

Abstract
Enteropathogenic E. coli (EPEC) is a major cause of infantile diarrhea, but the pathophysiology underlying associated diarrhea is poorly understood. We examined the role of the luminal membrane Cl–/OH– exchange process in EPEC pathogenesis using in vitro and in vivo models. Cl–/OH– exchange activity was measured as OH– gradient–driven 36Cl– uptake. EPEC infection (60 minutes–3 hours) inhibited apical Cl–/OH– exchange activity in human intestinal Caco-2 and T84 cells. This effect was dependent upon the bacterial type III secretory system (TTSS) and involved secreted effector molecules EspG and EspG2, known to disrupt the host microtubular network. The microtubule-disrupting agent colchicine (100 μM, 3 hours) also inhibited 36Cl– uptake. The plasma membrane expression of major apical anion exchanger DRA (SLC26A3) was considerably reduced in EPEC-infected cells, corresponding with decreased Cl–/OH– exchange activity. Confocal microscopic studies showed that EPEC infection caused a marked redistribution of DRA from the apical membrane to intracellular compartments. Interestingly, infection of cells with an EPEC mutant deficient in espG significantly attenuated the decrease in surface expression of DRA protein as compared with treatment with wild-type EPEC. EPEC infection in vivo (1 day) also caused marked redistribution of surface DRA protein in the mouse colon. Our data demonstrate that EspG and EspG2 play an important role in contributing to EPEC infection–associated inhibition of luminal membrane chloride transport via modulation of surface DRA expression.
Introduction
Electroneutral NaCl absorption in the human ileum and colon involves coupling of luminal Na+/H+ exchange (NHE) and Cl–/OH–(HCO3–) exchange activities (1, 2). Disturbances in these electrolyte transport systems have been implicated in the pathophysiology of diarrhea associated with inflammatory diseases or infection by enteric pathogens (1, 2). Enteropathogenic E. coli (EPEC) is a food-borne pathogen and a major cause of watery infantile diarrhea worldwide that is associated with high rates of morbidity and mortality. A unique feature of EPEC is that it neither expresses toxins nor is invasive but instead attaches intimately to host intestinal epithelial cells (3, 4). The pathogenicity island of EPEC houses a number of virulence proteins including those of a type III secretion system (TTSS) through which bacteria directly deliver their proteins into the host cell (5, 6). While the genes conferring pathogenicity to EPEC have been cloned and fully sequenced, the mechanisms by which infection by this pathogen cause diarrhea in the infected host remain unclear.
Previous studies show that the mechanism(s) of EPEC-induced diarrhea are multifactorial. For instance, we have reported previously that EPEC infection induces inflammation and disrupts the structure and barrier function of tight junctions (6, 7). In addition, EPEC induces IL-8 release to stimulate neutrophil transmigration (8) and the release of 5′-AMP, which is converted to the secretagogue adenosine (9). The pathogen-induced activation of NF-κB also upregulates expression of the galanin-1 receptor, resulting in increased Cl– secretion after activation by its ligand (10). However, to date, the mechanism(s) of EPEC-induced diarrhea, particularly during early infection, are not fully defined.
Diarrhea generally results from either increased secretion or impairment in NaCl absorption or both. Earlier studies have addressed the potential secretory effects of early EPEC infection. For example, studies by Collington et al. (11) indicated that EPEC infection resulted in a small increase in Cl–-dependent short-circuit current (Isc) in Caco-2 cells. In contrast, studies by our laboratory utilizing T84 cells showed that EPEC infection failed to stimulate chloride secretion (10). In fact, EPEC infection diminished the effects of secretagogues on these cells (10). Therefore, we focused on examining the effects of EPEC on the host cell NaCl-absorptive activities. We previously showed that EPEC infection of host intestinal epithelial cells stimulated the activity of NHE2, whereas the activity of NHE3, the predominant NHE isoform involved in Na+ absorption, was inhibited (12). Thus, it was also critical to examine the effects of EPEC on intestinal apical membrane Cl–/OH– exchange activity to better understand the pathogenesis of associated diarrhea. The role of apical anion exchangers SLC26A3 (down regulated in adenoma [DRA]) and SLC26A6 (putative anion transporter 1 [PAT-1]) (13–17) in mediating luminal Cl–/HCO3–(OH–) exchange in the human intestine was also examined in response to EPEC infection using in vitro and in vivo models of EPEC infection.
Our studies revealed the mechanistic basis for microbial-epithelial interactions that might underlie pathophysiology of EPEC-induced diarrhea. We demonstrated that EPEC infection of intestinal epithelial cells inhibited Cl–/OH– exchange activity via the involvement of the effector molecules EspG and EspG2. Our results also reveal that optimal Cl–/OH– exchange activity requires an intact tubulin network. In addition, EPEC infection in in vitro and in vivo models was shown to modulate DRA protein expression on the apical plasma membrane. Alterations in the level of DRA expression at the apical membrane in response to EPEC infection represent what we believe to be a novel and distinct mechanism for its acute regulation.
Results
EPEC infection inhibits apical Cl–/OH– exchange activity in human intestinal epithelial cells.
To determine the effects of EPEC infection on Cl–/OH– exchange activity, Caco-2 monolayers were infected with EPEC for 3 hours and 4,4′-diisothiocyanateo-stilbene-2,2′-disulfonic acid–sensitive (DIDS-sensitive) 36Cl– uptake was then assessed after cells were base loaded. Figure 1A shows that Cl–/OH– exchange activity was significantly decreased (by approximately 70%) in response to EPEC infection. Nonpathogenic E. coli, however, caused no significant alteration in 36Cl– uptake. A time course of EPEC infection was assessed by infecting Caco-2 cells with EPEC for different periods ranging from 60 to 120 minutes. The inhibition of 36Cl– uptake occurred as early as 60 minutes after infection and persisted for up to 90 and 120 minutes (Figure 1B). All subsequent experiments were therefore performed at 60 minutes after infection. In order to determine whether the effects of EPEC on Cl–/OH– exchange activity are cell line specific, experiments were also performed in another human intestinal cell line, T84, representing colonic crypts. Similar to the effects in Caco-2 cells, EPEC infection of T84 cells also resulted in significant inhibition of apical Cl–/OH– exchange activity as early as 60 minutes after infection (Figure 1C).
Figure 1. EPEC inhibits Cl–/OH– exchange activity.

(A) Caco-2 cells were infected with EPEC or nonpathogenic E. coli in the cell culture medium for 3 hours. Cl–/OH– exchange activity was measured in base-loaded cells as DIDS–sensitive (300 μM) 36Cl– uptake. Results represent mean ± SEM of 9 separate experiments performed in triplicate. *P < 0.05 compared with control. (B) Time course of Cl–/OH– exchange activity inhibition by EPEC in Caco-2 cells. Results represent mean ± SEM of 6 separate experiments performed in triplicate. *P < 0.05 compared with control. (C) Effects of EPEC are not cell line specific. T84 cells were infected with EPEC for 60, 90, or 120 minutes, and Cl–/OH– exchange activity was measured as DIDS-sensitive (300 μM) 36Cl– uptake. Results represent mean ± SEM of 3 separate experiments performed in triplicate. *P < 0.05 compared with control.
Inhibition of Cl–/OH– exchange by EPEC is TTSS dependent.
