

Abstract
Background: Chronic fatigue syndrome (CFS) is a multisystem disease, the pathogenesis of which remains undetermined.
Aims: To test the hypothesis that there are reproducible abnormalities of gene expression in patients with CFS compared with normal healthy persons.
Methods: To gain further insight into the pathogenesis of this disease, gene expression was analysed in peripheral blood mononuclear cells from 25 patients with CFS diagnosed according to the Centers for Disease Control criteria and 25 normal blood donors matched for age, sex, and geographical location, using a single colour microarray representing 9522 human genes. After normalisation, average difference values for each gene were compared between test and control groups using a cutoff fold difference of expression ⩾ 1.5 and a p value of 0.001. Genes showing differential expression were further analysed using Taqman real time polymerase chain reaction (PCR) in fresh samples.
Results: Analysis of microarray data revealed differential expression of 35 genes. Real time PCR confirmed differential expression in the same direction as array results for 16 of these genes, 15 of which were upregulated (ABCD4, PRKCL1, MRPL23, CD2BP2, GSN, NTE, POLR2G, PEX16, EIF2B4, EIF4G1, ANAPC11, PDCD2, KHSRP, BRMS1, and GABARAPL1) and one of which was downregulated (IL-10RA). This profile suggests T cell activation and perturbation of neuronal and mitochondrial function. Upregulation of neuropathy target esterase and eukaryotic translation initiation factor 4G1 may suggest links with organophosphate exposure and virus infection, respectively.
Conclusion: These results suggest that patients with CFS have reproducible alterations in gene regulation.
Keywords: chronic fatigue syndrome, pathogenesis, gene expression, T cell activation, mitochondrion, neurone, organophosphate, virus infection, interleukin 10 receptor
Chronic fatigue syndrome (CFS)/myalgic encephalomyelitis is a disease that is characterised by severe and debilitating fatigue, sleep abnormalities, impaired memory and concentration, and musculoskeletal pain.1 In the Western world, the population prevalence is estimated to be in the order of 0.5%.1,2 Although CFS is now recognised as a genuine clinical entity, a considerable research effort has failed to identify quantifiable parameters that consistently exhibit abnormal results in well documented cases. Therefore, the pathological basis for CFS remains poorly understood. Although there is no known aetiology and no known diagnostic marker, a large number of diverse factors such as viral infection; immune activation; exposure to toxins, chemicals, and pesticides; stress; hypotension; lymphocyte abnormalities; and neuroendocrine dysfunction have been proposed as factors in the pathogenesis of CFS.3
“Although chronic fatigue syndrome is now recognised as a genuine clinical entity, a considerable research effort has failed to identify quantifiable parameters that consistently exhibit abnormal results in well documented cases”
Three previous reports have studied gene expression in the peripheral blood of patients with CFS.4–6 Although there was little agreement between these studies as to the genes identified, only one used quantitative polymerase chain reaction (PCR) to confirm the initial findings4 and the initial method in this study was differential display. To address this discrepancy and to investigate the hypothesis that abnormalities of gene regulation occur in CFS, we studied gene expression in peripheral blood mononuclear cells (PBMC) of patients with CFS and normal blood donors using a microarray, and used Taqman real time PCR to confirm those genes identified as being differentially expressed between the groups. Sixteen genes were thus confirmed as having an expression profile associated with the CFS.
METHODS
Subject enrolment
Patients with CFS (n = 25) were enrolled from the Dorset CFS service in South East England. These cases were diagnosed according to the criteria of Fukuda and colleagues.2 Additional clinical information was recorded and is presented in table 1. This includes measurements of physical and mental fatigue using the Chalder Fatigue Scale7 and measurements of anxiety and depression using the Hospital Anxiety and Depression scales.8 None of these patients had undergone previous treatment for psychiatric disorders. Patients were sampled at two time points, six months apart, between which their symptoms did not vary significantly; the first sample from each patient was used for microarray analysis whereas the second was used for real time PCR. This approach provides an additional safeguard against attaching particular importance to genes that may be differentially expressed at a single time point, but not reproducibly. Normal blood donors were enrolled from the East Dorset division of the National Blood Service (NBS); 25 age and sex matched normal blood donors were used as a comparison group for the microarray part of the study, and 21 normal blood donors (age and sex matched as a group) were used as a comparison group for the real time PCR part of the study.
Table 1.
Patient information including summary of aspects used for the CFS case definition and scores for physical fatigue, mental fatigue, anxiety, and depression
| Clinical parameter | N |
| Sex | 9 male/16 female |
| Mean age | 40.6 years |
| Mean duration of disease | 2.3 years |
| CFS began with flu-like illness | 15 |
| Impaired memory | 22 |
| Impaired concentration | 21 |
| Sore throat | 17 |
| Myalgia | 20 |
| Arthralgia | 13 |
| Headache | 19 |
| Unrefreshing sleep | 21 |
| Post exertional malaise | 23 |
| Tender lymphadenopathy | 18 |
| Autonomic features | 20 |
| Atopic features | 13 |
| Physical fatigue (Chalder) score (most severe, 21) | Mean, 17.2; range, 12–21 |
| Mental fatigue (Chalder) score (most severe, 12) | Mean, 9.28; range, 5–12 |
| Anxiety score (HAD) (most severe, 21) | Mean, 9.88; range, 3–18 |
| Depression score (HAD) (most severe, 21) | Mean, 7.96; range, 1–20 |
CFS, chronic fatigue syndrome; HAD, Hospital Anxiety and Depression.
