

Abstract
Aims: To determine whether allelic variants of the cystatin C gene CST3 are genetically associated with exudative age related macular degeneration (ARMD). Cystatin C is a cysteine protease inhibitor that regulates the activity of cathepsin S, a protease with central regulatory functions in retinal pigment epithelial cells.
Methods: CST3 of 167 patients with exudative ARMD was genotyped by using polymerase chain reaction of genomic DNA and restriction enzyme digestion with KspI and compared with those of 517 control subjects. Patients and controls were white.
Results: There was a significant difference in genotype counts between patients and controls (χ2 = 7.158, df = 2; Fisher's exact test: p = 0.037). There was no significant difference in allele frequencies between patients and controls and between controls from Germany, Switzerland, Italy, and United States. The significant difference in genotype counts between patients and controls could be explained completely by an excess of the homozygous CST3 genotype B/B in patients with exudative ARMD (6.6%) over controls (2.3%), suggesting an odds ratio for ARMD in association with CST3 B/B of 2.97 (95% CI: 1.28–6.86). The results also suggest a stronger association of B/B with ARMD in males than in females. However, in both males and females there was a similar and significant effect of CST3 B/B on disease free survival assessed by Kaplan-Meier analysis. The mean disease free survival time in pooled males and females with genotypes A/A or A/B was 85 years (SE 1; 95% CI: 83–86) and 76 years (SE 2; 95% CI: 72–79) respectively in B/B homozygotes (log rank p = 0.0006).
Conclusion: Genotyping data, the absence of a significant difference in allele frequencies between patients and controls, and survival analyses suggest an increased susceptibility for ARMD in CST3 B/B homozygotes. Therefore, CST3 B may be a recessive risk allele, significantly contributing to disease risk in up to 6.6% of German ARMD patients. Functional correlates of the allelic CST3 variants A and B remain to be investigated.
Keywords: age related macular degeneration, proteinases, cystatin C, CST3 gene
A ge related macular degeneration (ARMD) is the leading cause of legal blindness in Western Europe and in the United States. Among the elderly, the overall prevalence is estimated between 5.7% and 30% depending on the definition of early ARMD, and its differentiation from features of normal ageing, a distinction that remains poorly understood.1,2 The hallmark of early ARMD is the presence of drusen, yellow deposits beneath the retinal pigment epithelium (RPE), and, on histology, diffuse thickening of Bruch's membrane because of deposition of debris in the inner collagenous layer. Advanced stages of ARMD can be subdivided into geographic atrophy and exudative ARMD. Geographic atrophy is characterised by progressive atrophy of the RPE. In exudative ARMD the key phenomenon is the occurrence of choroidal neovascularisation (CNV). Eyes with CNV have varying degrees of reduced visual acuity, depending on location, size, type, and age of the neovascular lesion. The development of choroidal neovascular membranes can be considered a late complication in the natural course of the disease possibly due to tissue disruption (Bruch's membrane) and decompensation of the underlying longstanding processes of ARMD.
Many pathophysiological aspects, as well as vascular and environmental risk factors, are known to be associated with a progression of the disease, but little is known about the aetiology of ARMD itself as well as about the underlying processes of complications like the occurrence of CNV.
Family, twin, segregation, and case-control studies suggest an involvement of genetic factors in the aetiology of ARMD.3–7 However, the extent of heritability, number of genes involved, and mechanisms underlying phenotypic heterogeneity are unknown. The search for genes related to ARMD faces challenges: the onset is late in life, and there is usually only one generation available for studies. The parents of patients are often dead, and the children are too young to manifest the disease. Generally, the heredity of late onset diseases has been difficult to estimate because of the uncertainties of the diagnosis in previous generations and the inability to diagnose ARMD among the children of an affected individual.8 Even in the absence of the ambiguities in the diagnosis of ARMD in previous generations, the late onset of the condition itself, natural death rates, and small family sizes result in underestimation of genetic forms of ARMD, and in overestimation of rates of sporadic disease. Moreover, the phenotypic variability is considerable, and it is conceivable that the currently used diagnostic entity of ARMD in fact represents a spectrum of underlying conditions with various genetic and environmental factors involved. The search for genetic factors related to ARMD has to address these challenges.
