

Abstract
Multigenic influences are major contributors to human genetic disorders. Since humans are highly polymorphic, there are a high number of possible detrimental, multiallelic gene pairs. The actin cytoskeleton of yeast was used to determine the potential for deleterious bigenic interactions; ∼4800 complex hemizygote strains were constructed between an actin-null allele and the nonessential gene deletion collection. We found 208 genes that have deleterious complex haploinsufficient (CHI) interactions with actin. This set is enriched for genes with gene ontology terms shared with actin, including several actin-binding protein genes, and nearly half of the CHI genes have defects in actin organization when deleted. Interactions were frequently seen with genes for multiple components of a complex or with genes involved in the same function. For example, many of the genes for the large ribosomal subunit (RPLs) were CHI with act1Δ and had actin organization defects when deleted. This was generally true of only one RPL paralog of apparently duplicate genes, suggesting functional specialization between ribosomal genes. In many cases, CHI interactions could be attributed to localized defects on the actin protein. Spatial congruence in these data suggest that the loss of binding to specific actin-binding proteins causes subsets of CHI interactions.
Keywords: Actin, cytoskeleton, genetic interactions, haploinsufficiency
Genetic influences on human disease have been the subject of many investigations that have naturally focused first on the simple cases of monogenic disorders resulting from either dominant or recessive loss of function alleles. However, dominant alleles are fairly rare, and the incidence of homozygosing even common recessive alleles (allele frequencies of ∼1:1000) is fairly rare, but clearly observable in large populations. For an increasing number of disorders, researchers have been able to show that there are clear genetic influences that are polygenic in nature. Estimates of sequence polymorphisms within human genomes range from one in 100–300 base pairs (bp) (The Human Genome Project Web site, http://www.ornl.gov/sci/techresources/Human_Genome/faq/snps.shtml) to one in ∼1200 bp (The International HapMap project, http://hapmap.org/whatishapmap.html). Given the size of human genes, these estimates suggest that any given individual has two different alleles for almost all genes. With a lower estimate of 30,000 genes in the human genome, this means that in a single person there are (60,000-choose-2) − 30,000 or ∼1.8 × 109 possible binary, complex genetic interactions. Estimates derived from consanguineous matings indicate that we each have three to 10 recessive loss of function alleles, meaning that every person could have as many as 45 (10-choose-2) complex haploinsufficient (CHI) interactions negatively influencing phenotype. In the work presented here, we employed the simplest eukaryotic model, Saccharomyces cerevisiae, to determine the prevalence of deleterious, binary, CHI interactions that can be observed for a major conserved cellular system, the actin cytoskeleton.
The actin cytoskeleton is incredibly diverse in function, participating in many cytoplasmic activities including membrane trafficking, polarized cell growth, cytokinesis, and organelle positioning and segregation. More recently, the long suspected involvement of actin in nuclear functions such as chromatin remodeling, nuclear structure, and transcription have been confirmed (Bettinger et al. 2004; Percipalle and Visa 2006). This functional diversity positions the actin cytoskeleton to be an ideal integrator of subcellular systems. In addition, the actin cytoskeleton is superbly sensitive to disturbances in stoichiometry/actin concentration (Wertman et al. 1992); these two facts, and the extremely high level of conservation of actin in structure and function, make it an ideal model for testing the potential for deleterious complex genetic interactions in the eukaryotic genome.
What we are referring to as complex haploinsufficiency is a special case of what has been previously called dominant enhancers or dominant modifiers by Drosophila researchers and unlinked noncomplementation for yeast researchers. Dominant enhancer screens in Drosophila have been employed to great utility for many years for the purpose of identifying additional players in pathways of interest. They have been particularly useful for studying the roles of signaling pathways in development. Such screens have been used to identify genes involved in position–effect variegation (Reuter and Wolff 1981), homeotic genes (Duncan 1982), the roles of the Ras and sevenless pathways in eye development (Cutforth and Rubin 1994; Karim et al. 1996; Verheyen et al. 1996), and genes involved in segmentation (Kankel et al. 2004) to name but a few examples. In comparison, unlinked noncomplementation screens have been underutilized in yeast. The first applications were on cytoskeletal genes, including screens using mutant alleles of the α-tubulin and β-tubulin genes (Stearns and Botstein 1988) and a point mutant in actin, act1-4 (Vinh et al. 1993; Welch et al. 1993). The actin screen identified four genes: ANC1, ANC2, ANC3, and ANC4. Vinh et al. (1993) went on to show genetic interactions between anc1 and the fimbrin and tropomyosin encoding genes SAC6 and TPM1, between anc3 and sac6, and between anc4 and tpm1. This network of interactions provides good support for the functional specificity of the screen. In fact, Anc1p/Taf14p is known to be in the TFIID transcription complex with actin (Sanders et al. 2002). The only previous application of unlinked noncomplementation screens to the yeast ordered knockout collection was a screen using the ctf13-30 allele that encodes for a mutant core kinetochore protein (Baetz et al. 2004). This screen was extremely successful for identifying genes involved in chromosome segregation, lending further support to the argument that this class of genetic interaction can be used to uncover functionally related genes. On a related note, randomly generated (and presumably functionally unrelated) mutations have been tested for epistasis/synergy as complex heterozygotes in yeast, and little evidence was found for interactions between such alleles (Szafraniec et al. 2003).
In this study, we screened the nonessential genes of yeast for CHI interactions with an actin-null allele. Remarkably, we found >200 genes that display CHI interactions, resulting in varying degrees of phenotypic consequence from lethality to reduced growth rates. Although functionally diverse, the collection of CHI genes is highly enriched for subsets of genes with specific functions, including several related to known actin functions.
Results
Identification of the actin CHI gene network
The yeast actin gene is an ideal candidate for performing genetic screens and selections as S. cerevisiae has a single and essential actin gene, ACT1. Furthermore, it is well suited to an analysis of genetic interactions resulting from haploinsufficiency as diploids bearing a single wild-type ACT1 allele opposite a null allele (a hemizygote) are viable but display a strong haploinsufficient phenotype (Wertman et al. 1992). We have confirmed that this haploinsufficiency is the result of lowered actin expression as the hemizygote expresses one-half the actin of a wild-type diploid as normalized to total protein (data not shown). The mechanics of a complex haploinsufficiency screen in yeast are relatively straightforward. We constructed a haploid strain deleted for the genomic copy of the single conventional actin gene (ACT1) in which the actin open reading frame was replaced with the selectable NATr (nourseothricin) resistance gene (the act1Δ∷NATr allele). Viability of this strain was maintained by a low-copy-number plasmid carrying ACT1wt. This plasmid also carried the URA3 gene, allowing selection for plasmid loss by adding 5-fluoro-orotic acid (5-FOA) to the medium. This query strain was mated robotically to an ordered array of ∼4800 haploid strains, each of which was deleted for a nonessential gene by replacement with the G418/kanamycin resistance gene (G418r) (Winzeler et al. 1999). The complex heterozygous-null diploids were selected on medium containing both nourseothricin (Nat) and G418. Diploids were then spotted to media with and without 5-FOA (5-FOA selected for cells that had lost the ACT1wt-bearing plasmid), and their relative growth rates were compared at 25°C and 35°C; 35°C is semipermissive for the act1Δ hemizygote (Wertman et al. 1992). CHI strains that appeared to have a growth deficit were confirmed by mating them manually, streaking for single colonies on rich medium (YPD) that contained Nat and G418, then replica plating to 5-FOA medium. Comparisons were made to each single mutant, and CHI scores were assigned based on the severity of the double mutants’ growth defects. These growth defects ranged from slight to completely inviable and are reported in Supplementary File S1.
Our experience with these primary CHI screens indicated that they are subsaturating and can give an appreciable number of false positives. For example, in three separate screens, 35% overlap was seen per screen. Of those genes identified in at least two screens, ∼35% reconfirmed by manual testing and of those identified in a single screen only 20% reconfirmed. These results point out the necessity for performing the screen multiple times and the need to individually and manually confirm all potential interactions. In our experience, other automated genetic interaction screening methods can have much higher false positive and negative rates. Fortunately, the CHI screen is easier to perform and the confirmation process is much less labor intensive than, for example, the tetrad dissection required for systematic synthetic lethal analysis.