The NBS restricts donors to those who fulfil the following criteria: aged between 17 and 59 years; have not given blood in the previous 16 weeks; are not currently suffering from an infection; are not pregnant; are not currently taking (or within three months of taking) antibiotics, steroids, or antidepressants; have not had hepatitis, jaundice, body piercing, acupuncture, or blood transfusion in the past year; do not have two family members who suffered from Creutzfeldt-Jakob Disease; are not known to be positive for human immunodeficiency virus, hepatitis B or C; are not currently or previously abusers of injecting or body building drugs; are not post-vaccination; and do not suffer a chronic illness including malaria. Patients and controls gave written consent according to the guidance of the ethics committees of both the East Dorset NBS, the Royal Brompton and Harefield NHS Trusts, and the National Heart and Lung Institute. The human experimentation guidelines of the US Department of Health and Human Services were followed in our study.
Sample collection and processing
A 20 ml sample of blood was immediately placed in cell preparation tubes containing density gradient solution and EDTA (BD Biosciences, Manchester, UK). PBMCs were isolated by density gradient centrifugation. Total RNA was extracted using Trizol (Invitrogen, Carlsbad, California, USA), washed in phosphate buffered saline, and the quality and amount confirmed by microspectrophotometry (Nanodrop, Rockland, Delaware, USA). Total RNA samples had an absorbance ratio (A260/280) of 1.85–1.95. A 20 ml sample of peripheral blood is estimated to contain a total of approximately 15 × 106 PBMCs and buffy coat from this sample yielded 10–25 µg total RNA, which was shipped to Nimblegen, USA, at room temperature in ethanol, for array testing. Total RNA was converted to double stranded cDNA by the SuperScript Choice System (Invitrogen) with an oligo dT primer containing the T7 RNA polymerase promoter sequence (5′-GGCCAGTGAATTGTAATACGACTCACTATAGGGAGGCGG-T24-3′). A 15 µg aliquot of total RNA was converted into cDNA. In vitro transcription was used to produce biotin labelled cRNA from cDNA using the Ambion MEGAscript T7 kit (Ambion, Austin, Texas, USA). Before hybridisation, cRNA was fragmented to an average size of 50–200 bp by incubation in 100mM potassium acetate, 30mM magnesium acetate, and 40mM Tris/acetate at 94°C for 35 minutes. Fragmentation was checked by gel electrophoresis in 1% agarose.
Microarray design and analysis
A custom microarray was manufactured by Nimblegen (Madison, Wisconsin, USA) using maskless array synthesis (http://www.nimblegen.com). The human genes on this design (n = 9522) were selected from the Homo sapiens entries in the RefSeq collection of sequences as of August 2002. Each gene was compared with all others using the BLAST program to remove redundancies. Ten probe pairs for each target were selected from the 3′ 1 kb of each target. Probes were spaced evenly over the length of the target region (⩽1 kb), so that the exact spacing depended on the length of the target sequence. Each probe was 24 nucleotides in length. For each perfect match probe there was also a mismatch probe, which differed by a single nucleotide.
Labelled cRNA was hybridised to the oligonucleotide probes on the microarray. After washing, arrays were stained with streptavidin–cy3 conjugate (Amersham Biosciences, Piscataway, New Jersey, USA) for 25 minutes at room temperature, followed by washing and a blow dry step using high pressure grade 5 Argon (Badger Welding, Madison, Wisconsin, USA). Slides were scanned using a GenePix 4000B microarray scanner (Axon Instruments, Union City, California, USA), and the feature intensities extracted from the TIF files were calculated by the scanner software using a proprietary application developed at NimbleGen (Madison, Wisconsin, USA).9 This application calculates mean signal intensities for the pixels that define each feature (3 × 3 grid of pixels). The intensities for each gene are calculated by taking the mean of the intensities for the perfect match probes specific to each target minus the mean of the intensity of the mismatch probes. Probes that differed from the mean for the set by more than 3 SD were removed from the set and the mean recalculated. Average differences (recalculated mean) were used for subsequent analysis.
Data analysis was performed using BRB ArrayTools version 3.02 (Molecular Statistics and Bioinformatics Section, National Cancer Institute, Bethesda, Maryland, USA) developed by Dr R Simon and A Peng (http://linus.nci.nih.gov/BRB-ArrayTools.html). Average difference values were normalised to median over the array. The data were filtered so that only those genes that were adequately measured on 75% of the arrays were included. A class comparison protocol was used to identify genes whose degree of expression differed significantly by ⩾ 1.5 fold between the two groups. This consisted of a multivariate permutation test, which was computed based on 1000 random permutations using the following parameters: nominal significance level = 0.001; confidence level of false discovery rate assessment = 50%; maximum allowed number of false positive genes = 10; maximum allowed proportion of false positive genes = 0.1. Values for differentially expressed genes were used to cluster all 50 subjects using Genepilot software (http://www.genepilot.com) (TG Services, El Sobrante, California, USA).
Taqman real time PCR
Taqman real time PCR (Applied Biosystems, Foster City, California, USA) was used to confirm the importance of genes identified by array experiments in the same group of CFS cases (n = 17) and a different group of normal controls (n = 21); the controls were age and sex matched. cDNA was prepared from total RNA using the random hexamer method of reverse transcription, according to the instructions of the kit manufacturer (Applied Biosystems, Warrington, UK). Experiments were performed in triplicate in a custom 384 well low density array format using the ABI PRISM 7900HT instrument (Applied Biosystems) incorporating 38 target gene assays (including three instances where two assays were used for a single gene to include transcript variants) (table 2) along with endogenous controls, namely: HMBS (hydroxymethylbilane synthase), HPRT-1 (hypoxanthine phosphoribosyltransferase 1), GAPDH (glyceraldehyde-3-phosphate dehydrogenase), and eukaryotic 18S rRNA. Fluorogenic probes were 5′ labelled with 6-carboxyfluorescein (FAM) and 3′ labelled with MGB non-fluorescent quencher. Reactions were performed in a 1 µl reaction volume in Taqman Universal PCR Mastermix (Applied Biosystems UK). For each card channel (48 PCR reactions), 50 ng total RNA from PBMCs converted to cDNA was used as inoculum. Fluorescent signal detection used “ROX” as the internal passive reference dye.