Several results on genetic studies with ARMD have been published. In a recent study, a genetic association of the manganese superoxide dismutase with exudative ARMD was reported.9 Contradictory results exist concerning a possible role of ApoE polymorphisms in ARMD. Some authors have reported a lower frequency of the ɛ4 allele in subgroups of ARMD, while other reports could not confirm this association.10,11 The ɛ2 allele was reportedly more frequent in ARMD patients.12 Further, allelic variations in the ABCR gene were proposed to be associated with advanced atrophic ARMD,13 but again, other authors found no evidence to support this hypothesis.14 Recently, Allikmets et al have reported additional data in support of an association of ABCR with ARMD15,16; however, other studies have given negative results.17,18 Together, these studies illustrate the challenge to identify susceptibility genes in a most likely complex genetic disorder with the influence of unknown extents of environmental factors.
Familial forms of another macular dystrophy point to the role of the extracellular matrix (ECM) in the pathophysiology of the disease. Mutations in the TIMP3 gene, which encodes a metalloprotease inhibitor that is involved in ECM degradation, are linked to Sorsby's fundus dystrophy, a rare hereditary disease with striking similarities to ARMD in clinical phenotype.19 However, TIMP3 is not associated with ARMD.20 Proteases and protease inhibitors are good candidates for pathophysiological factors since extracellular deposits may be related to impaired ECM turnover. One of these protease inhibitors is cystatin C, a ubiquitous secretory cysteine protease inhibitor which is present in various tissues and body fluids.21 Cystatin C is a strong inhibitor of several cathepsins, among them cathepsin S, a lysosomal enzyme present in retinal pigment epithelial cells where it supposedly functions in the processing of rod outer segments through an incompletely understood mechanism. Inhibition of cathepsin S has been shown to lead to accumulation of debris, when RPE cells are challenged with rod outer segments.22
A mutation of the cystatin C gene (CST3) causes the Icelandic form of hereditary cerebral haemorrhage with amyloidosis (HCHWA-I), a dominant disease characterised by brain haemorrhage and death in young adults. In this disease, a leucine to glutamine mutation at position 68 gives rise to a mutant variant of cystatin C which readily forms amyloid deposits in the walls of cortical arteries and causes these fatal manifestations.23
CST3 maps to chromosome 20p11.2. Two KspI polymorphisms are known in the 5' untranslated sequence, a further KspI polymorphism results in an amino acid substitution in the penultimate position of the signal peptide.24 Through a strong linkage disequilibrium between the three polymorphisms only two haplotypes are observed: CST3 A and CST3 B. The CST3 B/B genotype has recently been shown to be associated with late onset Alzheimer's disease.25
Allelic variation of a gene may be related to the biological function of its encoded protein. We therefore hypothesised a potential association of allelic variants of CST3 in patients with exudative ARMD and determined if any of the CST3 genotypes are associated with exudative ARMD.
METHODS
Patients and controls
Patients (n = 167, age range 51–94 years), 114 females and 53 males, with mean ages at presentation of 75.3 (SD 7.6) and 73.6 (7.4) years, respectively, were recruited between February and October 1998 at the University Eye Hospital Hamburg-Eppendorf, Germany. Comprehensive ophthalmological examinations in all patients included visual acuity measurements, fundus examination, and fluorescein angiography. All patients were diagnosed with unilateral or bilateral neovascular ARMD based on fluorescein angiography findings and had no other retinal dystrophy or disease that may be associated with the development of CNV. Fluorescein angiographic photographs were evaluated by two independent graders (JZ, GR) according to the guidelines of the international classification and grading system of the International ARM Epidemiological Study Group.26 Grading results were subsequently reviewed by an additional independent grader. Inclusion criteria for the study were the presence of any form of neovascular maculopathy secondary to ARMD on fluorescein angiography including classic CNV, occult CNV, and disciform scarring, or a combination of any of the above in one or both eyes. If only one eye was affected by neovascular ARMD, the other eye had to present with phenomena of early ARMD including soft drusen and pigmentary abnormalities, or with advanced atrophic ARMD (geographic atrophy) to ensure that ARMD was the cause of neovascular maculopathy. In cases of disagreement between the two initial graders the conclusive grading was done by the reviewing grader. All graders were masked with respect to the genotyping results.