The results from these initial CHI screens, following manual confirmation, remarkably led to the identification of 129 genes that are CHI with actin. These genes are shown in Figure 1, with dark-green-colored lines connecting them to the central actin hub. This includes several known actin-binding protein genes (labeled ABPs on the right of Fig. 1), including the genes for the Arp2/3 complex component Arc18p, the Arp2/3 regulator Abp1p, the V-ATPase component Vma5p, yeast fimbrin (Sac6p), yeast adenylyl cyclase-associated protein Srv2p, the formin Bnr1p, and the transcription factor Taf14p/Anc1p, which was previously found to show unlinked noncomplementation with a mutant actin allele (Welch et al. 1993). Other genes that were found to be CHI with actin have been directly implicated in actin function, including SAC1 and SAC7 (identified as suppressors of an actin mutant) (Novick et al. 1989; Dunn and Shortle 1990); the polarity regulators BEM1, PEA2, and GIC1; the catalytic subunit for phosphatase 2A (PPH21); and the folding facilitators GIM3 and GIM4. We identified seven of the 64 gene pairs curated with actin in the Saccharomyces Genome Database (SGD). The fitness of monogenic haploinsufficient strains (from the heterozygous diploid knockout collection) was recently analyzed in competition growth assays (Deutschbauer et al. 2005), providing a useful comparison to the results of our CHI screen. One question relates to whether the CHI screen with actin was biased to identify other genes that display simple haploinsufficiency. This does not seem to be the case, as only eight genes were identified in both screens (PAF1, HSL7, RPP1A, RPL6A, RPL13B, RPL14A, RPL35B, and RPL43A). The overlap is clearly biased toward translation, as six of the eight encode for components of the large ribosomal subunit. The results from the monogenic haploinsuffiency study were interpreted as having a general bias toward genes required for optimal biosynthetic capacity (e.g., translation and transcription). However, this does not seem to be the case for the actin CHI screen, as evidenced by the low amount of overlap between the CHI and simple haploinsufficiency screens, and the diversity of functional groups identified in the CHI screens.
Figure 1.

Actin shows complex haploinsufficiency with a large number of nonessential yeast genes. Hemizygosity of the indicated genes causes a growth deficit if the strain is also hemizygous for the actin gene ACT1 (example genotype yfg1Δ/YFG1wt ACT1wt/act1Δ). Some of these genes were identified by robotic screening of the nonessential gene knockout collection (indicated by dark-green connecting lines to the central hub) while others were tested because they were implicated by the results of the robotic screens (indicated by light-green connecting lines to the central hub). Functional associations between CHI gene products are indicated by blue connecting lines. Genes that encode known actin-associated proteins are grouped and labeled as “ABPs” on the right. The genes are color-coded based on their primary Gene Ontology assignment (key on the bottom right). This figure was generated largely through the use of the program Osprey (Breitkreutz et al. 2003).
Expansion of the actin CHI gene network through inference
As mentioned previously, the preliminary CHI screens were clearly subsaturating, possibly due to the ability of yeast to generate and tolerate at high frequency, 2N + 1 aneuploidies (rate ∼10−4) (Hartwell and Smith 1985; Meeks-Wagner and Hartwell 1986). Given this rate, and the pinning density of our robotics (∼10,000 cells per spot), we would expect a high incidence of suppression due to either the gain of the chromosome bearing the wild-type actin allele or the chromosome bearing the wild-type copy of the gene knocked out in the array. Therefore, the initially identified CHI genes were analyzed, with the help of the SGD, to determine additional genes that could be expected to be CHI with actin. The criteria included shared functions and/or being present in a defined complex with the product of the primary CHI gene or having a known biological function that is related to actin. In this way another 172 nonessential genes were targeted for CHI testing against the act1Δ allele; 79 of these were found to be CHI with act1Δ. This resulted in a current total of 208 genes that are CHI with act1Δ. To confirm that this enrichment is actually due to biological relationships, we screened a random set of 165 knockout strains from the collection, and only six genes (ROM1, ROM2, YLL047w, YNL205c, SPS19, and YPR147c) displayed CHI interactions with actin. Given the subsaturating nature of the screen, and the prevalence of actin CHI interactions in the genome from our screens, this is approximately the number of new CHI interactions we would have expected from these 165 genes. This also suggests that our screens may have missed as many as 138 genes that are CHI with actin, bringing the possible total to nearly 350 genes. In addition, we backcrossed 10 CHI alleles and found in all 10 cases that the CHI interaction with actin was linked to G418 resistance, confirming that neither spurious background mutations nor aneuploidies are responsible for the size of the CHI gene network.
Interconnecting blue lines have been added to Figure 1 to show the physical interactions between gene products so that functionally related clusters can be recognized. In many instances, several genes of shared function also showed a CHI interaction with actin. Some examples include the CCR4–NOT transcription complex (CCR4, NOT3, NOT5, and POP2, shown on the right of Fig. 1), the tubulin prefoldin complex (GIM1, GIM3, GIM5, PAC10, PFD1, and YKE2, shown at the left of Fig. 1), the INO80 complex (ANC1, ARP8, IES1, and IES3, shown in the lower right of Fig. 1), and the ESCRT complexes involved in multivesicular body protein sorting (DID4, STP22, VPS20, VPS22, VPS24, VPS25, VPS28, VPS32, and VPS36, shown on the upper right of Fig. 1). This observation was not universally true even for complexes in which multiple genes are CHI with actin. For example, for the CCR4–NOT complex the MOT2 gene was not CHI with actin, for the INO80 complex ARP5 was not CHI with actin, and for the ESCRT complexes SRN2 was not CHI with actin. These results suggest, as one might expect, that not all components of these complexes are sufficiently limiting to see the interaction when gene copy is lowered by one-half. Note that many essential genes that might be inferred to be CHI with actin were not tested.
These functional enrichments in the CHI network are statistically significant given the background of ∼4800 nonessential genes used for testing. We calculated the statistical enrichment of Gene Ontology (Ashburner et al. 2000) terms in our set of genes that are CHI with actin (see Supplementary File S2 for full details and results) (Sealfon et al. 2006). Many complexes, including those seen in Figure 1, are significantly enriched: the CCR4–NOT complex (P value = 2 × 10−5), the prefoldin complex (P value = 6 × 10−9), the INO80 complex (P value = 4 × 10−5), the ESCRT III complex (P value = 3 × 10−6), the endosome (P value = 3 × 10−6), the V-type ATPase complex (P value = 5 × 10−8), and the large ribosomal subunit (P value = 1 × 10−5). Additionally, several of the molecular functions associated with these complexes are also statistically overrepresented.
There is actin-related functional specialization within the ribosome
Due to an ancient genome duplication event, the yeast genome contains many paralogous gene pairs. In particular, almost all of the large ribosomal subunit genes have very closely related paralogs. These gene pairs have generally been considered to be functionally equivalent, and yet in the primary CHI screen we only identified one of each pair. We tested both paralogs for several of the large ribosomal genes for a CHI interaction with actin. We were very surprised to find that in nearly all cases, only one of the paralogs for each gene pair is CHI with actin. The exceptions are RPL7 and RPL35, for which both paralogs gave a strong CHI interaction.
To examine possible involvement in actin function, we visualized the actin cytoskeleton by rhodamine-phalloidin staining in haploid deletion strains for each paralog of a set of ribosomal genes. In nearly all cases, the paralog that is CHI with actin has severe actin and cell morphology defects while the paralog that is not CHI with actin has a normal actin cytoskeleton. The one exception was RPL43, for which both the rpl43aΔ and rpl43bΔ strains have moderate actin organization defects. As an example, Figure 2 shows the CHI testing (panel A) and rhodamine-phalloidin staining (panel B) for the RPL6 and RPL13 gene pairs. Note that rpl6aΔ and rpl13bΔ are CHI with actin and have severe actin organization defects, while rpl6bΔ and rpl13aΔ are normal in both regards, and yet the proteins encoded by these loci are predicted to be 94% and 99% identical for the RPL6 and RPL13 gene pairs, respectively.
Figure 2.

Large ribosomal subunit gene paralogs make differential contributions to actin function. (A) Strains of the genotypes ACT1wt/act1Δ rpl6aΔ/RPL6Awt, ACT1wt/act1Δ rpl6bΔ/RPL6Bwt, ACT1wt/act1Δ rpl13aΔ/RPL13Awt, and ACT1wt/act1Δ rpl13bΔ/ RPL13Bwt were constructed by mating act1Δ strains carrying a low-copy plasmid expressing wild-type actin and the URA3 gene product to strains deleted for the RPL6 and RPL13 paralogs. These complex heterozygotes were then plated to medium containing 5-FOA to select for cells that have lost the Ura3p-expressing plasmid. RPL6A and RPL13B were strongly CHI with actin while RPL6B and RPL13A were not CHI with actin. (B) Haploid strains deleted for the RPL6 and RPL13 paralogs were stained with rhodamine-phalloidin and their actin cytoskeletons were visualized by fluorescence microscopy.