Table 2.
Genes showing a significant ⩾1.5 fold change between cases and controls in microarray experiments
| GenBank accession number | Gene name (mRNA) | Gene symbol | Fold difference (array) | p Value | Taqman assay ID | Fold difference (PCR) | p Value |
| NM_020325 | ATP binding cassette, subfamily D (ALD), member 4, tv-4 | ABCD4 | 3.398 | 0.00257 | Hs00245340_m1 | 1.825 | 0.0019 |
| NM_002741 | Protein kinase C-like 1 | PRKCL1 | 2.288 | 0.00438 | Hs00177028_m1 | 2.669 | 1.09×10−5 |
| NM_021134 | Mitochondrial ribosomal protein L23 | MRPL23 | 2.175 | 0.00135 | Hs00221699_m1 | 4.032 | 1.25×10−6 |
| NM_001558 | Interleukin 10 receptor α | IL10RA | 0.252 | 0.00225 | Hs00387004_m1 | 0.395 | 2.34×10−12 |
| NM_006110 | CD2 antigen (cytoplasmic tail) binding protein 2 | CD2BP2 | 2.311 | 0.00158 | Hs00272036_m1 | 2.411 | 2.6×10−4 |
| NM_000177 | Gelsolin (amyloidosis, Finnish type) | GSN | 2.141 | 0.00105 | Hs00609276_m1 | 1.83 | 6.23×10−7 |
| NM_006702 | Neuropathy target esterase | NTE | 2.877 | 0.00043 | Hs00198648_m1 | 6.409 | 1.31×10−12 |
| NM_002696 | Polymerase (RNA) II (DNA directed) polypeptide G | POLR2G | 2.264 | 0.00372 | Hs00275738_m1 | 3.407 | 3.69×10−6 |
| NM_004813 | Peroxisomal biogenesis factor 16, tv-1 | PEX16 | 3.004 | 0.00213 | Hs00191337_m1 | 1.758 | 0.0126 |
| NM_015636 | Eukaryotic translation initiation factor 2B, subunit 4δ, tv-1 | EIF2B4 | 2.414 | 0.00125 | Hs00248984_m1 | 1.882 | 1.8×10−5 |
| NM_004953 | Eukaryotic translation initiation factor 4γ1, tv-5 | EIF4G1 | 3.081 | 0.00177 | Hs00191933_m1 | 2.964 | 7.63×10−13 |
| NM_016476 | APC11 anaphase promoting complex subunit 11 homologue | ANAPC11 | 3.278 | 0.00219 | Hs00212858_m1 | 2.366 | 3.5×10−6 |
| NM_002598 | Programmed cell death 2, tv-1 | PDCD2 | 2.052 | 0.00266 | Hs00751277_sH | 1.887 | 1.3×10−4 |
| NM_003685 | KH-type splicing regulatory protein (FUSE binding protein 2) | KHSRP | 2.366 | 0.00139 | Hs00269352_m1 | 1.64 | 0.0022 |
| NM_015399 | Breast cancer metastasis suppressor 1 | BRMS1 | 2.246 | 0.00219 | Hs00363036_m1 | 1.598 | 0.003 |
| NM_031412* | GABA(A) receptor associated protein-like 1 | GABARAPL1 | 2.358 | 0.00046 | Hs00744468_s1 | 1.838 | 0.0097 |
| NM_004655 | Axin 2 (conductin, axil) | AXIN2 | 0.351 | 0.00056 | Hs00610344_m1 | 0.780 | 0.023 |
| NM_019051* | Mitochondrial ribosomal protein L50 | MRPL50 | 0.427 | 0.00409 | Hs00745120_s1 | 0.815 | 0.0019 |
| NM_005103* | Fasciculation and elongation protein ζ1 (zygin I), tv-1 | FEZ1 | 0.336 | 0.00007 | Hs00192714_m1 | 3.485 | 2.3×10−6 |
| NM_002093 | Glycogen synthase kinase 3β | GSK3B | 2.294 | 0.00238 | Hs00275656_m1 | 0.763 | 0.0015 |
| NM_005098 | Musculin (activated B cell factor 1) | MSC | 0.374 | 0.00121 | Hs00231955_m1 | 1.118 | 0.036 |
| NM_138325 | Paired basic amino acid cleaving system 4, tv-6 | PACE4 | 0.473 | 0.00414 | Hs00159844_m1 | 2.093 | 1.2×10−4 |
| NM_003584 | Dual specificity phosphatase 11 | DUSP11 | 0.366 | 0.00005 | Hs00186058_m1 | 1.725 | 0.134 |
| NM_002483 | Carcinoembryonic antigen related cell adhesion molecule 6 | CEACAM6 | 0.301 | 0.00048 | Hs00366002_m1 | 1.585 | 0.182 |
| NM_019051* | Mitochondrial ribosomal protein L50 | MRPL50 | 0.427 | 0.00409 | Hs00747929_m1 | 1.136 | 0.178 |
| NM_030575 | Hypothetical protein MGC10334 | MGC10334 | 2.584 | 0.00118 | Hs00257998_s1 | 1.724 | 0.067 |
| NM_031412* | GABA(A) receptor associated protein-like 1 | GABARAPL1 | 2.358 | 0.00046 | Hs00740588_mH | 0.93 | 0.127 |
| NM_007175 | Chromosome 8 open reading frame 2 | C8orf2 | 2.554 | 0.00206 | Hs00200360_m1 | 1.198 | 0.052 |
| NM_032118 | Hypothetical protein FLJ12953 | FLJ12953 | 1.931 | 0.00309 | Hs00259557_m1 | 1.139 | 0.203 |
| NM_004879 | Etoposide induced 2.4 mRNA | EI24 | 0.45 | 0.00181 | Hs00747550_m1 | 1.03 | 0.085 |
| NM_079834 | Secretory carrier membrane protein 4 | SCAMP-4 | 2.8 | 0.00069 | Hs00365263_m1 | 1.586 | 0.173 |
| NM_022145 | Leucine zipper protein FKSG14 | FKSG14 | 0.345 | 0.00205 | Hs00259557_m1 | 1.212 | 0.345 |
| NM_005103* | Fasciculation and elongation protein ζ1 (zygin I), tv-1 | FEZ1 | 0.336 | 0.00007 | Hs00363763_m1 | 1.062 | 0.41 |
| NM_003409 | Zinc finger protein 161 homologue | ZFP161 | 0.415 | 0.00146 | NA | NT | NT |
| NM_007075 | JM5 protein | JM5 | 2.383 | 0.00172 | NA | NT | NT |
| NM_005345 | Heat shock 70 kDa protein 1A | HSPA1A | 3.198 | 0.00003 | NA | NT | NT |