The role of EPEC virulence proteins on Cl–/OH– exchange activity was next investigated. First, the involvement of the TTSS required to inject effector molecules into host cells was assessed. The EPEC protein EscN is the putative ATPase that drives type III secretion. As shown in Figure 2A, there was no inhibition of apical Cl–/OH– exchange activity in Caco-2 cells infected with the escN mutant strain, indicating that the effects of EPEC on apical Cl–/OH– exchange activity are TTSS dependent. In order to further analyze the role of the TTSS in EPEC-modulated Cl–/OH– exchange activity, cells were infected with either espA, espB, or espD mutant strains, which are incapable of forming translocation channels and pores. The results, as shown in Figure 2B, demonstrated that infection of cells with either the espA, espB, or espD mutant strain failed to inhibit Cl–/OH– exchange activity. These results suggest the involvement of either the structural components of the translocation pore itself or the secreted effector molecules that are delivered by the translocation pore into the host cytosol.
Figure 2. Effects of EPEC are TTSS dependent.

(A) Caco-2 cells were infected with EPEC or escN mutant (mutated for ATPase driving TTSS; ΔescN) for 60 minutes. Cl–/OH– exchange activity was measured in base-loaded cells as DIDS-sensitive (300 μM) 36Cl– uptake. Results represent mean ± SEM of 4 separate experiments performed in triplicate. *P < 0.05 compared with control. (B) Role of type III secretion mutants. Cl–/OH– exchange activity was measured in Caco-2 cells subsequent to infection with EPEC or different TTSS mutants that are incapable of forming translocation pores. The effects of EPEC on Cl–/OH– exchange activity were dependent on TTSS. Results represent mean ± SEM of 4 separate experiments performed in triplicate. *P < 0.05 compared with control.
Role of EPEC effector proteins.
Several EPEC effector proteins, including EspF, EspG, EspH, and Map, are delivered into host cytosol following infection. The role of these effector molecules in EPEC-induced Cl–/OH– exchange inhibition was further investigated by infecting Caco-2 cells with the mutant strains deficient in the expression of espF, espG, espH, or map for 60 minutes. Figure 3A shows that mutation of espF, espH, and map inhibited the apical Cl–/OH– exchange activity to the same degree as wild-type EPEC. In contrast, the espG mutant affected 36Cl– uptake significantly less than wild-type EPEC. Interestingly, complementation of the espG mutant with espG restored the inhibition in apical Cl–/OH– exchange activity to levels similar to those resulting from infection with wild-type EPEC (Figure 3B). These results suggest that EspG contributes to the observed decrease in apical Cl–/OH– exchange activity by EPEC. An espG paralog, encoded by espG2, has been recently identified (18). Therefore, in order to determine whether the presence of espG2 in EPEC dampened the effects of espG deletion on Cl–/OH– exchange activity by EPEC, we also examined the effects of espG2 mutant and the espG/espG2 double mutant on apical Cl–/OH– exchange activity. Similar to that of the espG mutant, the inhibitory effect of the espG2 mutant on 36Cl– uptake was significantly attenuated as compared with wild-type EPEC (Figure 3B). As shown in Figure 3B, deletion of both espG and espG2 ablated the inhibition in Cl–/OH– exchange activity. This activity was partially restored when the double mutant was complemented with espG (Figure 3B).
Figure 3. The role of EPEC effector proteins.

(A) Involvement of espF, espG, espH, or map in the effects of EPEC. The role of secreted effector molecules was investigated by infecting Caco-2 cells with EPEC or espF, espG, espH, and map mutants for 60 minutes. Cl–/OH– exchange activity was measured in base-loaded cells as DIDS-sensitive (300 μM) 36Cl– uptake. espF, espH, or map mutants inhibited Cl–/OH– exchange activity to the same extent as wild-type EPEC. The EspG mutant caused a relatively attenuated response as compared with wild-type EPEC. Results represent mean ± SEM of 3 separate experiments performed in triplicate. *P < 0.05 compared with control; **P < 0.05 compared with wild-type EPEC. (B) Role of espG and espG2. Caco-2 monolayers were infected with either the espG mutant, espG paralog espG2 mutant, or double mutant containing deletion of espG and espG2 for 60 minutes. Cl–/OH– exchange activity was measured in base-loaded cells as DIDS-sensitive (300 μM) 36Cl– uptake. espG and espG2 mutants significantly attenuated the inhibition in Cl– uptake compared with wild type EPEC. The double mutant completely abolished the inhibitory response. Complementation of espG in the espG mutant or in the double mutant restored the inhibition in Cl–/OH– exchange activity. Results represent mean ± SEM of 3–6 separate experiments performed in triplicate. *P < 0.05 compared with control; **P < 0.05 compared with wild-type EPEC.
Effect of colchicine on Cl–/OH– exchange activity.
Earlier studies regarding the role of E. coli–secreted proteins on the host cytoskeletal structure demonstrated that EspG and EspG2 disrupt the microtubular network of the host intestinal epithelial cells (19, 20). Therefore, it was of interest to examine whether the disruption of microtubules was involved in the loss of apical Cl–/OH– exchange activity. For this, specific inhibitors of tubulin polymerization, colchicine and nocadozole, were used. Incubation of Caco-2 cells with colchicine (100 μM) for 3 hours significantly decreased Cl–/OH– exchange activity as compared with treatment of cells with vehicle alone (Table 1). There was no effect of colchicine treatment at a concentration of 50 μM. Time-course studies of the effect of colchicine (100 μM) on apical Cl–/OH– exchange activity showed that maximum inhibition occurred at 3 hours, with no significant changes at 1 hour and 2 hours of incubation (values represent 36Cl uptake expressed as nmol/mg protein/5 min: control, 2.49 ± 0.25; colchicine [1 hour], 2.53 ± 0.3; colchicine [2 hours], 2.22 ± 0.29; colchicine [3 hours], 1.34 ± 0.12). This is consistent with previous observations (21) demonstrating the breakdown of the microtubular network in epithelial cells, apparent at the concentration and time point used in the present study. Similar results were also observed with another microtubule-disrupting agent, nocodazole (33 μM for 3 hours; data not shown). These results suggest that optimal Cl–/OH– exchange activity requires an intact tubulin network. In order to determine whether EPEC and colchicine mediate their effects on Cl–/OH– exchange activity via similar mechanism(s), Caco-2 cells were treated with colchicine (100 μM for 2 hours) and then infected with EPEC in the presence of colchicine for another 1 hour. As shown in Table 1, colchicine treatment and EPEC infection alone inhibited the apical Cl–/OH– exchange activity by approximately 50% and 80%, respectively; however, the presence of both colchicine and EPEC did not exert any synergistic effects on this inhibition (80%). These results suggest that both colchicine and EPEC inhibit Cl–/OH– exchange activity via disruption of the microtubule network.
Table 1 .
Disruption of microtubule network inhibits apical Cl–/OH– exchange activity

Is Cl–/OH– exchange activity inhibition secondary to NHE3 inhibition?
Since EPEC inhibits NHE3 activity in Caco-2 cells, it could be argued that the observed effects of EPEC on Cl–/OH– exchange activity are secondary to inhibition of NHE3. To examine this possibility, Caco-2 cells were pretreated with the NHE inhibitor ethyl-isopropyl amiloride (EIPA) at a concentration of 50 μM and then infected with EPEC in the presence of either EIPA or DMSO (vehicle). As shown in Table 2, blocking of NHE by EIPA modestly decreased the apical Cl–/OH– exchange activity in control monolayers; however, the magnitude of EPEC-mediated inhibition of 36Cl uptake (70%) was not significantly altered in the absence or presence of EIPA. These results indicate that effects of EPEC on apical Cl–/OH– exchange activity were not completely secondary to inhibition of NHE3 activity.