The 167 patients with advanced exudative ARMD represented a subset of 200 ARMD patients that included patients with geographic atrophy (n = 26) or early ARMD (n = 7) in one or both eyes and did not have CNV. They were therefore excluded from this study. Thus, 88% of all ARMD patients had advanced exudative ARMD in at least one eye, a typical rate for our tertiary care hospital.
We also genotyped 517 unrelated white control subjects (age range 19–99 years), 283 females and 234 males with mean age of 69.5 (12.7) and 66.3 (11.5) years, respectively. In order to allow the assessment of possible regional or ethnic differences in allele frequencies, the controls consisted of an international collection of adult volunteers originating from Hamburg (n = 235), Basle, Switzerland (n = 164), Brescia, Italy (n = 56), and Boston, USA (n = 62). Control subjects were recruited in several hospitals in these four centres without specific requirements in order to form a large control group that can be used for different association studies. The controls were not examined for ophthalmological disorders and were expected to develop ARMD at the population rate, and there were no exclusion criteria with respect to macular appearance in the control group.
The study was conducted according to the tenets of the Declaration of Helsinki. Informed consent was obtained from all subjects.
Genotyping
Genomic DNA was isolated from peripheral blood leucocytes using a standard salt precipitation technique.27 Polymerase chain reaction (PCR) products (318 bp) from genomic DNA were generated by using primer 024, TGGGAGGGACGAGGCGTTCC,24 and 1206R, TCCATGGGGCCTCCCACCAG. A 10 μl polymerase chain reaction was performed, containing 0.4 μl of suspended genomic DNA, 500 nM of each forward and reverse primer, 1.5 mM magnesium chloride, 1 μl 10× buffer, 200 μM deoxyribonucleoside triphosphate, 0.4 units Taq DNA polymerase (Gibco, Gaithersburg, MD, USA), 0.5 μl of 5% dimethyl sulphoxide, and 6.7 μl water. The thermoprofile was 95°C 45 seconds, 13 × (95°C 15 seconds, 68°C 30 seconds −1°C per cycle, 72°C 30 seconds), 23 × (95°C 15 seconds, 55°C 30 seconds, 72°C 30 seconds), and 72°C 5 minutes. Three polymorphic KspI restriction sites in the 5' region of CST3 were covered by the 318 bp PCR fragment. Through a strong linkage disequilibrium between the three polymorphisms only two haplotypes were observed. The haplotypes are defined by either concomitant KspI restriction endonuclease cleavage both 80 bp upstream of the mRNA transcription start site and in the penultimate codon of the signal peptide (haplotype A), or by an exclusive cleavage downstream of the transcription start site (haplotype B). Haplotypes were confirmed by direct sequencing of PCR products from individuals with the genotypes A/A, A/B, and B/B. Restriction digestion of the PCR product with KspI (MBI Fermentas, Vilnius, Lithuania) at 37°C overnight revealed fragment sizes of 41/226/51 bp (homozygote haplotype A), or 127/191 bp (homozygote haplotype B), or all five fragments in A/B heterozygotes. The digestion products were electrophoresed on a 2.5% agarose gel, stained with ethidium bromide, and visualised under ultraviolet light.
Statistical analysis
All statistical association analyses were done with spss, version 8.0 (SPSS Inc, Chicago, IL, USA). p Values less than 0.05 were considered significant. Statistical analyses of deviations from Hardy Weinberg equilibrium (HWE) were done by
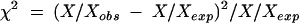 |
where X/Xobs is the observed genotype count of the respective CST3 genotypes (A/A, A/B, or B/B) and X/Xexp is the respective genotype count expected under HWE which is calculated based on the frequencies (FA and FB) of the observed allelic variants (here: A and B) at a given locus: FA + FB = 1, therefore (FA + FB)2 = 1 = (FA)2 + (FB)2 + 2FAFB. Note: (FA)2 = A/Aexp, (FB)2 = B/Bexp, 2FAFB = A/Bexp.