| NM_017616 | Hypothetical protein FLJ20004 | FLJ20004 | 2.679 | 0.00237 | NA | NT | NT |
| NM_020663 | Ras homologue gene family, member J | ARHJ | 2.277 | 0.00346 | NA | NT | NT |
mRNA transcripts identified by array and differential expression confirmed by real time PCR are shown in bold.
The table also shows confirmatory Taqman assay numbers (Applied Biosystems UK) and fold changes by real time PCR.
*Transcript variants of particular genes.
NA, not available; NT, not tested; PCR, polymerase chain reaction.
The inoculum (100 µl) was a mixture of Taqman universal PCR master mix (Applied Biosystems) (50 µl), RNase/DNase free sterile water (45 µl), and cDNA (5 µl). Cycling times and temperatures were as follows. Initial denaturation was carried out for 10 minutes at 95°C, followed by 40 cycles of denaturation at 95°C for 15 seconds and combined primer annealing/extension at 60°C for one minute. Data was displayed using SDS 2.1 software (Applied Biosystems). GAPDH gave the least variable results in all samples and was used as the endogenous reference control. The threshold cycle (Ct) for each gene/sample pair was compared with a calibrator sample and a ΔCt value used to calculate a relative quantity of gene expression compared with the calibrator. RQ values were finally normalised to GAPDH expression. The F test for equality of standard deviations was used to compare mean RQ values for each gene in test versus control groups.
RESULTS
CFS disease phenotype
All 25 patients with CFS were diagnosed according to the Centers for Disease Control criteria of Fukuda et al.2 Table 1 summarises the patient and clinical details. Our study included patients with CFS whose disease was severe and necessitated bed rest for much of the day, and also several patients whose disease was of a milder nature (table 1).
Microarray analysis
Analysis of microarray data identified 35 genes that showed significantly different expression in patients with CFS compared with normal controls (table 2). Hierarchical clustering of subjects on the basis of their degree of expression of these 35 genes revealed a cluster of 18 subjects, comprising 17 CFS patients and one normal person, whose expression profiles were very similar but distinctly different from the other patients and controls (fig 1). However, this patient cluster did not differ significantly with regard to other clinical variables shown in table 1, so that we were unable to explain this clustering.
Figure 1.

Hierarchical clustering experiment of differentially expressed gene profiles among patients with chronic fatigue syndrome (CFS; n = 25) and normal persons (n = 25) identified by analysis in BRB Array Tools. The figure was generated using Genepilot software. Each column represents the expression profile for each of the 35 genes. Each row represents a single gene, with its GenBank accession number to the right hand side of the figure. Coloured pixels represent the magnitude of the response for any gene. Shades of red and green represent induction and repression, respectively, relative to the mean value for each respective gene among the normal persons. This figure shows a cluster of 18 subjects (from P29–P9) consisting of predominantly patients with CFS (n = 17) and one normal person, who have a similar profile of expression of these 35 genes. P, patients; C, controls.
Taqman real time PCR
TaqMan real time PCR analysis was used to confirm the importance of genes that were identified using microarray analysis. Significantly different expression, with the same profile as in gene arrays, was confirmed for 16 of 33 genes in the 17 patients tested. This method revealed upregulation of 15 genes and downregulation of one gene (table 2). Figure 2 illustrates the concurrent fold difference in gene expression between test and control groups for the 16 differentially expressed genes for both microarray and real time PCR analysis. The standard deviation from the mean is also shown in fig 2B and provides an indirect measure of the probable reproducibility of differential expression for each gene. In general, the standard deviation of these 16 genes in normal persons is very much lower than in patients with CFS, except for IL-10RA, in which the opposite is the case.
Figure 2.
