autofluorescence of living cells. J Microsc 1998;191:1–7. [DOI] [PubMed] [Google Scholar]
- 28.Wolbers F, Andersson H, van den Berg A, et al. Apoptosis induced kinetic changes in autofluorescence of cultured HL60 cells—possible application for single cell analysis on chip. Apoptosis 2004;9:749–55. [DOI] [PubMed] [Google Scholar]
- 29.Berry R, Powell S, Paraskeva C. In vitro culture of human foetal colonic epithelial cells and their transformation with origin minus SV40 DNA. Br J Cancer 1988;57:287–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baten A, Sakamoto K, Shamsuddin M. Long-term culture of normal human colonic epithelial cells in vitro. FASEB J 1992;6:2726–34. [DOI] [PubMed] [Google Scholar]
- 31.Fonti R, Latella G, Bises G, et al. Human colonocytes in primary culture: a model to study epithelial growth, metabolism and differentiation. Int J Colorectal Dis 1994;9:13–22. [DOI] [PubMed] [Google Scholar]
- 32.Chopra D, Yeh K. Long-term culture of epithelial cells from the normal rat colon. In Vitro 1981;17:441–9. [DOI] [PubMed] [Google Scholar]
- 33.Buset M, Winawer S, Friedman E. Defining conditions to promote the attachment of adult human colonic epithelial cells. In Vitro Cell Dev Biol 1987;23:403–12. [DOI] [PubMed] [Google Scholar]
- 34.Osborn M, Debus E, Weber K. Monoclonal antibodies specific for vimentin. Eur J Cell Biol 1982;34:137–43. [PubMed] [Google Scholar]
- 35.Makin C, Bobrow L, Bodmer W. Monoclonal antibody to cytokeratin for use in routine histopathology. J Clin Pathol 1984;37:975–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moll R, Franke W, Schiller D, et al. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumours, and cultured cells. Cell 1982;31:11–24. [DOI] [PubMed] [Google Scholar]
- 37.Harris DM, Werkhaven J. Endogenous porphyrin fluorescence in tumors. Lasers Surg Med 1987;7:467–72. [DOI] [PubMed] [Google Scholar]
- 38.DaCosta RS, Andersson H, Wilson BC. Molecular fluorescence excitation–emission matrices relevant to tissue spectroscopy. Photochem Photobiol 2003;78:384–92. [DOI] [PubMed] [Google Scholar]
- 39.Araki K, Ogata T. Three-dimensional configuration of crypts of different types of colorectal adenomas. Scanning Microsc 1995;9:149–58. [PubMed] [Google Scholar]
- 40.Furuya Y, Ogata T. Scanning electron microscopic study of the collagen networks of the normal mucosa, hyperplastic polyps, tubular adenoma, and adenocarcinoma of the human large intestine. Tohoku J Exp Med 1993;169:1–19. [DOI] [PubMed] [Google Scholar]
- 41.Fujimitsu Y, Nakanishi H, Inada K, et al. Development of aberrant crypt foci involves a fission mechanism as revealed by isolation of aberrant crypts. Jpn J Cancer Res 1996;87:1199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meijer GA, Fleege JC, Baak JPA. Stereological assessment of architectural changes in dysplastic epithelium of colorectal adenomas. Pathol Res Pract 1994;190:333–41. [DOI] [PubMed] [Google Scholar]
- 43.Fisher E, Hellstrom R. Ceroid-like colonic histiocytosis. Am J Clin Pathol 1964;42:581–93. [PubMed] [Google Scholar]
- 44.Buchner A, David R. Lipofuscin in salivary glands in health and disease. Oral Surg Oral Med Oral Pathol 1978;46:79–86. [DOI] [PubMed] [Google Scholar]
- 45.Winterbourne DJ, Weingast-Johnson J. Purines induce lipofuscin formation in a colon carcinoma cell line. Biochem J 1994;301:373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brennick JB, O’Connell JX, Dickersin GR, et al. Lipofuscin pigmentation (so-called “melanosis”) of the prostate. Am J Surg Pathol 1994;18:446–54. [DOI] [PubMed] [Google Scholar]
- 47.Emmelkamp J, Wolbers F, Andersson H, et al. The potential of autofluorescence for the detection of single living cells for label-free cell sorting in microfluidic systems. Electrophoresis 2004;25:3740–5. [DOI] [PubMed] [Google Scholar]
- 48.Kiesslich R, Burg J, Vieth M, et al. Confocal laser endoscopy for diagnosing intraepithelial neoplasias and colorectal cancer in vivo. Gastroenterology 2004;127:706–13. [DOI] [PubMed] [Google Scholar]
- 49.MacAulay C, Lane P, Richards-Kortum R. In vivo pathology: microendoscopy as a new endoscopic imaging modality. Gastrointest Endosc Clin N Am 2004;14:595–620. [DOI] [PubMed] [Google Scholar]