Table 2 .
EPEC effects on Cl–/OH– exchange activity were not secondary to alteration in NHE activity

EPEC reduces surface DRA expression.
Two members of the SLC26 gene family, namely DRA, which is impaired in congenital chloride diarrhea, and PAT-1, have been implicated as candidate genes for apical Cl–/OH–(HCO3–) exchange in the human intestine (13). Therefore, to examine the effect of EPEC infection on DRA and PAT-1 protein expression, cell-surface biotinylation studies were performed to determine the changes in cell-surface antigen, defined as the biotin-accessible fraction of total cellular antigen. Biotinylated proteins from control and infected cells were separated from the cell lysate by avidin, and proteins were probed with affinity-purified human anti-DRA or ant–PAT-1 antibodies.
The surface expression of DRA in infected cells was considerably decreased, paralleling the loss in Cl–/OH–(HCO3–) exchange activity, while total cellular DRA (sum of apical and intracellular DRA) remained unchanged (Figure 4B). Densitometric analysis suggested that EPEC reduced surface DRA expression by approximately 70% compared with uninfected or nonpathogenic E. coli (Figure 4C). These data indicate that reduced surface expression of DRA contributes to early EPEC-induced diarrhea. Interestingly, deletion of espG in EPEC resulted in an approximately 40% decrease in surface expression of DRA as compared with wild-type EPEC (Figure 4, B and C), whereas infection of Caco-2 cells with espG/espG2 double mutant caused surface DRA expression comparable to that in uninfected or nonpathogenic E. coli–infected cells (data not shown). In order to ensure that EPEC infection would not alter the efficiency of biotinylation, the surface proteins were also probed with avidin-peroxidase antibody. Results, as shown in Figure 4A, suggest that the efficiency of biotinylation was similar in control and EPEC-infected cells. In contrast, the expression of PAT-1 on the surface was not significantly altered in response to EPEC infection, as shown in Figure 5B. Densitometric analysis (Figure 5C) showed no quantitative difference in the surface expression of PAT-1 between control and EPEC-infected cells. Also, the efficiency of biotinylation remained similar in uninfected cells and cells infected with wild-type or nonpathogenic E. coli (Figure 5A).
Figure 4. Cell-surface expression of DRA.

Caco-2 monolayers grown on plastic supports were infected with nonpathogenic E. coli, EPEC, or espG mutant for 60 minutes and subjected to biotinylation at 4°C using sulfo-NHS-biotin. After solubilization, biotinylated proteins were extracted with streptavidin-agarose from equal amounts of total cellular protein. Surface and intracellular fractions were run on 10% SDS–polyacrylamide electrophoresis gels, followed by transfer to nitrocellulose membrane. The blot was immunostained with an avidin-peroxidase antibody (A) or rabbit anti-DRA (B). Representative blot of 5 different experiments are shown. (C) Scanning densitometry of the DRA protein band was performed and the results expressed as surface/total DRA (surface plus intracellular DRA). The intensities of EPEC, nonpathogenic E. coli, and espG mutant were calculated in relation to those of the uninfected cells, and the value of each time control was arbitrarily set to 100. Values represent mean ± SEM of 5 different blots. *P < 0.01 versus control; **P < 0.05 compared with EPEC.
Figure 5. Cell-surface expression of PAT-1.

Caco-2 monolayers grown on plastic supports were infected with nonpathogenic E. coli or EPEC for 60 minutes and subjected to biotinylation at 4°C using sulfo-NHS-biotin. Surface and intracellular fractions were run on 10% SDS-polyacrylamide electrophoresis gels, followed by transfer to nitrocellulose membrane. The blot was immunostained with an avidin-peroxidase antibody (A) or rabbit anti–PAT-1 (B). Representative blots of 3 different experiments are shown. (C) Scanning densitometry of the PAT-1 protein band was performed and expressed as surface/total PAT-1 (surface plus intracellular PAT-1). The intensities of EPEC and nonpathogenic E. coli were calculated in relation to those of the uninfected cells, and the value of each time control was arbitrarily set to 100. Values represent mean ± SEM of 3 different blots.
EPEC causes redistribution of DRA.
Immunofluorescence studies were performed to confirm the regulation of DRA by internalization of the DRA protein from surface to intracellular compartment in response to EPEC infection. DRA or PAT-1 expression was determined in control and EPEC-infected cells after immunostaining with antibodies against DRA. The primary antibody was washed, and then cells were incubated with goat anti-rabbit Alexa Fluor conjugate 568 (red). To visualize the outline of the monolayers, actin was labeled with Alexa Fluor 488–conjugated phalloidin (green). As shown in Figure 6A, DRA protein was predominantly expressed on the apical membrane in Caco-2 cells, as judged by both the vertical xy image and the horizontal xz image. Similar studies were performed in monolayers infected with EPEC or the double mutant espG/espG2. As shown in the in xz images in Figure 6A, EPEC infection caused dramatic reorganization of the actin cytoskeleton and caused redistribution of DRA from apical to intracellular compartments. However, in Caco-2 monolayers infected with the double espG/espG2 mutant, DRA staining was predominantly restricted to the apical surface, similar to what occurred in the uninfected monolayers, paralleling the results of the studies on Cl–/OH–(HCO3–) exchange activity (Figure 6A).
Figure 6. EPEC infection causes a reduction in the expression of DRA on the plasma membrane.

(A) Confocal microscopic localization of DRA (red) and Alexa Fluor 488–conjugated phalloidin (actin; green) showing vertical (xy; near the apical plasma membrane) and horizontal (xz) sections. Hoechst dye labeled the nuclei (blue). At the basal level, DRA protein (red) was primarily localized in the apical membrane, with no staining in the basolateral membranes in control monolayers. EPEC-infected monolayers showed absence of DRA from the apical membrane, with marked redistribution to intracellular compartments, whereas double mutant espG/espG2 showed predominant localization of DRA on the apical membrane. Nuclear staining with Hoechst dye also shows the bacterial attachment sites as indicated by the diffused pattern of nuclei (blue) in EPEC- and double mutant–infected monolayers compared with the uniform staining pattern of the nuclei in uninfected monolayers. Scale bars: 10 μm (control and EPEC), 5 μm (ΔespG/espG2). (B) Caco-2 monolayers infected with nonpathogenic E. coli (A) or EPEC (B) were stained with DRA and phalloidin, and images were converted on 3D projections generated using AutoVisualize. In monolayers infected with nonpathogenic E. coli, DRA can be seen at the apical border associated with brush border F-actin and apical plasma membrane (arrow). As described in previous studies, EPEC infection causes a dramatic reorganization of F-actin such that it is concentrated beneath bacterial attachment sites. While some apical plasma membrane DRA could still be detected (arrow), the majority of the DRA was found in the basal cytoplasm (seen as purple due to overlap with blue nuclear stain).