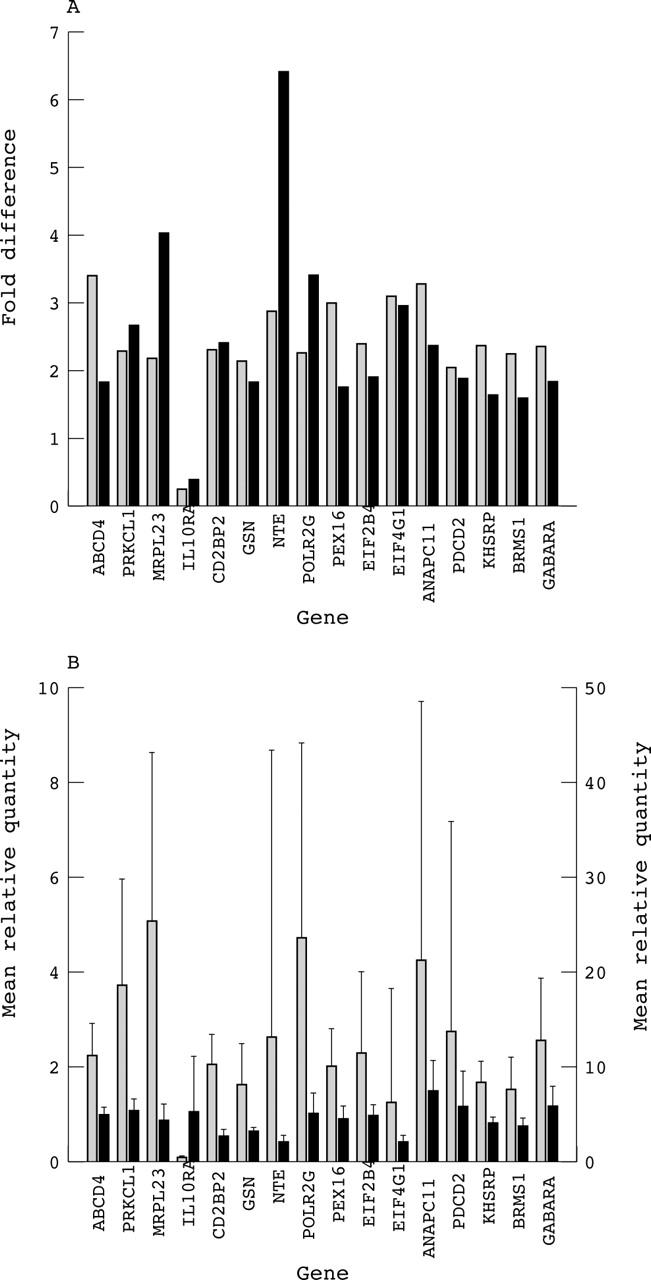
(A) Bar chart showing the fold difference in gene expression between test and control groups by microarray (shaded) and real time polymerase chain reaction (solid black) for 16 genes that are differentially expressed in chronic fatigue syndrome (CFS). (B) Bar chart showing the mean relative quantity of mRNA transcripts in test (shaded) and control (solid black) groups for 16 genes that are differentially expressed in CFS. Error bars indicate the standard deviation from the mean in each case. All values for the mean relative quantity mRNA transcript are shown on the left y axis, except those for NTE and EIF4G1, which are shown on the right y axis.
Table 3 provides information on the chromosomal location, expression, subcellular localisation, and function of these 16 genes. Although these genes do not fit neatly into known metabolic pathways, several broad themes are apparent. For example, T cell activation and neuronal and mitochondrial function.
Table 3.
Chronic fatigue syndrome associated genes: chromosomal location, expression, subcellular localisation, and function
| GenBank accession number | Gene name (mRNA) | Gene symbol* | Chromosomal location | Expression | Subcellular localisation | Gene function |
| NM_020325 | ATP binding cassette, subfamily D (ALD), member 4, tv-4 | ABCD4 | 14q24.3 | Ubiquitous | Peroxisomal membrane | ALDP expression |
| NM_002741 | Protein kinase C-like 1 | PRKCL1 | 19p13.1–p12 | Ubiquitous | Cytoplasm | Regulation of cell motility |
| NM_021134 | Mitochondrial ribosomal protein L23 | MRPL23 | 11p15.5 | Widely expressed | Mitochondrial matrix | Protein biosynthesis |
| NM_001558 | Interleukin 10 receptor α | IL10RA | 11q23.3 | – | Unknown | Cytokine signalling |
| NM_006110 | CD2 antigen (cytoplasmic tail) binding protein 2 | CD2BP2 | 16p12.1 | – | Unknown | – |
| NM_000177 | Gelsolin (amyloidosis, Finnish type) | GSN | 9q33.3 | – | Unknown | Severing and capping of actin |
| NM_006702 | Neuropathy target esterase | NTE | 19p13.3–p13.2 | Nervous system | Plasma membrane | Neurodegenerative disease |
| NM_002696 | Polymerase (RNA) II (DNA directed) polypeptide G | POLR2G | 11q13.1 | Nervous system | Nucleus | Transcription from Pol II |
| NM_004813 | Peroxisomal biogenesis factor 16, tv-1 | PEX16 | 11p11.11 | – | Peroxisomal membrane | Peroxisomal biogenesis |
| NM_015636 | Eukaryotic translation initiation factor 2B, subunit 4δ, tv-1 | EIF2B4 | 2p23.3 | Ubiquitous | Mitochondrion | Initiation of translation |
| NM_004953 | Eukaryotic translation initiation factor 4γ, 1, tv-5 | EIF4G1 | 3q27–qter | Ubiquitous | Mitochondrion | Initiation of translation |
| NM_016476 | APC11 anaphase promoting complex subunit 11 homologue | ANAPC11 | 17q25.3 | Brain, heart, pancreas, lung | Nucleus | Ubiquitin ligase activity |
| NM_002598 | Programmed cell death 2, tv-1 | PDCD2 | 6q27 | Ubiquitous | Cytoplasm | Apoptosis and regulation of cell proliferation |
| NM_003685 | KH-type splicing regulatory protein (FUSE binding protein 2) | KHSRP | 19p13.3 | Nervous system | Nucleus | Neurone specific splicing of the N1 exon of SRC; assembly of other proteins |
| NM_015399 | Breast cancer metastasis suppressor 1 | BRMS1 | 14q13.1 | Brain | Unknown | – |
| NM_0314122 | GABA(A) receptor associated protein-like 1 | GABARAPL1 | 12p12.3 | Ubiquitous | Unknown | – |
DISCUSSION
In our present study, we studied transcript profiles from patients with CFS and from sex and age matched normal controls from the same area of South East England. The expression of 16 genes was significantly different in patients compared with controls in both microarray analysis and real time PCR. These genes may be important in the pathogenesis of CFS and can be grouped according to immune, neuronal, mitochondrial, and other functions that have particular relevance to our present knowledge of the epidemiology of CFS (table 4). Our present study has certain parallels with two published studies in this area, summarised in table 4.