We theorized that perhaps the more highly expressed paralog would be the one that displays the CHI interaction. To address this we examined the calculated protein levels based on GFP fluorescence from the integrated GFP fusions (Ghaemmaghami et al. 2003). Although this is an imperfect way to infer exact protein expression levels, they are expressed under the control of their normal promoters and therefore should be useful for comparisons. We found that there is no correlation for a paralog’s relative expression level and for it being CHI with actin. For the case of RPL13, it was estimated that there are 133,000 copies of Rpl13Ap per cell and 54,200 copies of Rpl13Bp per cell; thus the one that is CHI with actin in fact shows relatively less expression.
It has been noted that for many gene paralogs in yeast, one of the pair appears to have evolved more rapidly than its counterpart, suggesting a mechanism for functional specialization (Kellis et al. 2004). This does not appear to explain the functional divergence we are observing for the ribosomal gene pairs, as they are, in general, very highly conserved, and therefore are not evolving rapidly. Even given this slow rate of evolution, no clear correlation was observed that would suggest the CHI ribosomal genes as a group are evolving more slowly or rapidly than their non-CHI counterparts.
Although we cannot say at this time why these intriguing differences between ribosomal genes arise, our observation is that many of the large ribosomal gene paralogs are not functionally equivalent: One set appears to differentially contribute to the function and proper organization of the actin cytoskeleton.
A high percentage of CHI genes affect actin assembly in vivo; excessive actin assembly with limiting actin is lethal
In the most general sense, the CHI interactions with actin could be interpreted from two perspectives: (1) The interacting gene could impact on the function and/or organization of the actin cytoskeleton. For example, it could encode for an actin-binding protein that regulates actin assembly and dynamics. (2) The process in which the interacting gene participates could rely on the actin cytoskeleton for its efficient operation. Addressing possibility #1, experience has shown us that most actin regulators are required for proper organization of the actin cytoskeleton, as measured by rhodamine-phalloidin staining. To evaluate possibility #1 for the CHI gene set, we visualized the actin cytoskeleton in haploid knockout strains, reasoning that if a gene is involved in regulating actin, then obvious defects are most likely to be observed in the complete absence of gene activity. Surprisingly, nearly 75% of the CHI gene knockout strains had discernible defects in actin organization that were, as expected, usually accompanied by cell morphology defects. In fact, ∼40% had severe actin and cell morphology defects, and in most cases this aspect of their phenotypes had not previously been reported. Although the nature of the observed defects was diverse, many appeared to share a common feature of excessive actin filament elaboration accompanied by an elongated cell phenotype. Figure 3 shows the rhodamine-phalloidin staining for the knockouts of several genes that had this elongated cell phenotype: YDR049w is a gene of unknown function, UAF30 is involved in RNA Polymerase I transcription, SHP1 is involved in ubiquitin-mediated protein degradation, SET5 encodes a SET domain-containing protein of unknown function, and DBF2 encodes a kinase involved in exit from mitosis. Although we were surprised at the high percentage of strains displaying actin defects, these results suggest that the CHI set is enriched for genes required for proper actin organization. In comparison, a random sampling of 40 strains showed that 25% had discernible defects in actin organization with only two strains (5%) scored as having severe defects, including a cla4Δ strain that was previously known to have severe actin and cell morphology defects.
Figure 3.

Many of the genes that are CHI with actin are required for proper organization of the actin cytoskeleton. A wild-type haploid strain versus haploid strains deleted for the CHI genes YDR049W, UAF30, SHP1, SET5, and DBF2 were stained with rhodamine-phalloidin and their actin cytoskeletons were visualized by fluorescence microscopy.
Our initial assumption had been that the lowered expression in actin hemizygotes would result in a lowered capacity to make essential filamentous actin structures, and that anything that further reduced the capacity to make filaments would create a filament crisis, resulting in loss of viability. The observation that many of the null strains for CHI genes show excessive actin assembly seems to argue the converse; that the CHI diploids do not have trouble making sufficient filamentous structures but perhaps have a critically limiting pool of monomeric actin, and that excessive actin assembly under conditions of limiting actin concentration compromises cell viability. To our knowledge, this is a novel hypothesis to explain the potential deleterious effects of haploinsufficiency, so we engineered a relatively simple test of this model. The act1-159p mutant (V159N) forms more stable actin filaments by reducing the rate of filament disassembly (Belmont and Drubin 1998). We generated a diploid strain that had the act1-159 allele over an actin-null allele at the genomic actin loci, and also carried the ACT1wt, URA3 plasmid. This strain was inviable on 5-FOA medium (that selected against the ACT1wt- bearing plasmid), confirming that under conditions of limiting actin concentration, filamentous actin stabilization is detrimental.
Distinct steps in endocytosis are sensitive to actin stoichiometry
Another example of inappropriate and excessive actin assembly was observed in the strains deleted for genes encoding components of the ESCRT I, II, and III complexes. These multiprotein complexes are sequentially recruited to endosomal membranes where they are involved in sorting and degradation of endosomal membrane proteins (Slagsvold et al. 2006). As shown in Figure 4, F-actin was observed to accumulate on an intracellular compartment found in the mother cell body of these vps36Δ cells. All of the strains deleted for any of the genes encoding components of the ESCRT complexes displayed an identical pattern of F-actin accumulation. This F-actin-containing structure was found in virtually 100% of the mutant cells; it was usually not cortical, and was always found in the mother cell body (not polarized), and therefore seems distinct from the actin cortical patches. This localization strongly resembled the “class E body” that is an intermediate, prevacoular endocytic compartment known to accumulate in these mutants (Babst 2005). Although actin’s involvement in endocytic trafficking is well established (Ayscough 2004), F-actin accumulation on class E bodies has not been previously reported. Given our results with the act1-159 allele, it is tempting to theorize that excessive F-actin assembly on accumulating class E bodies leads to a consumption of the monomer pool and the observed CHI interactions with ESCRT complex encoding genes. Alternatively, the cumulative defects in this and other actin-directed endocytic pathways may result in the observed growth defects.
Figure 4.
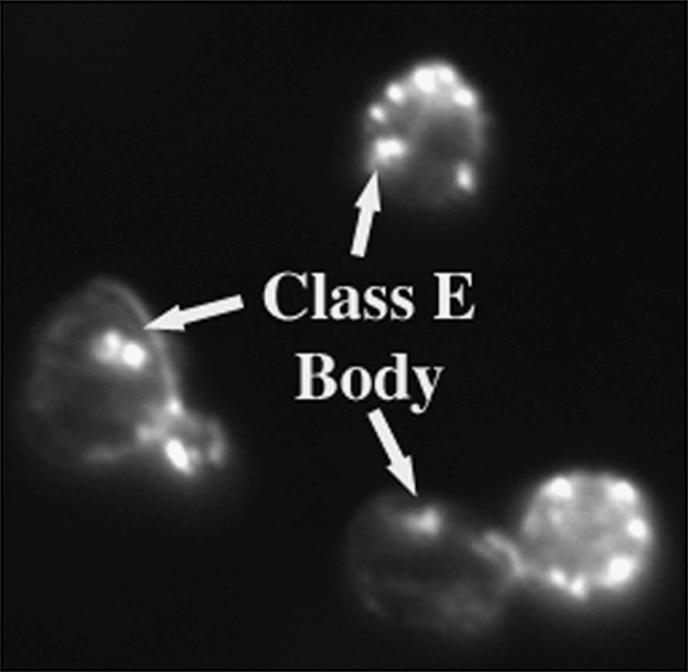
Deletion of genes encoding ESCRT complex components causes F-actin to accumulate on the Class E endocytic compartment. A vps32Δ strain was stained with rhodamine-phalloidin and its actin cytoskeleton was visualized by fluorescence microscopy.