To compare the cellular distribution of DRA in control and EPEC-infected cells, 3D images were created (Figure 6B). These confocal images demonstrated that DRA (red) is predominantly expressed on the surface of cells infected with nonpathogenic E. coli (Figure 6B, upper panel); however, infection with EPEC resulted in marked internalization of the DRA to intracellular compartments (Figure 6B, lower panel). It should be noted that PAT-1 antibody was not suitable for immunofluorescence studies.
In vivo effects of EPEC on Cl–/OH–(HCO3–) exchanger.
The in vitro studies are advantageous in allowing for a direct investigation of EPEC-modulated intestinal electrolyte transport processes. However, these studies are limited in their ability to provide a comprehensive model for assessing the net impact of EPEC infection on the native intestinal tissue. Therefore, we also examined effects of EPEC on DRA expression in the intestine utilizing our recently characterized in vivo murine model of EPEC infection (22, 23). For these studies, mice were infected with EPEC for 1 day, and DRA expression was examined in mouse ileum and colonic tissues using immunofluorescence microscopy. The expression of DRA was more abundant in the colon than the ileum in the control mice (data not shown). As shown in Figure 7, DRA (red) was predominantly expressed on the surface of cells and colocalized with actin (green) near the apical membrane in the control colon. However, following infection with EPEC, DRA was primarily located in subapical pools reflected as numerous punctate inclusions throughout the cytoplasm. These findings correlate with the results obtained in in vitro cultured intestinal epithelial cell models.
Figure 7. In vivo effects of EPEC on DRA localization.

Immunofluorescent localization of DRA (red) and phalloidin (actin; green) in the colonic epithelium of control mice and mice infected with EPEC for 1 day. Control colon shows localization of DRA protein predominantly to apical and subapical compartments. However, EPEC infection led to marked redistribution to intracellular sites, with more diffused localization in the basal and perinuclear cytoplasm. Scale bars: 5 μm. A representative of 3 different experiments is shown.
Discussion
Cl–/OH–(HCO3–) exchangers or anion exchangers (AEs) are responsible for the electroneutral exchange of Cl– for HCO3– across the plasma membranes of polarized epithelial cells and thus play an important role in the vectorial transport of Cl–, regulation of intracellular pH, and maintenance of intracellular Cl– concentration and cell volume (2). The coupled operation of Na+/H+ and Cl–/HCO3– exchangers represents a major route of electroneutral NaCl absorption in the mammalian intestine. Disturbances in these electrolyte ion transport mechanism(s) generally accompany diarrhea associated with inflammatory bowel diseases. We have previously demonstrated the modulation of apical Cl–/OH– exchange activity by the inflammatory mediators nitric oxide and serotonin, as well as phorbol esters in Caco-2 cells (24–26). However, the pathophysiology of early diarrhea associated with EPEC, an important food-borne pathogen, has not yet been fully defined. In this regard, our previous studies utilizing human intestinal epithelial cells showed that EPEC infection failed to stimulate Cl– secretion (10) but significantly inhibited the activities of intestinal NHE3 and monocarboxylate 1 transporter, the predominant Na+-absorbing isoform and short-chain fatty acid transporter, respectively (12, 27). The current studies were aimed at examining in detail the role of the luminal Cl–/OH–(HCO3–) exchange process in the pathophysiology of EPEC-induced diarrhea.
We showed that EPEC infection of confluent differentiated Caco-2 monolayers caused a significant decrease in apical Cl–/OH– exchange activity. These effects were not cell line specific, as EPEC infection in T84 cells showed similar results. T84 cells represent secretory crypt cells derived from the colon and have been used previously to study the regulation of Cl–/HCO3– exchanger (28). Consistent with these previous studies, our current studies utilizing T84 cells showed modest expression of apical Cl–/OH– exchange activity (approximately 2-fold less than in Caco-2 cells).
EPEC is known to deliver its virulence factors directly into the host cells through the TTSS. Several mutational studies performed previously have specifically identified the individual components of the TTSS (29, 30). Previous studies have demonstrated that EspA forms a filamentous organelle on the bacterial surface that acts as a channel to deliver secreted proteins into the host cells. EspB and EspD are thought to form the translocation pore, with both of these being translocated into the host cell membrane, although EspB is also found in the cytoplasm (31). The effects of EPEC on apical Cl–/OH– exchange activity were found to be dependent upon an intact TTSS, as mutation of the putative ATPase (escN) ablated the alteration in Cl–/OH– exchange activity. These results are consistent with our previous studies in which escN mutant also failed to influence NHE activity in Caco-2 cells (12). Further, espA, espB, and espD mutants, all of which lack the type III secretion ability, failed to inhibit the apical Cl–/OH– exchange activity in Caco-2 cells.
The secreted effector molecules including EspF, EspG, EspH, and Map produce a wide array of phenotypes. EPEC-secreted effector proteins include EspF, known to disrupt tight junctions (5); EspH, which alters pedestal morphology and filopodia formation (32); EspG and its paralog from the gene espG2, which disrupt host microtubule network (18, 19); Map, which alters mitochondrial membrane potential (30); and the translocated intimin receptor (Tir), which mediates adherence to the bacterial outer membrane protein intimin and pedestal formation (33). The decreased apical Cl–/OH– exchange activity in response to EPEC, however, was not dependent upon the secreted effector molecules EspF, EspH, or Map. EspG, on the other hand, partially mediated the effects of EPEC on apical Cl–/OH– exchange activity. Elliot et al. (18) recently reported the identification of another EspG paralog that is encoded by espG2. The partial attenuation of the effects of EPEC on Cl–/OH– exchange activity by deletion of espG could be due to the presence of espG2 in the espG mutant (18). Deletion of espG2, like deletion of espG partially contributed to the inhibitory effects of EPEC on apical Cl–/OH– exchange activity. In fact, our studies with the espG/espG2 double mutant showing the complete loss of inhibition in Cl–/OH– exchange activity suggest an important role for both EspG and EspG2 in the inhibitory effects of EPEC. It is plausible that the observed effects of EspG mutants are due to polar effects of mutating EspG or EspG2 on the corresponding isoform. Since espG and espG2 are located in different regions of the chromosome, it is unlikely that mutations in one have any effects on expression of the other. Interestingly, the complementation of espG gene restored the inhibition in Cl– uptake to wild-type EPEC levels, indicating that the effects of EspG and EspG2 mutants are specific to the effector molecules. Thus, the restoration of the phenotype by complementation rules out the possibility that defects observed in the mutants are due to polar effects caused by dysregulated expression of genes downstream of espG/espG2. In addition, the failure of nonpathogenic E. coli and type III secretion mutants escN, espA, espB, and espD to decrease Cl–/OH– exchange activity indicates that the effects are specific to secreted effector molecules. Also, the effects of EPEC on Cl–/HCO3–(OH–) exchange activity appear to be specific and not secondary to alterations in other transporters, as EPEC effects were similar in magnitude in the absence or presence of the NHE inhibitor EIPA (50 μM). Additionally, our previous studies showed that EspG was not involved in EPEC-mediated effects on NHE and monocarboxylate 1 transporter activity (12, 27).