RESULTS
There was no significant difference in allele frequency of haplotype B (FB) in the control groups of the four centres Hamburg, Basle, Brescia, and Boston with FB of 0.18, 0.20, 0.21, and 0.12, respectively (p = 0.22, df = 3). The similar FB between the German controls (FB = 0.181) and those pooled from the other three centres with a mean FB = 0.184 (p = 0.88, df = 1) suggested widespread population similarity of FB and allowed us to pool all controls.
Table 1 shows the genotype counts of all patients and controls. None of the genotype counts significantly deviated from those expected under HWE. There was a significant difference in genotype counts between patients and controls (χ2 = 7.16, exact p = 0.028, df = 2; two sided Fisher's exact test: p = 0.037). The strongest difference between patients and controls was observed in the B/B homozygotes with 6.6% and 2.3%, respectively, suggesting an odds ratio (OR) for ARMD in association with CST3 B/B of 2.97 (95% CI 1.28; 6.86). The respective proportion of A/B heterozygotes was almost identical in patients and controls. The difference in FB between patients and controls with FB = 0.23 and 0.18, respectively, failed to reach statistical significance (χ2 = 2.83, p = 0.093, df = 1) and may be explained by the higher proportion of B/B homozygotes in the patients. In accordance with this there were slightly less (B/Bobs = 12) than expected under HWE (B/Bexp = 17.3) B/B homozygotes in the controls (χ2 = 1.62) and more than expected (B/Bobs = 11 v B/Bexp = 8.4, χ2 = 0.81) B/B homozygotes in the patients (Σχ2 = 2.43, ns).
Table 1.
CST3 genotype counts and frequencies in patients with ARMD and control subjects
| No | CST3 genotype counts and frequencies (%) | |||
| No | A/A | A/B | B/B | |
| Patients | 167 | 103 (61.7) | 53 (31.7) | 11 (6.6) |
| Female | 114 | 69 (60.5) | 40 (35.1) | 5 (4.4) |
| Male | 53 | 34 (64.2) | 13 (24.5) | 6 (11.3) |
| All controls | 517 | 340 (65.8) | 165 (31.9) | 12 (2.3) |
| All controls age matched | 349 | 239 (68.5) | 102 (29.2) | 8 (2.3) |
| Age matched, female | 211 | 150 (71.1) | 56 (26.5) | 5 (2.4) |
| Age matched, male | 138 | 89 (64.5) | 46 (33.3) | 3 (2.2) |
| All controls Hamburg (HH) | 235 | 175 (66.0) | 55 (39.9) | 5 (2.1) |
| Controls HH age matched | 175 | 115 (65.7) | 57 (32.6) | 3 (1.7) |
| Age matched, female | 124 | 79 (63.7) | 42 (33.9) | 3 (2.4) |
| Age matched, male | 51 | 36 (70.6) | 15 (29.4) | 0 |
Logistic regression analysis entering CST3 genotype (B/B v A/A or A/B), sex, and age revealed significant coefficients (β) for CST3 (β =1.26, p = 0.005), for sex (β = 0.41, p = 0.038), and for age (β = 0.06 , p <0.0001). Therefore we reanalysed the association between B/B homozygosity and ARMD both in separately age matched males (53 patients, 138 controls) and females (114 patients, 211 controls). For age matching, among control subjects males less than 64 years and females less than 62 years were excluded.
In addition, the association was reanalysed for the age matched controls from Hamburg only as well as for all controls from Hamburg since these control subjects represent the best ethnic match available. Among the age matched controls from Hamburg, males less than 64 years and females less than 58 years were excluded. The results shown in Table 2 suggest a stronger association of B/B with ARMD in males than in females. Little difference was found between the whole control group and the control group from Hamburg, again suggesting population similarity and that ethnic matching did not influence results. Interestingly, among the 51 age matched male controls from Hamburg no subject carrying the B/B genotype was found, whereas there were six B/B homozygotes among the 53 male patients from Hamburg.
Table 2.