Table 4.
Possible mechanisms of disease in CFS based on three gene expression studies, each on patients with CFS and normal controls
| Mechanism | Present study | Powell and colleagues4 | Vernon and colleagues5 |
| Microarray (9522 genes)/qPCR; 25 CFS; 25 normal | Differential display/qPCR; 7 CFS; 4 normal | Filter array (1764 genes); 5 CFS; 17 normal | |
| T cell activation | CD2BP2, IL-10RA | Moesin, CTSC | ITGA, NFATC3 |
| Immune response | PRKCL1 | TNF, MAIL | IL-8, CMRF35, ICAM2, ITGB, IER2, PLA2G2A, LCP-1, PRKCL |
| Neurone | PRKCL1, GSN, KHSRP, NTE, GABARAPL1 | HD, PRKCL, ataxin, ADRA2A, DCTN1 | |
| Mitochondrion | EIF4G1, EIF2B4, MRPL23 | SLC25A16, MCFP | |
| Skeletal muscle | GSN | DCTN1 | |
| Thyroid | GSN | THRAP2, SLC25A16 | GDC |
| Cell cycle | ANAPC11 | MAD1L1 | |
| Apoptosis | PDCD2 | ||
| Transcription | POLR2G, BRMS1 | RCOR3, POLR1B | |
| Peroxisome | ABCD4, PEX16 |
CSF, chronic fatigue syndrome; qPCR, quantitative polymerase chain reaction.
T cell activation is suggested by upregulation of CD2BP2 and downregulation of IL-10RA10–12; in addition, PRKCL1 plays a role in the immune response. Genes that are active in the immune response have been found to be differentially expressed in all studies of gene expression in CFS (table 4). Furthermore, genes that are crucial for T cell activation10 have also been found to be upregulated in all three studies, namely: CD2BP2 and IL-10RA (present study); moesin and cathepsin C4; ITGA and NFATC3.5 These findings are consistent with previous work showing that patients with CFS have evidence of immune activation, such as increased numbers of activated T cells and cytotoxic T cells, and raised circulating cytokine concentrations.3,13–19
A neuronal component is suggested by the upregulation of PRKCL1, NTE, GSN, GABARAPL1, KHSRP, and EIF2B4. Protein kinase C family members are implicated in various psychiatric and affective disorders, and have been implicated in previous gene studies of CFS.5 NTE is a target for organophosphates and chemical warfare agents, both of which may precipitate CFS,20 on the basis of a neuropathy resulting from inactivation of serine esterase activity.21 GSN regulates cell growth and plays a role in amyloidosis (Finnish type), which may result in dysfunction of neurones, skeletal muscle, and thyroid gland.22,23 GABARAPL1 is a microtubule associated anchor protein with increased expression in neuronal cells.24 KHSRP facilitates splicing of the N1 exon of the SRC protooncogene in neuronal but not other cells.25 EIF2B4 is a mitochondrial translation initiation factor and one of the EIF2B family, within which mutations have been shown to be associated with central nervous system hypomyelination and encephalopathy.26 Powell and colleagues4 have reported upregulation of an EIF2B3 gene homologue (BQ580379). These findings are interesting in that abnormalities in the white matter of the frontal lobes have been found in patients with CFS using magnetic resonance imaging and have been suggested to account for the cognitive defect in CFS.27 Neuronal gene involvement in CFS has also been reported by Vernon and colleagues.5
Mitochondrial involvement is suggested by the upregulation of EIF2B4, EIF4G1 (see above), and MRPL23. Mitochondrial gene upregulation has also been reported by Powell et al.4
The cell cycle is implicated by upregulation of ANAPC11, which regulates the onset of anaphase by mediation of degradation of mitotic cyclins. Powell and colleagues4 reported upregulation of MAD1L1, which prevents the onset of anaphase until all chromosomes are aligned at the metaphase plate.
“The upregulation of EIF4G1 identified in our present study may represent a common host response to persistent infection with several different viruses”
Transcriptional perturbation is suggested by the upregulation of POLR2G and BRMS1. Powell and colleagues4 reported the upregulation of genes homologous with POLR1B (BQ580386) and RCOR3 (BQ580388), which are each involved in transcriptional regulation.