EspG and EspG2 have been shown to play an important role in the disruption of host microtubular network (19, 20). Our studies using EPEC mutants EspG and EspG2 strongly suggest that regulation of Cl–/HCO3–(OH–) exchange activity by EPEC might also occur via disruption of microtubules. Additionally, the failure of EPEC and colchicine to act synergistically suggests that EPEC and colchicine act via a similar mechanism, i.e., disruption of microtubules to inhibit apical Cl–/OH– exchange activity in Caco-2 cells. The integrity of microtubules is an essential factor for proper sorting and targeting of proteins to specific membrane domains (34). Previous studies by Fuller et al. (21) showed that Cl– secretion in T84 cells and rat distal colon was also dependent on intact microtubules. Cell-surface biotinylation studies clearly suggested reduced surface expression of DRA protein in response to EPEC infection. This decrease in surface expression of DRA was not observed with either nonpathogenic E. coli or the TTSS mutant escN. Studies with the espG mutant showed that espG might play an important role in redistribution of DRA from apical to intracellular compartments. These biochemical studies were complemented with qualitative immunofluorescence studies that also showed redistribution of DRA protein from surface to intracellular compartments. Further, the role of intact microtubules was confirmed by the finding that optimal Cl–/HCO3–(OH–) exchange activity required an intact microtubular network, as the microtubule-disrupting agents colchicine and nocodazole decreased the Cl–/OH– exchange activity in Caco-2 cells. The expression of PAT-1, however, was unaltered in response to EPEC infection.
The exact contribution of DRA and PAT-1 to luminal chloride absorption in the human small intestine and colon is not fully known. Studies have suggested that in the intestine, DRA is predominantly expressed in the colon (35–37), whereas PAT-1 is predominantly expressed in the small intestine (38). Therefore, although both DRA and PAT-1 can contribute to the electroneutral Cl–/HCO3–(OH–) exchange process in the human intestine, EPEC seems to have differential effect on these 2 apical anion exchangers. Similarly, our previous studies showed that the activities of the apical Na+/H+ exchanger isoforms NHE2 and NHE3 were reciprocally regulated in response to EPEC infection, with an increase in the activity of NHE2 but a decrease in NHE3 activity (12).
The intestinal epithelial cells also express other anion exchangers, e.g., AE2 and AE3, expressed on the basolateral side (1, 2). Therefore, it could be argued that the observed inhibition of apical Cl–/OH– exchange activity in response to EPEC infection could also be contributed by the DIDS-sensitive basolateral anion transporters. In this regard, our previous studies have suggested that infection of human intestinal epithelial cells with EPEC does not cause any transepithelial electrical resistance (TER) changes until 3 hours (5, 39). Thus, in the absence of permeability changes, apical 36Cl– uptake would be primarily facilitated by DRA and PAT-1.
Further, previous studies reported conflicting findings with regard to the stilbene sensitivity of the SLC26 family of anion exchangers, especially for DRA. For example, DRA-mediated sulfate transport in Xenopus oocytes (40) and Sf9 cells (41) has been suggested to be highly sensitive to inhibition by DIDS. Also, the Cl– transport function of DRA was inhibited almost completely by 0.5 mM DIDS (15). On the other hand, some studies suggest extremely low sensitivity of DRA-mediated Cl– transport to DIDS, when expressed in Xenopus oocytes (42) or HEK 293 cells (14). It should be noted that, in our current studies, both the total and the DIDS-sensitive apical Cl– uptake showed a marked decrease in response to EPEC infection. These functional studies, complemented with the biochemical and immunofluorescence studies, suggest that DRA plays an important role in observed alterations in Cl– uptake and thus contributes to the pathophysiology of early EPEC-induced diarrhea.
The results obtained in in vitro cell culture models were validated in an in vivo model using our recently established mouse model of EPEC infection (22). The salient features of this model include: (a) colonization of EPEC to murine intestine; (b) formation of attachment and effacement (A/E) lesions and actin rearrangement; (c) soft stool in proximal colon of infected mice compared with hard pellets in control mice; (d) increased number of lamina propria neutrophils with occasional crypt abscesses. The results of the current studies suggested that EPEC infection for 1 day caused marked redistribution of DRA from apical to subapical compartments in the colon, similar to the results obtained in the in vitro model.
Based on these studies, we propose a model showing the mechanism of EPEC-induced decrease in Cl–/OH– exchange activity in intestinal epithelial cells (Figure 8). We have shown significant inhibition of Cl–/OH– exchange activity in human intestinal epithelial cells infected with EPEC, which is mediated via TTSS and effector molecules EspG and EspG2. EspG and EspG2 might impair the membrane targeting event(s) of DRA via disruption of the microtubular network, thereby decreasing its expression on the apical plasma membrane. We speculate that a decrease in Cl–/OH– exchange and NHE3 activities might impair luminal NaCl absorption and may explain the pathophysiology of early EPEC-induced diarrhea. The findings of our current studies enhance our knowledge about the normal physiology of intestinal NaCl absorption and also about the pathophysiology of early diarrhea associated with infection by food-borne enteric pathogens such as EPEC.
Figure 8. Proposed model of the potential mechanism of EPEC-induced decrease in Cl–/OH– exchange activity in intestinal epithelial cells.
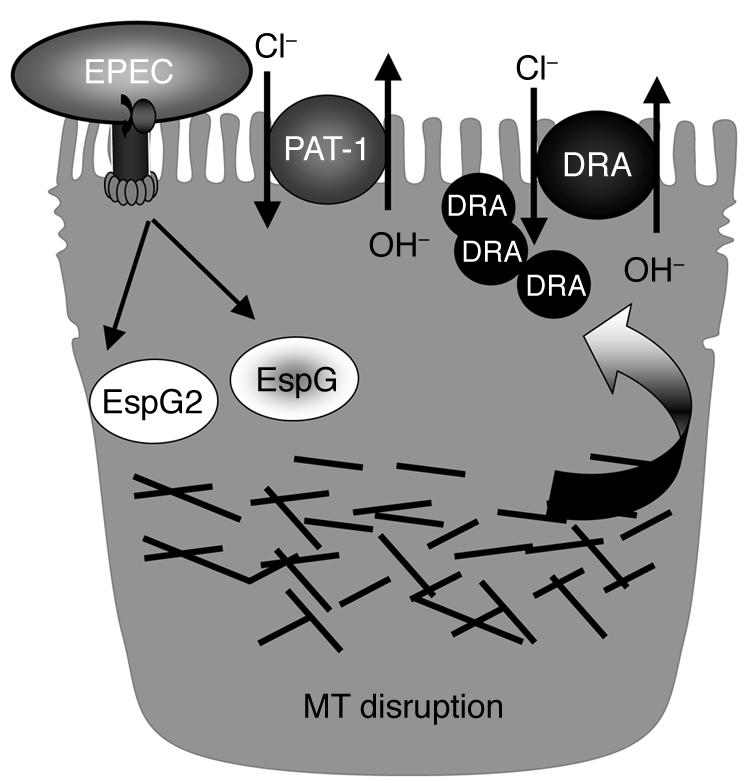
MT, microtubule.
Methods
Materials.
Radionuclide 36Cl was obtained from PerkinElmer. Caco-2 cells were obtained from ATCC. DIDS, 2-(N-Morpholino)ethanesulfonic acid (MES), colchicine, and EIPA were obtained from Sigma-Aldrich. All reagents for SDS-PAGE such as acrylamide, Bis acrylamide, and ammonium persulfate were procured from Fisher Scientific.
Cell culture.