As can be observed in Figure 1, the CHI screen with actin identified a large number of yeast genes of unknown or unassigned function (indicated in gray). Certainly one of the goals of genome level screens is to assign functions to the still considerably large number of yeast genes for which little is known. We might expect some of these genes to fall within the well-represented functional categories shown in Figure 1. For example, we might expect one to two new actin-binding proteins within this set of genes of unknown function. Since most actin-binding proteins display a distinctive localization pattern, we again turned to GFP localization data for functional clues (Huh et al. 2003). Two proteins in particular had very similar localization patterns to actin cortical patch localization: Lsp1p and Pst2p. The cortical spots of Lsp1p and Pst2p were different from actin cortical patches in that they were not strictly polarized to the bud or neck cortex. LSP1 has a paralog (PIL1) whose protein shows a similar localization pattern to Lsp1p, and PST2 has two paralogs (YCP4 and RFS1) whose products both show similar localization patterns to Pst2p and Lsp1p. All of the paralogs were found to have CHI interactions with actin, and the null strains showed modest defects in actin organization and a low percentage of cells with aberrant cell morphologies. Subsequent to these analyses, the laboratory of Peter Walter (Walther et al. 2006) identified pre-endocytic structures (termed eisosomes) that contain the Lsp1 and Pil1 proteins. The CHI interactions with actin suggest that the cytoskeleton has a role in the function of these structures. In addition, the localization data further suggests that the Pst2p, Ycp4p, and Rfs1p homologs may also function within the eisosomes.
Further bioinformatic analysis of the CHI gene set
The initial set of genes identified to be CHI with actin were also analyzed using the Genomica Web site (http://genomica.weizmann.ac.il) to identify possible similarities/correlations. Beside the previously mentioned bias toward cytoskeletal and polarity genes, these programs found a nonrandom distribution of predicted Sum1p transcription factor-binding sites in the promoter regions of several CHI genes (P value = 10−4) (Segal and Sharan 2005). This observation predicted that sum1Δ should also be CHI with actin, and this was, in fact, confirmed to be the case. Also consistent with a role in the regulation of cytoskeletal genes are results from the Boone laboratory (Tong et al. 2004) reporting that the sum1Δ allele is synthetic lethal/sick with several prefoldin complex genes identified in our CHI screen (GIM3, GIM5, PAC10, and YKE2), and several polarity and cytoskeletal genes (ARP2, ARC40, CDC42, RVS161, and RVS167). Collectively, these observations suggest that Sum1p has a particularly important role in the regulation of genes that impact the actin cytoskeleton.
In addition to this analysis, we observed statistical enrichment of many biological process GO terms related to actin, such as vacuolar transport (P value = 2 × 10−13), actin filament-based process (P value = 2 × 10−6), and actin filament organization (P value = 4 × 10−6). We also compared our CHI results with a Bayesian integration of functional genomic data (such as copurification, two-hybrid, synthetic lethality, and other physical and genetic interactions) using the bioPIXIE system (Myers et al. 2005). A significant proportion of the genes identified as CHI in our study appear in the list of actin interaction partners based on data integration (see Supplementary File S3 for full details and results). However, many genes CHI with actin are not represented in existing data sources, which indicates that these important, complex interactions require study beyond the existing high-throughput experimental literature. The statistical analyses reported above were performed on the final network and therefore accurately reflect its composition. However, these biological processes are also significantly enriched in the original network of 129 genes obtained through random screening, which indicates that this functional enrichment was present in the primary screens even prior to expansion of the network with functionally related genes. Complete enrichment analysis of both the original and final networks is available in Supplementary File S2.
The examples presented above (the ribosomal genes, the ESCRT genes, the eisosome genes, and SUM1) have been highlighted to point out just some of the novel biological information that can be derived from our data set. We certainly expect that more intensive bioinformatics analysis will suggest additional, biologically relevant hypotheses.
The loss of distinct actin functions recapitulates CHI interactions
We have at our disposal an extremely useful set of reagents for studying structure/function relationships within actin: a collection of actin mutants specifically designed by alanine scanning to neutralize clusters of charged residues on the surface of the actin protein (see Fig. 5A; Wertman et al. 1992). We theorize that many of the CHI interactions with the actin-null allele may not merely result from a 50% reduction in total actin activity, but in fact may be due to the loss of subsets of specific actin functions; for example, a reduction in the capacity to bind specific actin-binding proteins. The actin alanine scan alleles are perfectly suited to address this hypothesis.
Figure 5.

CHI interactions can be attributed to the loss of subsets of actin functions. (A) The front surface of actin is shown with the locations of actin alanine scan alleles color-coded by growth phenotype (green, wild type; yellow, recessive conditional; red, recessive lethal; blue, presumed dominant lethal). The locations of six alleles tested for unlinked noncomplementation with the CHI genes shown in Figure 1 are indicated with arrows; note that the act1-129 allele residues are on the back side of the actin. (B) The hubs show actin alleles tested against the CHI gene set for complex heterozygous interactions, and the nodes are those genes that were found to interact with these alleles. B was generated largely through the use of the program Osprey (Breitkreutz et al. 2003).
As a preliminary test of this idea, we constructed query haploid strains carrying six different alanine scan alleles at the normal actin locus and covered with the wild-type actin gene on the counterselectable plasmid. These strains were manually tested for deleterious complex heterozygous (unlinked noncomplementation) interactions with null alleles for the strains known to be CHI with the act1Δ allele from our primary screens. As can be seen in Figure 5B, some of the CHI interactions can be recapitulated as complex heterozygous interactions with these alleles. As one might expect, the severity of the alleles (as indicated by the growth deficits of strains in which the mutant is the only source of actin) correlates with the number of interactions. For example, strains bearing both the act1-105 and act1-111 alleles are severely impaired, while strains expressing act1-102 are wild type for growth (Wertman et al. 1992).
Our first indication that CHI interactions may reflect the loss of localized activities on the surface of actin came from an apparent spatial congruity in the data. Alleles that alter residues near each other on the surface of actin (such as act1-105 and act1-111) (see Fig. 5A) share a large number of interactions. This could be misleading, because these alleles recapitulate a large number of interactions (90 of 208 for act1-105 and 50 of 208 for act1-111). However, based on a hypergeometric distribution, the P value for this result occurring by random chance is 8 × 10−8, clearly allowing us to discard the null hypothesis. We theorize that this deviation from random behavior might be partially attributable to reductions in the binding of specific actin-interacting proteins, and would therefore be able to be phenocopied by null alleles of these actin-interacting proteins. In the case of act1-111, 37 of its 50 interactions (74%) are shared with act1-105, suggesting that most of the defects associated with act1-111p are shared with act1-105p. For example, the 29 genes that interact genetically only with the act1-111 and act1-105 alleles may predict that there are one or more actin-binding proteins that are unable to bind to either act1-111p or the act1-105p proteins, and that it is the failure of these physical interactions that could be the underlying cause of this subset of CHI genetic interactions. In contrast, RPB9 and PSD1 also interact with act1-102, possibly suggesting the existence of another actin-binding protein that cannot interact with act1-105p, act1-111p, and act1-102p, and that it is the loss of this interaction that is responsible for the RPB9 and PSD1 interactions. Some of the CHI gene alleles such as uaf30Δ and bud32Δ interact with all six alleles, indicating they are very sensitive to the loss of any actin function. In addition, some alleles that alter distant residues of actin show extensive overlaps in CHI interactions, suggesting that these mutants may cause more global defects in actin structure, or that the CHI genes identified are sensitive to diverse effects on actin function.
Discussion
The yeast model has proven to be extremely useful for determining the prevalence of multigenic influences on phenotype in a eukaryotic genome. Systematic screening for synthetic lethality has shown that most genes display multiple deleterious interactions with genes that are functionally related. However, synthetic lethality in the human genome would require homozygosing loss of function alleles for two genes, clearly a more rare event than inheriting heterozygous gene pairs.
In this study we modeled the extreme case of inheriting two null alleles, and found that quite frequently this can have an adverse effect on cell function. Actin is arguably one of the most centrally important proteins in eukaryotic cells, and it does display haploinsufficiency on its own, so one might have predicted it would have a large number of CHI interactions. Preliminary screens with two other genes (ORC1 and CDC12), whose products are found in important and complex structures, found relatively few CHI interactions (J. Lu, D. Sliter, and D. Amberg, unpubl.). Since neither of these genes show simple haploinsuffiency, their products may not be sufficiently limiting to make them good candidates for CHI analysis. Nevertheless, these results argue that complex heterozygous gene interactions may have significant effects on genetically based phenotypic diversity.