Influence of sex and age matching of controls on odds ratio (OR) for exudative ARMD in Hamburg cases and controls only (167 patients; 114 female, 53 male) in association with CST3 genotype B/B
| OR | 95% CI | p Value* | |
| All controls age matched (n=349) | 3.01 | 1.19 to 7.62 | 0.023 |
| All controls from Hamburg (n=235) | 4.04 | 1.11 to 14.76 | 0.028 |
| Age matched controls HH (n=175) | 3.24 | 1.11 to 9.52 | 0.036 |
| Females HH, controls age matched | 1.85 | 0.43 to 7.92 | 0.485 |
| Males HH, controls age matched† | 0.027 |
HH = Hamburg.
*Two sided Fisher's exact test; †OR cannot be calculated due to 0 age matched males from Hamburg carrying the B/B genotype.
In both males and females there was a significant effect of CST3 B/B on disease free survival analysed by Kaplan-Meier analysis. In males with genotypes A/A or A/B the mean disease free survival time was 86 years (SE 2; 95% CI: 82; 89) which was 74 years (SE 4; 95% CI: 67; 81) in B/B homozygotes (log rank p = 0.006). In females with genotypes A/A or A/B the mean disease free survival time was 85 years (SE 1; 95% CI: 83; 86) and 76 years (SE 2; 95% CI: 72; 80) in B/B homozygotes (log rank p = 0.041). The mean disease free survival time in pooled males and females with genotypes A/A or A/B was 85 years (SE 1; 95% CI: 83; 86) and 76 years (SE 2; 95% CI: 72; 79) in B/B homozygotes (log rank p = 0.0006). The graphic plot of the hazard function of the pooled survival analysis is shown in Figure 1. The log rank statistics were very similar (p = 0.0005) if a mean disease onset of 3 years before clinical presentation was assumed (data not shown). The oldest control subject homozygous B/B was 75 years, and 137 of 517 controls (26.5 %) were older than 75. The oldest patient homozygous B/B was 85, whereas only 10 of 167 patients (6 %) were older than 85.
Figure 1.

Kaplan-Meier hazard plot (log minus log) of survival analyses with cumulative hazard representing the probability per unit time that an individual who has survived to age x will experience the terminal event (manifest exudative ARMD) at age x. CST3 B/B homozygosity is associated with a strong increase in age associated risk of exudative ARMD whereas there is no difference between A/A and A/B genotypes.
A composite image of the late phase fluorescein angiographies of both eyes of all 11 patients with CST3 B/B is shown in Figure 2.
Figure 2.

Composite image shows late phase fluorescein angiography of both eyes of all 11 patients carrying the CST3 genotype B/B: choroidal neovascularisation or a disciform scar secondary to ARMD is present in all patients in at least one eye.
DISCUSSION
The homozygous CST3 genotype B/B was associated with exudative ARMD. These results establish CST3 as an interesting susceptibility gene for exudative ARMD. The association appeared to be stronger in males, but disease free survival time was significantly reduced in both B/B homozygous males and females. Our male patient sample may have been too small, however, to conclude an unambiguous male predominant effect of CST3 genotype on susceptibility for ARMD. On the other hand, natural history and clinical appearance are similar between male and female patients and therefore fail to provide a clinical explanation for a possible sex related difference.
For this study, we selected a clinically well defined group of patients with the exudative form of ARMD. We confirmed the diagnosis of neovascular ARMD at the time of presentation. Unfortunately, it is not possible to explore the exact age at onset of the initial signs because of the high variability of subjective histories provided by the patients. Therefore, our objective was to form a homogeneous patient group affected by advanced exudative ARMD proved by fluorescein angiography irrespective of the subjective patient history. If reliable data were available it would be tempting, however, to use them for the differentiation of specific subtypes, courses, or onsets of the disease. Nevertheless, the presented theoretical survival analysis based on an assumed 3 year interval between onset and presentation also revealed a similar strong effect of CST3 B/B on disease free survival, suggesting a robust effect of CST3 in a subset of patients.