Caco-2 cells were grown in T-75 cm2 plastic flasks at 37°C in a 5% CO2 environment. The culture medium consisted of high-glucose MEM, 20% FBS, 20 mM HEPES, 100 IU/ml penicillin, and 100 μg/ml streptomycin. Cells used for these studies were between passages 25 and 45 and were plated on 24-well plates at a density of 2 × 104 cells/well. Cells were used for experiments at day 10–14 after plating.
T84 cells were obtained from K. Barrett (UCSD, La Jolla, California, USA) and were grown in a 1:1 mixture of DMEM and Ham’s F-12 supplemented with 6% newborn calf serum, 14 mM NaHCO3, 15 mM HEPES, 65 IU/ml penicillin, 8 μg/ml ampicillin, and 60 μg/ml streptomycin in a 5% CO2 atmosphere at 37°C. Cells were plated in 24-well plates at a density of 8 × 104 cells/well and were used for experiments 2 weeks after plating.
Bacterial culture and cell infection.
The following EPEC strains were used: (i) wild-type EPEC strain E2348/69, (ii) CVD452 (E2348/69 escN:Km) (43), (iii) UMD864 (E2348/69 Δ48-759 espB1) (44), (iv) UMD870 (E2348/69 espD1:aph-3(Km)) (45), (v) a nonpathogenic E. coli strain (HB101), (vi) espG (19), (vii) ΔespG/ΔespG2, espG complements (ΔespG/+espG and ΔespG/ΔespG2 +espG) (19), (viii) espF (UMD874) (5), (ix) espH (SE874), and (x) map (SE882). espH and map were kindly provided by J. Kaper (University of Maryland, College Park, Maryland, USA). Strains were grown overnight in the presence of appropriate antibiotics. On the day of experimentation, 30 μl of bacterial culture was transferred to 1 ml of serum- and antibiotic-free T84 cell culture medium supplemented with 0.5% mannose. Bacteria were grown approximately 3 hours to an OD600 of 0.4. Cell monolayers were infected at an MOI of 100. Nonadherent bacteria were removed by washing in PBS after 30–90 minutes.
36Cl– uptake.
Chloride uptake experiments were performed essentially as described previously by us (24). Caco-2 cells were incubated with DMEM base medium containing 20 mM HEPES/KOH pH 8.5, for 30 minutes at room temperature. The medium was removed, and the cells were rapidly washed with 1 ml tracer-free uptake mannitol buffer containing 260 mM mannitol, 20 mM Tris/2-(N-Morpholino)ethanesulfonic acid pH 7.0. The cells were then incubated with the uptake buffer for 5 minutes in the absence or presence of 300 μM DIDS. This time period was chosen as it was within the linear range of Cl– uptake. The uptake buffer contained 1.4 μCi of 36Cl– (2.9 mM) of hydrochloric acid (specific activity: 17.12 mCi/g) in the mannitol buffer. The uptake was terminated by removing the buffer and washing the cells rapidly 2 times with 1 ml of ice-cold PBS, pH 7.2. Finally, the cells were solubilized by incubation with 0.5 N NaOH for 4 hours. The protein concentration was measured by the method of Bradford (46), and the radioactivity was determined by a Packard Tri-Carb 1600TR Liquid Scintillation Analyzer (Packard Instruments; PerkinElmer). The Cl–/OH– exchange activity was assessed as DIDS-sensitive 36Cl– uptake, and values are expressed as nanomoles per milligram protein per 5 minutes.
Cell-surface biotinylation studies.
Cell-surface biotinylation was performed using sulfo-NHS-biotin (0.5 mg/ml; Pierce Biotechnology) in borate buffer (in mM: 154 NaCl, 7.2 KCl, 1.8 CaCl2, 10 H3BO3, pH 9.0), as previously described, with labeling for 60 minutes at 4°C to stop endocytosis and internalization of antigens (47). After immunoprecipitation of biotinylated antigens with streptavidin agarose, biotinylated proteins were released by incubation in 100 μM dithiothreitol, reconstituted in Laemmli buffer, subjected to SDS-PAGE, and then labeled with anti-DRA or anti–PAT-1 purified antibody. The surface DRA or PAT-1 was compared with total cell antigen as determined by immunoblotting in solubilized cell extract and with the amount of DRA or PAT-1 not removed by the avidin precipitation method (intracellular pool). To determine the efficiency of biotinylation, the blots were also probed with avidin-peroxidase antibody (1:1,000 dilution for 1 hour).
Immunofluorescence staining in human intestinal epithelial cells.
Caco-2 cells grown on Transwell inserts were washed twice in 1× PBS containing 1 mM CaCl2, pH 7.4 and then fixed with 2% paraformaldehyde. Cells were then permeabilized with 0.08% saponin, washed, and blocked in 5% normal goat serum (NGS) for 30 minutes–2 hours at 25°C. These monolayers were incubated for 2 hours with rabbit anti-human DRA antibody (1:80) (custom synthesized against peptide sequence). Cells were then stained with anti-goat fluorescein isothiocyanate antibody diluted 1:100 and rhodamine-phalloidin (Invitrogen) diluted 1:60 and Hoechst 33342 (Invitrogen) for 60 minutes. Microscopy was performed using a Zeiss LSM 510 laser scanning confocal microscope equipped with a ×63 water-immersion objective. Beams of 488 nm and 534 nm from an Ag/Kr laser and 361 nm from a UV laser were used for excitation. Green and red fluorescence emissions were detected through LP505 and 585 filters, respectively. The 2 different fluorochromes were scanned sequentially using the multitracking function to avoid any bleed-through among these fluorescent dyes. Series sections were taken at z direction, and orthogonal sections were made in a z stack. For presentation of 3D data, confocal stacks were imported into AutoVisualize 9 (AutoQuant Imaging), and hardware-generated 3D maximum volume projections were created.
Immunofluorescence staining in mouse colon.
All experiments involving mice were approved by the University of Illinois at Chicago Animal Care Committee. Sections of ileal and colonic tissue from control and EPEC-infected mice were snap-frozen in optimal cutting temperature embedding medium (Tissue-Tek O.C.T compound; Sakura) and stored at –80°C. At least 3 sections from each of 2–3 animals for each condition were analyzed by immunofluorescence microscopy. Representative images are shown. For immunostaining, 4-μm frozen sections were fixed with 1% paraformaldehyde in PBS for 10 minutes at room temperature. Fixed sections were washed in PBS, permeabilized with 0.5% NP-40, and blocked with 5% NGS for 30 minutes. Tissues were incubated with anti-DRA antibody (1:100) in PBS with 1% NGS for 90 minutes at room temperature. After washing, sections were incubated with Alexa Fluor 594–conjugated goat anti-rabbit IgG, Alexa Fluor 488–conjugated phalloidin (5 U/ml; Invitrogen), and Hoechst 33342 (Invitrogen) for 60 minutes. Sections were then washed and mounted under coverslips using ProLong Gold antifade reagent (Invitrogen). Sections were imaged using a Leica DM4000 epifluorescence microscope (Leica Microsystems) equipped with appropriate filter cubes (Chroma Technology Corp.) and a Coolsnap HQ camera (Roper Scientific; Photometrics) controlled by MetaMorph 6 (Universal Imaging Corporation; Molecular Devices).
Statistics.
Results are expressed as mean ± SEM. Each independent set represents the mean ± SEM of data from at least 9 wells used on 3 separate occasions. Two-tailed Student’s t test was used for statistical analysis. P < 0.05 was considered statistically significant.