In fact, haploinsufficiency has been gaining in appreciation as an important influence on human disease. For example, simple haploinsufficiency in a large number of transcription factors has been found to contribute to severe and diverse genetic disorders (for a review, see Seidman and Seidman 2002). Haploinsufficiency has also been found to cause defects in liver regeneration in mice (Kalinichenko et al. 2003), disease-related defects in platelet production in humans (Deutsch et al. 2003), cardiomyopathy in mice and humans (Yu et al. 1998; Andersen et al. 2004; Carrier et al. 2004), and defects in neuronal migration (Kholmanskikh et al. 2003) and synaptogenesis (Boeckers et al. 2002). Complex haploinsufficiency is also gaining recognition as an important contributor to oncogenesis in humans (Santarosa and Ashworth 2004; Ma et al. 2005) and to cancer development in mouse models (Goss et al. 2002; Spring et al. 2002). Finally, a very recent paper reports that mice that are hemizygous for the Bub3 mitotic checkpoint gene in conjunction with being hemizygous for the Rae1 nucleoporin encoding gene display an early aging phenotype (Baker et al. 2006). Given the high levels of functional conservation between yeast and human genes, we might expect that CHI networks will be conserved as well. If this is the case, then modeling these networks in yeast could be highly informative for guiding human geneticists working on multigenic disorders for which the contributing genes have eluded identification.
Although estimates indicate that human individuals may have as many as 45 CHI gene pairs, partial loss-of-function alleles are likely to be much more common. Therefore, we examined whether our CHI interactions with actin could be recapitulated as complex heterozygous interactions with viable point mutations in actin. Although the number of interactions identified correlated with the severity of growth defects caused by the actin allele, many of the CHI interactions could be observed using actin mutants that partially compromise actin function, and even a few interactions could be observed with alleles that have only slight effects on actin function (Wertman et al. 1992). These mutants discriminated between the CHI genes, but also showed significant overlap in cases of spatial congruity between the residues changed by these alleles. This spatial congruity suggests to us that CHI interactions may result from the loss of interactions between actin and actin-binding proteins. If this is the case, then null alleles of actin-binding protein genes should phenocopy subsets of CHI interactions with the actin alleles. Conversely, the genetic interaction overlap between the actin alleles and actin-binding protein genes might be used to map the binding sites for these actin-binding proteins on actin, and may help to infer the binding sites for unidentified actin-binding proteins. By using the large set of actin mutants at our disposal, we expect to be able to derive a CHI-based structure–function map of the actin surface—a long-term goal of this project.
Standard genetic suppression and synthetic lethal analyses in yeast have been very fruitful for discovering functional relationships between genes. The data presented here indicate that CHI analysis will be equally powerful for ascertaining biologically relevant gene interaction networks. An additional advantage of CHI analysis is the ease with which essential genes can be analyzed; this is not so readily done with synthetic lethality. The screen was clearly biased toward genes already known to be involved in actin-related functions, and identified several well-known actin-binding protein genes. The set of CHI genes displayed in Figure 1 is functionally diverse, indicative of the importance of actin to a wide variety of cellular functions. The variety of functional categories that are overrepresented in this list likely reflects this functional diversity. Approximately 50% of these genes are required for normal actin organization, and although we have not examined the entire deletion collection in this manner, a random sampling of 40 deletion strains confirmed that there is an overrepresentation of genes in our list that impact actin function. In addition, if the screens lacked specificity, we would expect that genes with shared functions would not all be CHI with actin, but in fact, the converse was observed for several multigenic complexes. An interesting example of this specificity was the identification of genes encoding proteins of the large ribosomal subunit. Functional complexity for ribosomal proteins has been previously reported (for a review, see Wool 1996), but our finding that the paralogs for these genes are not equivalent for CHI interactions with actin, or their effects on the organization of the actin cytoskeleton, suggests further functional specialization between paralogs. A similar observation was previously reported in a screen for genes that affect bipolar bud site selection: Homozygous diploid knockouts of ribosomal protein genes were found to display a random bud site selection phenotype, but only one member of a paralog pair was defective (Ni and Snyder 2001). Unfortunately, there was no overlap between the RPL genes we identified in our primary CHI screens and their data. However, we have been analyzing all of the ribosomal protein gene knockouts for CHI interactions with actin, and it appears that many of the paralogs reported by the Snyder laboratory (Ni and Snyder 2001) to have bipolar bud site selection defects are also CHI with actin (B. Haarer, S. Viggiano, and D. Amberg, in prep.). Disruption of actin organization frequently causes bipolar bud site selection defects, so this correlation is not unexpected. We do not currently understand the biological basis for this functional specialization but two possibilities come to mind: (1) The cell constructs two (or more) types of ribosomes with different paralog compositions and some of these specialized ribosomes have a greater role in translating cytoskeletal proteins, or (2) RPL paralogs are under different regulatory constraints. For example, some may be able to autoregulate in response to reductions in expression of their paralog, or in response to cellular stresses, and that limiting production of functionally equivalent ribosomes selectively affects the actin cytoskeleton. We are currently investigating these possibilities and the underlying cause of this interesting genetic relationship.
We hypothesize that CHI analysis will not only be useful for uncovering disease relevant (networks of) genetic interactions, but that, like other methods of genetic interaction analysis, it will uncover novel aspects of cell biology. Two features that distinguish CHI analysis are (1) the relative ease with which these screens can be performed in contrast to the very labor intensive procedures that synthetic lethality or suppression analysis entail, and (2) a greater relevance to the polygenic influences that shape human phenotype.
Materials and methods
Yeast genetic methods, strain, and plasmid constructions
Growth and media for the manipulation and genetic analysis of yeast were as described (Amberg et al. 2005). Strains were manipulated with a Virtek Colony Arrayer (Virtek). The EUROSCARF collection of nonessential yeast deletion mutants arrayed in 384-colony-per-plate format was used for this study; the general genotype of these strains is MATa ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 yfgΔ∷G418r. The act1Δ0∷NATr query strain was generated by transformation of strain SGAY9 (MATa/MATα ura3Δ0/ura3Δ0 leu2Δ0/leu2Δ0 his3Δ1/his3Δ1 met15Δ0/MET15 lys2Δ0/LYS2 mfa1Δ∷PMFA1-sphis5+/mfa1Δ∷PMFA1-sphis5+) with a PCR fragment in which the ACT1 coding region had been replaced with the NATr gene. The resulting NATr diploid was transformed with plasmid pKFW29 (CEN, ACT1, URA3); Ura+ transformants were sporulated and tetrad dissection yielded haploid strain BHY336 (MATα ura3Δ0 leu2Δ0 his3Δ1 act1Δ0∷NATr mfa1Δ∷PMFA1-sphis5+ [pKFW29]; note that the mfa1Δ∷PMFA1-sphis5+ locus is irrelevant for this study, but was constructed for use in SGA-style screens).
A recipient strain for generating act1 alanine scan mutants was generated by transforming diploid strain BY4741 × BY4742 with the act1Δ0∷NATr PCR fragment, followed by introduction of plasmid pKFW29, then tetrad dissection to yield strain SVY12 (MATα ura3Δ0 leu2Δ0 his3Δ1 act1Δ0∷NATr [pKFW29]). Plasmids carrying act1 alanine scan alleles marked downstream with the HIS3 gene (Wertman et al. 1992) were digested with EcoRI, and the resulting act1:HIS3 fragment was used to transform SVY12 to His+. Transformants were tested for sensitivity to NAT (indicating replacement of the deletion allele with alanine scan mutant act1); the act1 loci were then amplified by PCR and sequenced to confirm the presence of appropriate act1 mutations. Finally, the act1-linked HIS3 gene was replaced with the NATr gene by transforming with a NAT-switcher PCR product; NATr transformants were confirmed to be His−.
Detailed media recipes and protocols for performing CHI screens can be found in Supplementary File S4.
Statistical analyses
All tests for statistical significance of GO term enrichment were performed using the hypergeometric distribution:
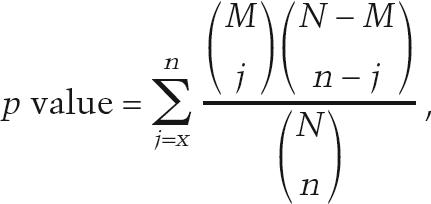 |
where N is the total number of background genes, M is the number of genes annotated to the GO term of interest, n is the number of query genes, and x is the number of query genes annotated to the GO term of interest. For the CHI enrichment analysis, only those genes present in the EUROSCARF haploid deletion collection were used as the background. In order to account for testing multiple hypotheses, significance levels were determined using a false discovery rate (FDR) of 0.05 (Benjamini and Yekutieli 2001). Other tests for significance of overlap also use the hypergeometric distribution with appropriate backgrounds.