Genotype distributions and allele frequencies in control groups are critical for candidate gene analyses in case-control association studies. The similar frequency of the allelic variants A and B in control samples from four different countries suggests a homogeneous distribution among white people and allowed us to pool control samples from several centres in order to increase the power to detect an association between ARMD and a relatively rare genotype. In addition, we observed no difference between the whole multicentre control group and the controls from Hamburg which lends support to the assumption of widespread population similarity as described above.
We decided not to chose an ARMD free control population, because the distinction between early ARMD and normal ageing is not well defined. This study design may indeed suffer from reduced statistical power, because the control group may well include subjects with ARMD, as well as subjects who could manifest the disease later in life as can be expected from the normal age dependent prevalence of ARMD.
Our genotypic association data, the absence of a significant difference in allele frequencies between patients and controls, and the survival analyses suggest an increased susceptibility for exudative ARMD in CST3 B/B homozygotes. Therefore, the CST3 haplotype B may be a recessive risk allele, significantly contributing to disease risk in up to 6.6% of German ARMD patients. Such rare risk gene may well escape whole genome scans even if several hundred sib pairs are analysed, as recently performed, particularly, if only dominant modes of inheritance are assumed.28 The heterogeneous and multifactorial aetiology of ARMD may well consist of recessive and dominant genetic risk factors acting in concert with non-genetic ones.
Interestingly, among our CST3 B/B homozygote patients there was a twin pair at the age of 62 years who presented at our hospital. One of them had classic CNV on her right eye and a disciform scar on her left eye. Her sister presented with occult CNV on her right eye and with early ARMD (confluent serous drusen and pigmentary abnormalities) on her left eye (only one of them is included in the data).
Our data do not exclude that CST3 B/B increases susceptibility for other age associated diseases or that it influences life expectancy. Indeed, none of the B/B homozygote controls was older than 75 years, an age survived by 26.5% of all controls.
It is important to identify the biological mechanisms that may link cystatin C to impairment of RPE function and macular degeneration. In RPE cells, a variety of lysosomal enzymes are known to digest phagocytosed rod outer segments. In the posterior segment of the eye, cystatin C is mainly localised to the RPE but can also be detected in the outer nuclear layer and in the inner retina.29,30 Cystatin C mRNA is present at fairly high abundance in cultured human RPE cells.31 It is known as an inhibitor of cathepsin S, a lysosomal cysteine protease also present in RPE cells. Until recently, cathepsin D was considered to be the most important protease because its activity by far exceeded the activity of other proteases, whereas cysteine proteases were considered less important.32 The interest in cysteine proteases increased by the demonstration that the cysteine protease inhibitor leupeptin caused a dramatic increase of debris accumulation in RPE cells in rats.33,34 The same phenomenon was demonstrated in human RPE cells: inhibition of cathepsin S with leupeptin caused the accumulation of autofluorescent debris in RPE cells when challenged with rod outer segments.22 All in all, experimental evidence demonstrates a role of cysteine proteases in the accumulation of debris as a result of impaired degradation in RPE cells, resembling the findings in the natural course of ARMD. This may be explained by functional abnormalities or differences of proteases and their physiological inhibitors expressed in RPE cells.
Cystatin C is synthesised with an N-terminal hydrophobic signal sequence which is removed during synthesis. One of the KspI polymorphisms leads to an alanine to threonine substitution in the penultimate amino acid of the signal peptide:
 |
This variation alters the hydrophobicity of the signal sequence near the signal peptidase cleavage site and could be associated with changes in secretory processing of the peptide. These changes are clearly different from the leucine to glutamine substitution at position 68 seen in the Icelandic form of hereditary cerebral haemorrhage with amyloidosis (HCHWA-I).35 The resulting aberrant cystatin C in HCHWA-I has a stronger tendency to dimerise and form aggregates, especially if the temperature is elevated. These aggregates then deposit as amyloid in these patients whereas the spinal fluid level is abnormally low. Deposits in brain vessels cause cerebral haemorrhages, strokes, paralysis, and death in young adults. The point mutation of the cystatin C gene in HCHWA-I resulting in this aberrant protein is different from the three polymorphisms of CST3 resulting in the haplotypes A and B. Instead, our genetic data provide evidence for a contribution of CST3 B/B to disease risk in the heterogeneous and multifactorial aetiology of exudative ARMD.