Acknowledgments
These studies were supported by the Department of Veterans Affairs Merit Award and Research Enhancement Awards Program (REAP) grants (to P.K. Dudeja, K. Ramaswamy, and G. Hecht); and NIDDK grants DK68324 and DK54016 (to P.K. Dudeja), DK67990 and DK33349 (to K. Ramaswamy), DK50694 (to G. Hecht), and P01 DK067887 (to P.K. Dudeja, G. Hecht, K. Ramaswamy, and J.R. Turner).
Footnotes
Nonstandard abbreviations used: DIDS, 4,4′-diisothiocyanateo-stilbene-2,2′-disulfonic acid; DRA, downregulated in adenoma; EIPA, ethyl-isopropyl amiloride; EPEC, enteropathogenic E. coli; NHE, Na+/H+ exchange; TTSS, type III secretory system.
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J. Clin. Invest. 117:428–437 (2007). doi:10.1172/JCI29625
Ravinder K. Gill, Gail Hecht, and Pradeep K. Dudeja are co-senior authors.
References
- 1. Dudeja, P.K., Gill, R.K., and Ramaswamy, K. 2003. Absorption-secretion and epithelial cell function. In Colonic diseases. T.R. Koch, editor. Humana Press. Totowa, New Jersey, USA. 3–24. [Google Scholar]
- 2. Gill, R.K., Alrefai, W.A., Ramaswamy, A., and Dudeja, P.K. 2003. Mechanisms and regulation of NaCl absorption in the human intestine. In Recent research developments in physiology. Volume 1. Research Signpost. Trivandrum, India. 643–677. [Google Scholar]
- 3.Gruenheid S., Finlay B.B. Microbial pathogenesis and cytoskeletal function. Nature. 2003;422:775–781. doi: 10.1038/nature01603. [DOI] [PubMed] [Google Scholar]
- 4.Celli J., Deng W., Finlay B.B. Enteropathogenic Escherichia coli (EPEC) attachment to epithelial cells: exploiting the host cell cytoskeleton from the outside. Cell. Microbiol. 2000;2:1–9. doi: 10.1046/j.1462-5822.2000.00033.x. [DOI] [PubMed] [Google Scholar]
- 5.McNamara B.P., et al. Translocated EspF protein from enteropathogenic Escherichia coli disrupts host intestinal barrier function. J. Clin. Invest. 2001;107:621–629. doi: 10.1172/JCI11138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hecht G. Microbes and microbial toxins: paradigms for microbial-mucosal interactions. VII. Enteropathogenic Escherichia coli: physiological alterations from an extracellular position. Am. J. Physiol. Gastrointest. Liver Physiol. 2001;281:G1–G7. doi: 10.1152/ajpgi.2001.281.1.G1. [DOI] [PubMed] [Google Scholar]
- 7.Spitz J., et al. Enteropathogenic Escherichia coli adherence to intestinal epithelial monolayers diminishes barrier function. Am. J. Physiol. 1995;268:G374–G379. doi: 10.1152/ajpgi.1995.268.2.G374. [DOI] [PubMed] [Google Scholar]
- 8.Savkovic S.D., Koutsouris A., Hecht G. Attachment of a noninvasive enteric pathogen, enteropathogenic Escherichia coli, to cultured human intestinal epithelial monolayers induces transmigration of neutrophils. . Infect. Immun. 1996;64:4480–4487. doi: 10.1128/iai.64.11.4480-4487.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Madara J.L., et al. 5′-Adenosine monophosphate is the neutrophil-derived paracrine factor that elicits chloride secretion from T84 intestinal epithelial cell monolayers. J. Clin. Invest. 1993;91:2320–2325. doi: 10.1172/JCI116462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hecht G., Koutsouris A. Enteropathogenic E. coli attenuates secretagogue-induced net intestinal ion transport but not Cl- secretion. Am. J. Physiol. 1999;276:G781–G788. doi: 10.1152/ajpgi.1999.276.3.G781. [DOI] [PubMed] [Google Scholar]
- 11.Collington G.K., Booth I.W., Knutton S. Rapid modulation of electrolyte transport in Caco-2 cell monolayers by enteropathogenic Escherichia coli (EPEC) infection. . Gut. 1998;42:200–207. doi: 10.1136/gut.42.2.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hecht G., et al. Differential regulation of Na+/H+ exchange isoform activities by enteropathogenic E. coli in human intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2004;287:G370–G378. doi: 10.1152/ajpgi.00432.2003. [DOI] [PubMed] [Google Scholar]
- 13.Mount D.B., Romero M.F. The SLC26 gene family of multifunctional anion exchangers. Pflugers Arch. 2004;447:710–721. doi: 10.1007/s00424-003-1090-3. [DOI] [PubMed] [Google Scholar]
- 14.Melvin J.E., Park K., Richardson L., Schultheis P.J., Shull G.E. Mouse down-regulated in adenoma (DRA) is an intestinal Cl(-)/HCO(3)(-) exchanger and is up-regulated in colon of mice lacking the NHE3 Na(+)/H(+) exchanger. J. Biol. Chem. 1999;274:22855–22861. doi: 10.1074/jbc.274.32.22855. [DOI] [PubMed] [Google Scholar]
- 15.Moseley R.H., et al. Downregulated in adenoma gene encodes a chloride transporter defective in congenital chloride diarrhea. Am. J. Physiol. 1999;276:G185–G192. doi: 10.1152/ajpgi.1999.276.1.G185. [DOI] [PubMed] [Google Scholar]
- 16.Lamprecht G., Baisch S., Schoenleber E., Gregor M. Transport properties of the human intestinal anion exchanger DRA (down-regulated in adenoma) in transfected HEK293 cells. Pflugers Arch. 2005;449:479–490. doi: 10.1007/s00424-004-1342-x. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z., Petrovic S., Mann E., Soleimani M. Identification of an apical Cl(-)/HCO3(-) exchanger in the small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2002;282:G573–G579. doi: 10.1152/ajpgi.00338.2001. [DOI] [PubMed] [Google Scholar]
- 18.Elliott S.J., et al. EspG, a novel type III system-secreted protein from enteropathogenic Escherichia coli with similarities to VirA of Shigella flexneri. Infect. Immun. 2001;69:4027–4033. doi: 10.1128/IAI.69.6.4027-4033.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomson F.L., et al. Enteropathogenic Escherichia coli EspG disrupts microtubules and in conjunction with Orf3 enhances perturbation of the tight junction barrier. Mol. Microbiol. 2005;56:447–464. doi: 10.1111/j.1365-2958.2005.04571.x. [DOI] [PubMed] [Google Scholar]
- 20.Matsuzawa T., Kuwae A., Yoshida S., Sasakawa C., Abe A. Enteropathogenic Escherichia coli activates the RhoA signaling pathway via the stimulation of GEF-H1. Embo J. 2004;23:3570–3582. doi: 10.1038/sj.emboj.7600359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fuller C.M., Bridges R.J., Benos D.J. Forskolin- but not ionomycin-evoked Cl- secretion in colonic epithelia depends on intact microtubules. Am. J. Physiol. 1994;266:C661–C668. doi: 10.1152/ajpcell.1994.266.3.C661. [DOI] [PubMed] [Google Scholar]