The set of predicted actin interaction partners was created through a Bayesian integration of many diverse data types (e.g., two-hybrid, affinity precipitation, synthetic lethality, microarrays, etc.) in a manner similar to Myers et al. (2005). This type of integration weighs each evidence type based on its reliability to recapitulate known biology, and then assigns a probabilistic score to each pair of genes indicating a confidence level of either direct or indirect functional interaction. We report only genes that interact with actin at a confidence score higher than the prior, which limits this list to contain only those genes that have some positive interaction evidence.
Acknowledgments
We thank Dan Burke, David Botstein, and members of the Amberg and Troyanskaya laboratories for advice and helpful discussions. This work was supported in the Amberg laboratory by NIH grant GM56189, and in the Troyanskaya laboratory by NIH grant R01 GM071966 and NSF grant IIS-0513552.
Footnotes
Supplemental material is available at http://www.genesdev.org.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.1477507
References
- Amberg D.C., Burke D.J., Strathern J.N., Burke D.J., Strathern J.N., Strathern J.N. Methods in yeast genetics: A Cold Spring Harbor Laboratory course manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2005. [Google Scholar]
- Andersen P.S., Havndrup O., Bundgaard H., Larsen L.A., Vuust J., Pedersen A.K., Kjeldsen K., Christiansen M., Havndrup O., Bundgaard H., Larsen L.A., Vuust J., Pedersen A.K., Kjeldsen K., Christiansen M., Bundgaard H., Larsen L.A., Vuust J., Pedersen A.K., Kjeldsen K., Christiansen M., Larsen L.A., Vuust J., Pedersen A.K., Kjeldsen K., Christiansen M., Vuust J., Pedersen A.K., Kjeldsen K., Christiansen M., Pedersen A.K., Kjeldsen K., Christiansen M., Kjeldsen K., Christiansen M., Christiansen M. Genetic and phenotypic characterization of mutations in myosin-binding protein C (MYBPC3) in 81 families with familial hypertrophic cardiomyopathy: Total or partial haploinsufficiency. Eur. J. Hum. Genet. 2004;12:673–677. doi: 10.1038/sj.ejhg.5201190. [DOI] [PubMed] [Google Scholar]
- Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Dolinski K., Dwight S.S., Eppig J.T., Dwight S.S., Eppig J.T., Eppig J.T., et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayscough K.R. Endocytosis: Actin in the driving seat. Curr. Biol. 2004;14:R124–R126. [PubMed] [Google Scholar]
- Babst M. A protein’s final ESCRT. Traffic. 2005;6:2–9. doi: 10.1111/j.1600-0854.2004.00246.x. [DOI] [PubMed] [Google Scholar]
- Baetz K.K., Krogan N.J., Emili A., Greenblatt J., Hieter P., Krogan N.J., Emili A., Greenblatt J., Hieter P., Emili A., Greenblatt J., Hieter P., Greenblatt J., Hieter P., Hieter P. The ctf13-30/CTF13 genomic haploinsufficiency modifier screen identifies the yeast chromatin remodeling complex RSC, which is required for the establishment of sister chromatid cohesion. Mol. Cell. Biol. 2004;24:1232–1244. doi: 10.1128/MCB.24.3.1232-1244.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D.J., Jeganathan K.B., Malureanu L., Perez-Terzic C., Terzic A., van Deursen J.M., Jeganathan K.B., Malureanu L., Perez-Terzic C., Terzic A., van Deursen J.M., Malureanu L., Perez-Terzic C., Terzic A., van Deursen J.M., Perez-Terzic C., Terzic A., van Deursen J.M., Terzic A., van Deursen J.M., van Deursen J.M. Early aging-associated phenotypes in Bub3/Rae1 haploinsufficient mice. J. Cell Biol. 2006;172:529–540. doi: 10.1083/jcb.200507081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont L.D., Drubin D.G., Drubin D.G. The yeast V159N actin mutant reveals roles for actin dynamics in vivo. J. Cell Biol. 1998;142:1289–1299. doi: 10.1083/jcb.142.5.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Yekutieli D., Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann. Statist. 2001;29:1165–1188. [Google Scholar]
- Bettinger B.T., Gilbert D.M., Amberg D.C., Gilbert D.M., Amberg D.C., Amberg D.C. Actin up in the nucleus. Nat. Rev. Mol. Cell Biol. 2004;5:410–415. doi: 10.1038/nrm1370. [DOI] [PubMed] [Google Scholar]
- Boeckers T.M., Bockmann J., Kreutz M.R., Gundelfinger E.D., Bockmann J., Kreutz M.R., Gundelfinger E.D., Kreutz M.R., Gundelfinger E.D., Gundelfinger E.D. ProSAP/Shank proteins—A family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J. Neurochem. 2002;81:903–910. doi: 10.1046/j.1471-4159.2002.00931.x. [DOI] [PubMed] [Google Scholar]
- Breitkreutz B.J., Stark C., Tyers M., Stark C., Tyers M., Tyers M. Osprey: A network visualization system. Genome Biol. 2003;4:R22. doi: 10.1186/gb-2003-4-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrier L., Knoll R., Vignier N., Keller D.I., Bausero P., Prudhon B., Isnard R., Ambroisine M.L., Fiszman M., Ross J., Jr., Knoll R., Vignier N., Keller D.I., Bausero P., Prudhon B., Isnard R., Ambroisine M.L., Fiszman M., Ross J., Jr., Vignier N., Keller D.I., Bausero P., Prudhon B., Isnard R., Ambroisine M.L., Fiszman M., Ross J., Jr., Keller D.I., Bausero P., Prudhon B., Isnard R., Ambroisine M.L., Fiszman M., Ross J., Jr., Bausero P., Prudhon B., Isnard R., Ambroisine M.L., Fiszman M., Ross J., Jr., Prudhon B., Isnard R., Ambroisine M.L., Fiszman M., Ross J., Jr., Isnard R., Ambroisine M.L., Fiszman M., Ross J., Jr., Ambroisine M.L., Fiszman M., Ross J., Jr., Fiszman M., Ross J., Jr., Ross J., Jr., et al. Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovasc. Res. 2004;63:293–304. doi: 10.1016/j.cardiores.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Cutforth T., Rubin G.M., Rubin G.M. Mutations in Hsp83 and cdc37 impair signaling by the sevenless receptor tyrosine kinase in Drosophila. Cell. 1994;77:1027–1036. doi: 10.1016/0092-8674(94)90442-1. [DOI] [PubMed] [Google Scholar]
- Deutsch S., Rideau A., Bochaton-Piallat M.L., Merla G., Geinoz A., Gabbiani G., Schwede T., Matthes T., Antonarakis S.E., Beris P., Rideau A., Bochaton-Piallat M.L., Merla G., Geinoz A., Gabbiani G., Schwede T., Matthes T., Antonarakis S.E., Beris P., Bochaton-Piallat M.L., Merla G., Geinoz A., Gabbiani G., Schwede T., Matthes T., Antonarakis S.E., Beris P., Merla G., Geinoz A., Gabbiani G., Schwede T., Matthes T., Antonarakis S.E., Beris P., Geinoz A., Gabbiani G., Schwede T., Matthes T., Antonarakis S.E., Beris P., Gabbiani G., Schwede T., Matthes T., Antonarakis S.E., Beris P., Schwede T., Matthes T., Antonarakis S.E., Beris P., Matthes T., Antonarakis S.E., Beris P., Antonarakis S.E., Beris P., Beris P. Asp1424Asn MYH9 mutation results in an unstable protein responsible for the phenotypes in May-Hegglin anomaly/Fechtner syndrome. Blood. 2003;102:529–534. doi: 10.1182/blood-2002-09-2783. [DOI] [PubMed] [Google Scholar]