Upregulated peroxisomal function is suggested by the upregulation of ABCD4 and PEX16, which may suggest enhanced defence to oxidative stress in CFS. Oxidative stress has already been suggested as a disease mechanism in CFS.28,29
Persistent virus infection is a recognised feature of CFS, which is interesting in the light of our finding of upregulation of EIF4G1 transcript variant 5, a mitochondrial translation initiation factor. Whistler and colleagues6 have also reported this finding in patients with CFS who have rapid (?triggered by virus infection) as compared with insidious onset. EIF4G1 is a component of the protein complex, EIF4F, which is crucial in translation through its involvement in the recognition of the mRNA cap, ATP dependent unwinding of 5′ terminal secondary structure, and recruitment of mRNA to the ribosome.30 Various viruses have developed strategies to divert EIF4G1 from its utilisation by the cellular machinery to facilitate production of viral proteins.30 The best characterised example is that of poliovirus,31,32 but this has also been demonstrated to occur with coxsackie virus,33 rhinoviruses,34 rotavirus,35 influenza virus,36 adenovirus,37 vesicular stomatitis virus,38 and human immunodeficiency virus 1.39 Therefore, the upregulation of EIF4G1 identified in our present study may represent a common host response to persistent infection with several different viruses. The vulnerability of EIF4G1 to virus modification may have particular importance for the development of CFS after an acute virus infection.40
Take home messages.
Sixteen genes were differentially expressed in patients with chronic fatigue syndrome compared with normal controls, as assessed by microarray and quantitative polymerase chain reaction
The involvement of genes from several disparate pathways suggests a complex pathogenesis involving T cell activation and abnormalities of neuronal and mitochondrial function
These results suggest possible molecular bases for the recognised contributions of organophosphate exposure and virus infection
In conclusion, we report the differential expression of 16 human genes in patients with CFS compared with normal controls. The involvement of genes from several disparate pathways suggests a complex pathogenesis involving T cell activation and abnormalities of neuronal and mitochondrial function, and suggests possible molecular bases for the recognised contributions of organophosphate exposure and virus infection, respectively.
Acknowledgments
We thank the CFS Research Foundation, Hertfordshire, UK, and Royal Brompton and Harefield NHS Trust for financial support. We thank Dr F Boulton, Ms J Williams, Mr P Rogers, Ms D Carr, and the NBS teams of East Dorset for their help in enrolment and sampling of normal blood donors; Dr A Bell, Consultant Haematologist, Poole Hospital, for use of laboratory facilities for processing of blood samples immediately after collection; and Dr J Sherlock, Applied Biosystems, for advice on Taqman PCR.
This paper is dedicated to Dr David Tyrrell CBE, FRS, who died on 2 May 2005, aged 75, and who gave so much of himself in support of this research program and all its participants.
Abbreviations
CFS, chronic fatigue syndrome
NBS, National Blood Service
PBMC, peripheral blood mononuclear cells
PCR, polymerase chain reaction
REFERENCES
- 1. A report of the CFS/ME working group. London: Department of Health, January 2002. (http://www.doh.gov.uk).
- 2.Fukuda K, Straus SE, Hickie I, et al. The chronic fatigue syndrome: a comprehensive approach to its definition and study. Ann Intern Med 1994;121:953–9. [DOI] [PubMed] [Google Scholar]
- 3.Komaroff AL, Buchwald D. Chronic fatigue syndrome; an update. Annu Rev Med 1998;49:1–13. [DOI] [PubMed] [Google Scholar]
- 4.Powell R, Ren J, Lewith G, et al. Identification of novel expressed sequences, upregulated in the leucocytes of chronic fatigue syndrome patients. Clin Exp Allergy 2003;33:1450–6. [DOI] [PubMed] [Google Scholar]
- 5.Vernon SD, Unger ER, Dimulescu IM, et al. Utility of the blood for gene expression and biomarker discovery in chronic fatigue syndrome. Dis Markers 2002;18:193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whistler T, Unger ER, Nisenbaum R, et al. Integration of gene expression, clinical and epidemiologic data to characterise chronic fatigue syndrome. J Transl Med 2003;1:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chalder T, Berelowitz G, Pawlikowska T, et al. J Psychosom Res 1993;37:147–53. [DOI] [PubMed] [Google Scholar]
- 8.Reeves WC, Lloyd A, Vernon SD, et al. International chronic fatigue syndrome study group. Identification of ambiguities in the 1994 chronic fatigue syndrome research case definition and recommendations for resolution. BMC Health Serv Res 2003;3:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nuwaysir EF, Huang W, Albert TJ, et al. Gene expression analysis using oligonucleotide arrays produced by maskless photolithography. Genome Res 2002;12:1749–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu P, Pardo J, Zhao H, et al. Systematic identification of regulatory proteins critical for T cell activation. J Biol 2003;2:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kofler M, Heuer K, Zech T, et al. Recognition sequences for the GYF domain reveal a possible spliceosomal function of CD2BP2. J Biol Chem 2004;279:28292–7. [DOI] [PubMed] [Google Scholar]
- 12.Izquierdo M, Cantrell DA. T-cell activation. Trends Cell Biol 1992;2:268–71. [DOI] [PubMed] [Google Scholar]