The exact impact of cystatin C in the RPE degradation process is not well known. Preliminary data indicate that cellular levels of cystatin C in fibroblasts differ among the genotypes A/A, A/B, and B/B (RM Nitsch, unpublished data), but it is unknown if RPE cells show the same behaviour. Functional correlates of the allelic CST3 variants A and B in the human eye remain to be investigated.
The results of our study imply a role of cystatin C in the pathophysiology of ARMD. They open novel avenues for the study of cathepsins and their inhibitors in ARMD.
Acknowledgments
The authors are grateful to Dr FG Holz, Department of Ophthalmology, University of Heidelberg for helping reading and classifying the fluorescein angiograms, and to Drs C Hock, H Staehelin, G Binetti, and JH Growdon for providing control samples.
REFERENCES
- 1.Ferris FL, Fine SL, Hyman L. Age-related macular degeneration and blindness due to neovascular maculopathy. Arch Ophthalmol 1984;102:1640–2. [DOI] [PubMed] [Google Scholar]
- 2.Klein R, Klein BEK, Jensen SC, et al. The five-year incidence and progression of age-related maculopathy. The Beaver Dam Eye Study. Ophthalmology 1997;104:7–21. [DOI] [PubMed] [Google Scholar]
- 3.Klein ML, Mauldin WM, Stoumbos VD. Heredity and age-related macular degeneration: observations in monozygotic twins. Arch Ophthalmol 1994;112:932–7. [DOI] [PubMed] [Google Scholar]
- 4.Meyers SM, Greene T, Gutman FA. A twin study of age-related macular degeneration. Am J Ophthalmol 1995;120:757–66. [DOI] [PubMed] [Google Scholar]
- 5.Heiba IM, Elston RC, Klein BEK, et al. Sibling correlations and segregation analysis of age-related maculopathy. The Beaver Dam Eye Study. Genetic Epidemiol 1994;11:51–67. [DOI] [PubMed] [Google Scholar]
- 6.Seddon JM, Ajani UA, Mitchell BD. Familial aggregation of age-related maculopathy. Am J Ophthalmol 1997;123:199–206. [DOI] [PubMed] [Google Scholar]
- 7.Klaver CC, Wolfs RC, Assink JJ, et al. Genetic risk of age-related maculopathy. Population-based familial aggregation study. Arch Ophthalmol 1998;116:1646–51. [DOI] [PubMed] [Google Scholar]
- 8.Rosenfeld PJ, Gorin MB. Genetics. In: Berger JW, Fine SL, Maguire MG, eds. Age-related macular degeneration. 1st ed. St Louis: Mosby, 1999:69–80.
- 9.Kimura K, Isashiki Y, Sonoda S, et al. Genetic association of manganese superoxide dismutase with exudative age-related macular degeneration. Am J Ophthalmol 2000;130:769–73. [DOI] [PubMed] [Google Scholar]
- 10.Souied EH, Benlian P, Amouyel P, et al. The ɛ4 allele of the apolipoprotein E gene as a potential protective factor for exudative age-related macular degeneration. Am J Ophthalmol 1998;125:353–9. [DOI] [PubMed] [Google Scholar]
- 11.De La Paz M, Pericak-Vance MA, Haines J, et al. Studies of apolipoprotein E (APOE) and age-related macular degeneration. [ARVO Abstract] Invest Ophthalmol Vis Sci 1997;38:S796. Abstract no 3695. [PubMed] [Google Scholar]
- 12.Cruickshanks KJ, Hamman RF, Nondahl DM, et al. Cardiovascular risk factors and early age-related maculopathy in Colorado non-Hispanic whites. [ARVO Abstract] Invest Ophthalmol Vis Sci 1997;38:S471. Abstract no 2187. [Google Scholar]
- 13.Allikmets R, Shroyer NF, Singh N, et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 1997;277:1805–7. [DOI] [PubMed] [Google Scholar]
- 14.Stone EM, Webster AR, Vandenburgh K, et al. Allelic variation in ABCR associated with Stargardt disease but not age-related macular degeneration. Nat Genet 1998;20:328–9. [DOI] [PubMed] [Google Scholar]