- Deutschbauer A.M., Jaramillo D.F., Proctor M., Kumm J., Hillenmeyer M.E., Davis R.W., Nislow C., Giaever G., Jaramillo D.F., Proctor M., Kumm J., Hillenmeyer M.E., Davis R.W., Nislow C., Giaever G., Proctor M., Kumm J., Hillenmeyer M.E., Davis R.W., Nislow C., Giaever G., Kumm J., Hillenmeyer M.E., Davis R.W., Nislow C., Giaever G., Hillenmeyer M.E., Davis R.W., Nislow C., Giaever G., Davis R.W., Nislow C., Giaever G., Nislow C., Giaever G., Giaever G. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics. 2005;169:1915–1925. doi: 10.1534/genetics.104.036871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan I.M. Polycomblike: A gene that appears to be required for the normal expression of the bithorax and antennapedia gene complexes of Drosophila melanogaster. Genetics. 1982;102:49–70. doi: 10.1093/genetics/102.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn T.M., Shortle D., Shortle D. Null alleles of SAC7 suppress temperature sensitive actin mutations in Saccharomyces cerevisiae. Mol. Cell. Biol. 1990;10:2308–2314. doi: 10.1128/mcb.10.5.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S., Huh W.K., Bower K., Howson R.W., Belle A., Dephoure N., O’Shea E.K., Weissman J.S., Huh W.K., Bower K., Howson R.W., Belle A., Dephoure N., O’Shea E.K., Weissman J.S., Bower K., Howson R.W., Belle A., Dephoure N., O’Shea E.K., Weissman J.S., Howson R.W., Belle A., Dephoure N., O’Shea E.K., Weissman J.S., Belle A., Dephoure N., O’Shea E.K., Weissman J.S., Dephoure N., O’Shea E.K., Weissman J.S., O’Shea E.K., Weissman J.S., Weissman J.S. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Goss K.H., Risinger M.A., Kordich J.J., Sanz M.M., Straughen J.E., Slovek L.E., Capobianco A.J., German J., Boivin G.P., Groden J., Risinger M.A., Kordich J.J., Sanz M.M., Straughen J.E., Slovek L.E., Capobianco A.J., German J., Boivin G.P., Groden J., Kordich J.J., Sanz M.M., Straughen J.E., Slovek L.E., Capobianco A.J., German J., Boivin G.P., Groden J., Sanz M.M., Straughen J.E., Slovek L.E., Capobianco A.J., German J., Boivin G.P., Groden J., Straughen J.E., Slovek L.E., Capobianco A.J., German J., Boivin G.P., Groden J., Slovek L.E., Capobianco A.J., German J., Boivin G.P., Groden J., Capobianco A.J., German J., Boivin G.P., Groden J., German J., Boivin G.P., Groden J., Boivin G.P., Groden J., Groden J. Enhanced tumor formation in mice heterozygous for Blm mutation. Science. 2002;297:2051–2053. doi: 10.1126/science.1074340. [DOI] [PubMed] [Google Scholar]
- Hartwell L.H., Smith D., Smith D. Altered fidelity of mitotic chromosome transmission in cell cycle mutants of S. cerevisiae. Genetics. 1985;110:381–395. doi: 10.1093/genetics/110.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh W.K., Falvo J.V., Gerke L.C., Carroll A.S., Howson R.W., Weissman J.S., O’Shea E.K., Falvo J.V., Gerke L.C., Carroll A.S., Howson R.W., Weissman J.S., O’Shea E.K., Gerke L.C., Carroll A.S., Howson R.W., Weissman J.S., O’Shea E.K., Carroll A.S., Howson R.W., Weissman J.S., O’Shea E.K., Howson R.W., Weissman J.S., O’Shea E.K., Weissman J.S., O’Shea E.K., O’Shea E.K. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Kalinichenko V.V., Bhattacharyya D., Zhou Y., Gusarova G.A., Kim W., Shin B., Costa R.H., Bhattacharyya D., Zhou Y., Gusarova G.A., Kim W., Shin B., Costa R.H., Zhou Y., Gusarova G.A., Kim W., Shin B., Costa R.H., Gusarova G.A., Kim W., Shin B., Costa R.H., Kim W., Shin B., Costa R.H., Shin B., Costa R.H., Costa R.H. Foxf1+/− mice exhibit defective stellate cell activation and abnormal liver regeneration following CCl4 injury. Hepatology. 2003;37:107–117. doi: 10.1053/jhep.2003.50005. [DOI] [PubMed] [Google Scholar]
- Kankel M.W., Duncan D.M., Duncan I., Duncan D.M., Duncan I., Duncan I. A screen for genes that interact with the Drosophila pair-rule segmentation gene fushi tarazu. Genetics. 2004;168:161–180. doi: 10.1534/genetics.104.027250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F.D., Chang H.C., Therrien M., Wassarman D.A., Laverty T., Rubin G.M., Chang H.C., Therrien M., Wassarman D.A., Laverty T., Rubin G.M., Therrien M., Wassarman D.A., Laverty T., Rubin G.M., Wassarman D.A., Laverty T., Rubin G.M., Laverty T., Rubin G.M., Rubin G.M. A screen for genes that function downstream of Ras1 during Drosophila eye development. Genetics. 1996;143:315–329. doi: 10.1093/genetics/143.1.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M., Birren B.W., Lander E.S., Birren B.W., Lander E.S., Lander E.S. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature. 2004;428:617–624. doi: 10.1038/nature02424. [DOI] [PubMed] [Google Scholar]
- Kholmanskikh S.S., Dobrin J.S., Wynshaw-Boris A., Letourneau P.C., Ross M.E., Dobrin J.S., Wynshaw-Boris A., Letourneau P.C., Ross M.E., Wynshaw-Boris A., Letourneau P.C., Ross M.E., Letourneau P.C., Ross M.E., Ross M.E. Disregulated RhoGTPases and actin cytoskeleton contribute to the migration defect in Lis1-deficient neurons. J. Neurosci. 2003;23:8673–8681. doi: 10.1523/JNEUROSCI.23-25-08673.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L., Teruya-Feldstein J., Behrendt N., Chen Z., Noda T., Hino O., Cordon-Cardo C., Pandolfi P.P., Teruya-Feldstein J., Behrendt N., Chen Z., Noda T., Hino O., Cordon-Cardo C., Pandolfi P.P., Behrendt N., Chen Z., Noda T., Hino O., Cordon-Cardo C., Pandolfi P.P., Chen Z., Noda T., Hino O., Cordon-Cardo C., Pandolfi P.P., Noda T., Hino O., Cordon-Cardo C., Pandolfi P.P., Hino O., Cordon-Cardo C., Pandolfi P.P., Cordon-Cardo C., Pandolfi P.P., Pandolfi P.P. Genetic analysis of Pten and Tsc2 functional interactions in the mouse reveals asymmetrical haploinsufficiency in tumor suppression. Genes & Dev. 2005;19:1779–1786. doi: 10.1101/gad.1314405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeks-Wagner D., Hartwell L.H., Hartwell L.H. Normal stoichiometry of histone dimer sets is necessary for high fidelity of mitotic chromosome transmission. Cell. 1986;44:43–52. doi: 10.1016/0092-8674(86)90483-6. [DOI] [PubMed] [Google Scholar]
- Myers C.L., Robson D., Wible A., Hibbs M.A., Chiriac C., Theesfeld C.L., Dolinski K., Troyanskaya O.G., Robson D., Wible A., Hibbs M.A., Chiriac C., Theesfeld C.L., Dolinski K., Troyanskaya O.G., Wible A., Hibbs M.A., Chiriac C., Theesfeld C.L., Dolinski K., Troyanskaya O.G., Hibbs M.A., Chiriac C., Theesfeld C.L., Dolinski K., Troyanskaya O.G., Chiriac C., Theesfeld C.L., Dolinski K., Troyanskaya O.G., Theesfeld C.L., Dolinski K., Troyanskaya O.G., Dolinski K., Troyanskaya O.G., Troyanskaya O.G. Discovery of biological networks from diverse functional genomic data. Genome Biol. 2005;6:R114. doi: 10.1186/gb-2005-6-13-r114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni L., Snyder M., Snyder M. A genomic study of the bipolar bud site selection pattern in Saccharomyces cerevisiae. Mol. Biol. Cell. 2001;12:2147–2170. doi: 10.1091/mbc.12.7.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P., Osmond B.C., Botstein D., Osmond B.C., Botstein D., Botstein D. Suppressors of yeast actin mutations. Genetics. 1989;121:659–674. doi: 10.1093/genetics/121.4.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percipalle P., Visa N., Visa N. Molecular functions of nuclear actin in transcription. J. Cell Biol. 2006;172:967–971. doi: 10.1083/jcb.200512083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter G., Wolff I., Wolff I. Isolation of dominant suppressor mutations for position–effect variegation in Drosophila melanogaster. Mol. Gen. Genet. 1981;182:516–519. doi: 10.1007/BF00293947. [DOI] [PubMed] [Google Scholar]