- 13.Chao CC, Janoff EN, Hu SX, et al. Altered cytokine release in PBMC cultures from patients with the chronic fatigue syndrome. Cytokine 1991;3:292–8. [DOI] [PubMed] [Google Scholar]
- 14.MacDonald KL, Osterholm MT, LeDell KH, et al. A case control study to assess possible triggers and cofactors in chronic fatigue syndrome. Am J Med 1996;100:548–54. [DOI] [PubMed] [Google Scholar]
- 15.Rasmussen AK, Nielsen H, Andersen V, et al. Chronic fatigue syndrome—a controlled cross sectional study. J Rheumatol 1994;21:1527–31. [PubMed] [Google Scholar]
- 16.Patarca-Montero R, Antoni M, Fletcher MA, et al. Cytokine and other immunologic markers in chronic fatigue syndrome and their relation to neuropsychological factors. Appl Neuropsychol 2001;8:51–64. [DOI] [PubMed] [Google Scholar]
- 17.Moss RB, Mercandetti A, Vojdani A. TNF-alpha and chronic fatigue syndrome. J Clin Immunol 1999;19:314–16. [DOI] [PubMed] [Google Scholar]
- 18.Penttila IA, Harris RJ, Storm P, et al. Cytokine dysregulation in the post-Q-fever fatigue syndrome. QJM 1998;91:549–60. [DOI] [PubMed] [Google Scholar]
- 19.Kerr JR, Barah F, Mattey DL, et al. Circulating tumour necrosis factor-alpha and interferon-gamma are detectable during acute and convalescent parvovirus B19 infection and are associated with prolonged and chronic fatigue. J Gen Virol 2001;82:3011–19. [DOI] [PubMed] [Google Scholar]
- 20.Khan F, Kennedy G, Spence VA, et al. Peripheral cholinergic function in humans with CFS, Gulf war syndrome and with illness following organophosphate exposure. Clin Sci (Lond) 2004;106:183–9. [DOI] [PubMed] [Google Scholar]
- 21.O’Callaghan JP. Neurotoxic esterase: not so toxic? Nat Genet 2003;33:437–8. [DOI] [PubMed] [Google Scholar]
- 22.McGough AM, Staiger CJ, Min JK, et al. The gelsolin family of actin regulatory proteins: modular structures, versatile functions. FEBS Lett 2003;552:75–81. [DOI] [PubMed] [Google Scholar]
- 23.Low PA, Vernino S, Suarez G. Autonomic dysfunction in peripheral nerve disease. Muscle Nerve 2003;27:646–61. [DOI] [PubMed] [Google Scholar]
- 24.Nemos C, Mansuy V, Vernier-Magnin S, et al. Expression of gec1/GABARAPL1 versus GABARAP mRNAs in human: predominance of gec1/GABARAPL1 in the central nervous system. Brain Res Mol Brain Res 2003;119:216–19. [DOI] [PubMed] [Google Scholar]
- 25.Min H, Turck CW, Nikolic JM, et al. A new regulatory protein, KSRP, mediates exon inclusion through an intronic splicing enhancer. Genes Dev 1997;11:1023–36. [DOI] [PubMed] [Google Scholar]
- 26.Leegwater PA, Pronk JC, van der Knaap MS. Leukoencephalopathy with vanishing white matter: from magnetic resonance imaging pattern to five genes. J Child Neurol 2003;18:639–45. [DOI] [PubMed] [Google Scholar]
- 27.Lange G, DeLuca J, Maldjian JA, et al. Brain MRI abnormalities exist in a subset of patients with chronic fatigue syndrome. J Neurol Sci 1999;171:3–7. [DOI] [PubMed] [Google Scholar]
- 28.Chaudhuri A, Behan PO. In vivo magnetic resonance spectroscopy in chronic fatigue syndrome. Prostaglandins Leukot Essent Fatty Acids 2004;71:181–3. [DOI] [PubMed] [Google Scholar]
- 29.Smirnova IV, Pall ML. Elevated levels of protein carbonyls in sera of chronic fatigue syndrome patients. Mol Cell Biochem 2003;248:93–5. [DOI] [PubMed] [Google Scholar]
- 30.Byrd MP, Zamora M, Lloyd RE. Generation of multiple isoforms of eukaryotic translation initiation factor 4GI by use of alternate translation initiation codons. Mol Cell Biol 2002;22:4499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prevot D, Darlix J - L, Ohlmann T. Conducting the initiation of protein synthesis: the role of EIF4G. Biol Cell 2003;95:141–56. [DOI] [PubMed] [Google Scholar]
- 32.Kuyumcu-Martinez NM, Van Eden ME, Younan P, et al. Cleavage of poly(A)-binding protein by poliovirus 3C protease inhibits host cell translation: a novel mechanism for host translation shutoff. Mol Cell Biol 2004;24:1779–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson SR, Sarnow P. Enterovirus 71 contains a type I IRES element that functions when eukaryotic initiation factor EIF4G is cleaved. Virology 2003;315:259–66. [DOI] [PubMed] [Google Scholar]
- 34.Glaser W, Triendl A, Skern T. The processing of eIF4GI by human rhinovirus type 2 2A(pro): relationship to self-cleavage and role of zinc. J Virol 2003;77:5021–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Groft CM, Burley SK. Recognition of EIF4G by rotavirus NSP3 reveals a basis for mRNA circularisation. Mol Cell 2002;9:1273–83. [DOI] [PubMed] [Google Scholar]
- 36.Burgui I, Aragon T, Ortin J, et al. PABP1 and eIF4GI associate with influenza virus NS1 protein in viral mRNA translation initiation complexes. J Gen Virol 2003;84:3263–74. [DOI] [PubMed] [Google Scholar]
- 37.Cuesta R, Xi Q, Schneider RJ. Adenovirus-specific translation by displacement of kinase Mnk1 from cap-initiation complex eIF4F. EMBO J 2000;19:3465–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Connor JH, Lyles DS. Vesicular stomatitis virus infection alters the EIF4F translation initiation complex and causes dephosphorylation of the EIF4E binding protein 4e-BP1. J Virol 2002;76:10177–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perales C, Carrasco L, Ventoso I. Cleavage of EIF4G by HIV-1 protease: effects on translation. FEBS Lett 2003;533:89–94. [DOI] [PubMed] [Google Scholar]
- 40.Chia JK. Diverse aetiologies for the chronic fatigue syndrome. Clin Infect Dis 2003;36:671–2. [DOI] [PubMed] [Google Scholar]