- 22.Savkovic S.D., Villanueva J., Turner J.R., Matkowskyj K.A., Hecht G. Mouse model of enteropathogenic Escherichia coli infection. Infect. Immun. 2005;73:1161–1170. doi: 10.1128/IAI.73.2.1161-1170.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shifflett D.E., Clayburgh D.R., Koutsouris A., Turner J.R., Hecht G.A. Enteropathogenic E. coli disrupts tight junction barrier function and structure in vivo. Lab. Invest. 2005;85:1308–1324. doi: 10.1038/labinvest.3700330. [DOI] [PubMed] [Google Scholar]
- 24.Saksena S., et al. Involvement of 5-HT3/4 receptor subtypes in the serotonin induced inhibition of Cl(-)/OH(-) exchange activity in Caco-2 cells. J. Biol. Chem. 2005;280:11859–11868. doi: 10.1074/jbc.M411553200. [DOI] [PubMed] [Google Scholar]
- 25.Saksena S., et al. Inhibition of apical Cl(-)/OH(-) exchange activity in Caco-2 cells by phorbol esters is mediated by PKCepsilon. Am. J. Physiol. Cell Physiol. 2002;283:C1492–C1500. doi: 10.1152/ajpcell.00473.2001. [DOI] [PubMed] [Google Scholar]
- 26.Saksena S., et al. Modulation of Cl-/OH- exchange activity in Caco-2 cells by nitric oxide. Am. J. Physiol. Gastrointest. Liver Physiol. 2002;283:G626–G633. doi: 10.1152/ajpgi.00395.2001. [DOI] [PubMed] [Google Scholar]
- 27.Borthakur A., et al. Enteropathogenic Escherichia coli inhibits butyrate uptake in Caco-2 cells by altering the apical membrane MCT1 level. Am. J. Physiol. Gastrointest. Liver Physiol. 2006;290:G30–G35. doi: 10.1152/ajpgi.00302.2005. [DOI] [PubMed] [Google Scholar]
- 28.Lee M.G., et al. Cystic fibrosis transmembrane conductance regulator regulates luminal Cl-/HCO3- exchange in mouse submandibular and pancreatic ducts. J. Biol. Chem. 1999;274:14670–14677. doi: 10.1074/jbc.274.21.14670. [DOI] [PubMed] [Google Scholar]
- 29.Goosney D.L., Gruenheid S., Finlay B.B. Gut feelings: enteropathogenic E. coli (EPEC) interactions with the host. Annu. Rev. Cell Dev. Biol. 2000;16:173–189. doi: 10.1146/annurev.cellbio.16.1.173. [DOI] [PubMed] [Google Scholar]
- 30.Kenny B., Jepson M. Targeting of an enteropathogenic Escherichia coli (EPEC) effector protein to host mitochondria. Cell. Microbiol. 2000;2:579–590. doi: 10.1046/j.1462-5822.2000.00082.x. [DOI] [PubMed] [Google Scholar]
- 31.Daniell S.J., et al. The filamentous type III secretion translocon of enteropathogenic Escherichia coli. . Cell. Microbiol. 2001;3:865–871. doi: 10.1046/j.1462-5822.2001.00168.x. [DOI] [PubMed] [Google Scholar]
- 32.Tu X., Nisan I., Yona C., Hanski E., Rosenshine I. EspH, a new cytoskeleton-modulating effector of enterohaemorrhagic and enteropathogenic Escherichia coli. Mol. Microbiol. 2003;47:595–606. doi: 10.1046/j.1365-2958.2003.03329.x. [DOI] [PubMed] [Google Scholar]
- 33.Kenny B., Warawa J. Enteropathogenic Escherichia coli (EPEC) Tir receptor molecule does not undergo full modification when introduced into host cells by EPEC-independent mechanisms. Infect. Immun. 2001;69:1444–1453. doi: 10.1128/IAI.69.3.1444-1453.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Christiansen J.J., et al. N-glycosylation and microtubule integrity are involved in apical targeting of prostate-specific membrane antigen: implications for immunotherapy. Mol. Cancer Ther. 2005;4:704–714. doi: 10.1158/1535-7163.MCT-04-0171. [DOI] [PubMed] [Google Scholar]
- 35.Jacob P., et al. Down-regulated in adenoma mediates apical Cl-/HCO3- exchange in rabbit, rat, and human duodenum. Gastroenterology. 2002;122:709–724. doi: 10.1053/gast.2002.31875. [DOI] [PubMed] [Google Scholar]
- 36.Hoglund P. Positional candidate genes for congenital chloride diarrhea suggested by high-resolution physical mapping in chromosome region 7q31. Genome Res. 1996;6:202–210. doi: 10.1101/gr.6.3.202. [DOI] [PubMed] [Google Scholar]
- 37.Hoglund P., et al. Mutations of the down-regulated in adenoma (DRA) gene cause congenital chloride diarrhoea. Nat. Genet. 1996;14:316–319. doi: 10.1038/ng1196-316. [DOI] [PubMed] [Google Scholar]
- 38.Saksena S., Gill R., Tyagi S., Alrefai W.A., Ramaswamy K., Dudeja P.K. Expression and membrane localization of PAT-1 in human intestine. Gastroenterology. 2004;122:W958. [Google Scholar]
- 39.Viswanathan V.K., et al. Comparative analysis of EspF from enteropathogenic and enterohemorrhagic Escherichia coli in alteration of epithelial barrier function. Infect. Immun. 2004;72:3218–3227. doi: 10.1128/IAI.72.6.3218-3227.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silberg D.G., Wang W., Moseley R.H., Traber P.G. The down-regulated in adenoma (dra) gene encodes an intestine-specific membrane sulfate transport protein. J. Biol. Chem. 1995;270:11897–11902. doi: 10.1074/jbc.270.20.11897. [DOI] [PubMed] [Google Scholar]
- 41.Byeon M.K., Frankel A., Papas T.S., Henderson K.W., Schweinfest C.W. Human DRA functions as a sulfate transporter in Sf9 insect cells. Protein Expr. Purif. 1998;12:67–74. doi: 10.1006/prep.1997.0809. [DOI] [PubMed] [Google Scholar]
- 42.Chernova M.N., et al. Acute regulation of the SLC26A3 congenital chloride diarrhoea anion exchanger (DRA) expressed in Xenopus oocytes. J. Physiol. 2003;549:3–19. doi: 10.1113/jphysiol.2003.039818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jarvis K.G., et al. Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc. Natl. Acad. Sci. U. S. A. 1995;92:7996–8000. doi: 10.1073/pnas.92.17.7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Donnenberg M.S., Yu J., Kaper J.B. A second chromosomal gene necessary for intimate attachment of enteropathogenic Escherichia coli to epithelial cells. . J. Bacteriol. 1993;175:4670–4680. doi: 10.1128/jb.175.15.4670-4680.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lai L.C., Wainwright L.A., Stone K.D., Donnenberg M.S. A third secreted protein that is encoded by the enteropathogenic Escherichia coli pathogenicity island is required for transduction of signals and for attaching and effacing activities in host cells. Infect. Immun. 1997;65:2211–2217. doi: 10.1128/iai.65.6.2211-2217.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 47.Akhter S., Cavet M.E., Tse C.M., Donowitz M. C-terminal domains of Na(+)/H(+) exchanger isoform 3 are involved in the basal and serum-stimulated membrane trafficking of the exchanger. Biochemistry. 2000;39:1990–2000. doi: 10.1021/bi991739s. [DOI] [PubMed] [Google Scholar]