- Sanders S.L., Jennings J., Canutescu A., Link A.J., Weil P.A., Jennings J., Canutescu A., Link A.J., Weil P.A., Canutescu A., Link A.J., Weil P.A., Link A.J., Weil P.A., Weil P.A. Proteomics of the eukaryotic transcription machinery: Identification of proteins associated with components of yeast TFIID by multidimensional mass spectrometry. Mol. Cell. Biol. 2002;22:4723–4738. doi: 10.1128/MCB.22.13.4723-4738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarosa M., Ashworth A., Ashworth A. Haploinsufficiency for tumour suppressor genes: When you don’t need to go all the way. Biochim. Biophys. Acta. 2004;1654:105–122. doi: 10.1016/j.bbcan.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Sealfon R.S.G., Hibbs M.A., Huttenhower C., Myers C.L., Troyanskaya O.G., Hibbs M.A., Huttenhower C., Myers C.L., Troyanskaya O.G., Huttenhower C., Myers C.L., Troyanskaya O.G., Myers C.L., Troyanskaya O.G., Troyanskaya O.G. GOLEM: An interactive graph-based gene-ontology navigation and analysis tool. BMC Bioinformatics. 2006;7:443. doi: 10.1186/1471-2105-7-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E., Sharan R., Sharan R. A discriminative model for identifying spatial cis-regulatory modules. J. Comput. Biol. 2005;12:822–834. doi: 10.1089/cmb.2005.12.822. [DOI] [PubMed] [Google Scholar]
- Seidman J.G., Seidman C., Seidman C. Transcription factor haploinsufficiency: When half a loaf is not enough. J. Clin. Invest. 2002;109:451–455. doi: 10.1172/JCI15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slagsvold T., Pattni K., Malerod L., Stenmark H., Pattni K., Malerod L., Stenmark H., Malerod L., Stenmark H., Stenmark H. Endosomal and non-endosomal functions of ESCRT proteins. Trends Cell Biol. 2006;16:317–326. doi: 10.1016/j.tcb.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Spring K., Ahangari F., Scott S.P., Waring P., Purdie D.M., Chen P.C., Hourigan K., Ramsay J., McKinnon P.J., Swift M., Ahangari F., Scott S.P., Waring P., Purdie D.M., Chen P.C., Hourigan K., Ramsay J., McKinnon P.J., Swift M., Scott S.P., Waring P., Purdie D.M., Chen P.C., Hourigan K., Ramsay J., McKinnon P.J., Swift M., Waring P., Purdie D.M., Chen P.C., Hourigan K., Ramsay J., McKinnon P.J., Swift M., Purdie D.M., Chen P.C., Hourigan K., Ramsay J., McKinnon P.J., Swift M., Chen P.C., Hourigan K., Ramsay J., McKinnon P.J., Swift M., Hourigan K., Ramsay J., McKinnon P.J., Swift M., Ramsay J., McKinnon P.J., Swift M., McKinnon P.J., Swift M., Swift M., et al. Mice heterozygous for mutation in Atm, the gene involved in ataxia–telangiectasia, have heightened susceptibility to cancer. Nat. Genet. 2002;32:185–190. doi: 10.1038/ng958. [DOI] [PubMed] [Google Scholar]
- Stearns T., Botstein D., Botstein D. Unlinked noncomplementation: Isolation of new conditional lethal mutations in each of the tubulin genes of Saccharomyces cerevisiae. Genetics. 1988;119:249–260. doi: 10.1093/genetics/119.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szafraniec K., Wloch D.M., Sliwa P., Borts R.H., Korona R., Wloch D.M., Sliwa P., Borts R.H., Korona R., Sliwa P., Borts R.H., Korona R., Borts R.H., Korona R., Korona R. Small fitness effects and weak genetic interactions between deleterious mutations in heterozygous loci of the yeast Saccharomyces cerevisiae. Genet. Res. 2003;82:19–31. doi: 10.1017/s001667230300630x. [DOI] [PubMed] [Google Scholar]
- Tong A.H., Lesage G., Bader G.D., Ding H., Xu H., Xin X., Young J., Berriz G.F., Brost R.L., Chang M., Lesage G., Bader G.D., Ding H., Xu H., Xin X., Young J., Berriz G.F., Brost R.L., Chang M., Bader G.D., Ding H., Xu H., Xin X., Young J., Berriz G.F., Brost R.L., Chang M., Ding H., Xu H., Xin X., Young J., Berriz G.F., Brost R.L., Chang M., Xu H., Xin X., Young J., Berriz G.F., Brost R.L., Chang M., Xin X., Young J., Berriz G.F., Brost R.L., Chang M., Young J., Berriz G.F., Brost R.L., Chang M., Berriz G.F., Brost R.L., Chang M., Brost R.L., Chang M., Chang M., et al. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- Verheyen E.M., Purcell K.J., Fortini M.E., Artavanis-Tsakonas S., Purcell K.J., Fortini M.E., Artavanis-Tsakonas S., Fortini M.E., Artavanis-Tsakonas S., Artavanis-Tsakonas S. Analysis of dominant enhancers and suppressors of activated Notch in Drosophila. Genetics. 1996;144:1127–1141. doi: 10.1093/genetics/144.3.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinh D.B., Welch M.D., Corsi A.K., Wertman K.F., Drubin D.G., Welch M.D., Corsi A.K., Wertman K.F., Drubin D.G., Corsi A.K., Wertman K.F., Drubin D.G., Wertman K.F., Drubin D.G., Drubin D.G. Genetic evidence for functional interactions between actin noncomplementing (Anc) gene products and actin cytoskeletal proteins in Saccharomyces cerevisiae. Genetics. 1993;135:275–286. doi: 10.1093/genetics/135.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther T.C., Brickner J.H., Aguilar P.S., Bernales S., Pantoja C., Walter P., Brickner J.H., Aguilar P.S., Bernales S., Pantoja C., Walter P., Aguilar P.S., Bernales S., Pantoja C., Walter P., Bernales S., Pantoja C., Walter P., Pantoja C., Walter P., Walter P. Eisosomes mark static sites of endocytosis. Nature. 2006;439:998–1003. doi: 10.1038/nature04472. [DOI] [PubMed] [Google Scholar]
- Welch M.D., Vinh D.B., Okamura H.H., Drubin D.G., Vinh D.B., Okamura H.H., Drubin D.G., Okamura H.H., Drubin D.G., Drubin D.G. Screens for extragenic mutations that fail to complement act1 alleles identify genes that are important for actin function in Saccharomyces cerevisiae. Genetics. 1993;135:265–274. doi: 10.1093/genetics/135.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertman K.F., Drubin D.G., Botstein D., Drubin D.G., Botstein D., Botstein D. Systematic mutational analysis of the yeast ACT1 gene. Genetics. 1992;132:337–350. doi: 10.1093/genetics/132.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler E.A., Shoemaker D.D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J.D., Bussey H., Shoemaker D.D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J.D., Bussey H., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J.D., Bussey H., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J.D., Bussey H., Anderson K., Andre B., Bangham R., Benito R., Boeke J.D., Bussey H., Andre B., Bangham R., Benito R., Boeke J.D., Bussey H., Bangham R., Benito R., Boeke J.D., Bussey H., Benito R., Boeke J.D., Bussey H., Boeke J.D., Bussey H., Bussey H., et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- Wool I.G. Extraribosomal functions of ribosomal proteins. Trends Biochem. Sci. 1996;21:164–165. [PubMed] [Google Scholar]
- Yu B., French J.A., Carrier L., Jeremy R.W., McTaggart D.R., Nicholson M.R., Hambly B., Semsarian C., Richmond D.R., Schwartz K., French J.A., Carrier L., Jeremy R.W., McTaggart D.R., Nicholson M.R., Hambly B., Semsarian C., Richmond D.R., Schwartz K., Carrier L., Jeremy R.W., McTaggart D.R., Nicholson M.R., Hambly B., Semsarian C., Richmond D.R., Schwartz K., Jeremy R.W., McTaggart D.R., Nicholson M.R., Hambly B., Semsarian C., Richmond D.R., Schwartz K., McTaggart D.R., Nicholson M.R., Hambly B., Semsarian C., Richmond D.R., Schwartz K., Nicholson M.R., Hambly B., Semsarian C., Richmond D.R., Schwartz K., Hambly B., Semsarian C., Richmond D.R., Schwartz K., Semsarian C., Richmond D.R., Schwartz K., Richmond D.R., Schwartz K., Schwartz K., et al. Molecular pathology of familial hypertrophic cardiomyopathy caused by mutations in the cardiac myosin binding protein C gene. J. Med. Genet. 1998;35:205–210. doi: 10.1136/jmg.35.3.205. [DOI] [PMC free article] [PubMed] [Google Scholar]