- 15.Allikmets R. Further evidence for an association of ABCR alleles with age-related macular degeneration. Am J Hum Genet 2000;67:487–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allikmets R. Simple and complex ABCR: genetic predisposition to retinal disease. Am J Hum Genet 2000;67:793–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De La Paz MA, Guy VK, Abou-Donia S, et al. Analysis of the Stargardt disease gene (ABCR) in age-related macular degeneration. Ophthalmology 1999;106:1531–6. [DOI] [PubMed] [Google Scholar]
- 18.Rivera A, White K, Stohr H, et al. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am J Hum Genet 2000;67:800–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weber BHF, Vogt G, Pruett RC, et al. Mutations in the tissue inhibitor metalloproteinases-3 (TIMP3) in patients with Sorsby`s fundus dystrophy. Nat Genet 1994;8:352–6. [DOI] [PubMed] [Google Scholar]
- 20.De La Paz MA, Pericak-Vance MA, Lennon F, et al. Exclusion of TIMP3 as a candidate locus in age-related macular degeneration. Invest Ophthalmol Vis Sci 1997;38:1060–5. [PubMed] [Google Scholar]
- 21.Katunuma N, Kominami E. Structure, properties, mechanisms and assays of cysteine protease inhibitors: cystatins and E-64 derivatives. Methods Enzymol 1995;251:382–97. [DOI] [PubMed] [Google Scholar]
- 22.Rakoczy PE, Mann K, Caveney DM, et al. Detection and possible functions of a cysteine protease involved in digestion of rod outer segments by retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 1994;35:4100–8. [PubMed] [Google Scholar]
- 23.Levy E, Lopez-Otin C, Ghiso J, et al. Stroke in Icelandic patients with hereditary amyloid angiopathy is related to a mutation in the cystatin C gene, an inhibitor of cysteine proteases. J Exp Med 1989;169:1771–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balbin M, Abrahamson M. SstII polymorphic sites in the promoter region of the human cystatin C gene. Hum Genet 1991;87:751–2. [DOI] [PubMed] [Google Scholar]
- 25.Finckh U, Von der Kammer H, Velden J, et al. Genetic association of a cystatin C gene polymorphism with late-onset Alzheimer disease. Arch Neurol 2000;57:1579–83. [DOI] [PubMed] [Google Scholar]
- 26.The International ARM Epidemiological Study Group. An international classification and grading system for age-related maculopathy and age-related macular degeneration. Surv Ophthalmol 1995;39:367–74. [DOI] [PubMed] [Google Scholar]
- 27.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucl Acids Res 1988;16:1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weeks D, Conley YP, Mah TS, et al. A full genome scan for age-related maculopathy. Hum Mol Genet 2000;9:1329–49. [DOI] [PubMed] [Google Scholar]
- 29.Wasselius J, Hakansson K, Johansson K, et al. Identification and localization of retinal Cystatin C. Invest Ophthalmol Vis Sci 2001;42:1901–6. [PubMed] [Google Scholar]
- 30.Barka T, van der Noen H. Expression of the cysteine proteinase inhibitor cystatin C mRNA in rat eye. Anat Rec 1994;239:343–8. [DOI] [PubMed] [Google Scholar]
- 31.Paraoan LI, Grierson I, Maden BEH. Analysis of expressed sequence tags of retinal pigment epithelium: cystatin C is an abundant transcript. Int J Biochem 2000;32:417–26. [DOI] [PubMed] [Google Scholar]
- 32.Rakoczy PE, Baines M, Kennedy CJ, et al. Correlation between autofluorescent debris accumulation and the presence of partially processed forms of cathepsin D in cultured retinal pigment epithelial cells challenged with rod outer segments. Exp Eye Res 1996;63:159–67. [DOI] [PubMed] [Google Scholar]
- 33.Ivy GO, Kanai S, Ohta M, et al. Lipofuscine-like substances accumulate rapidly with cysteine protease inhibition. Adv Exp Med Biol 1989;266:31–45. [DOI] [PubMed] [Google Scholar]
- 34.Katz ML, Shanker MJ. Development of lipofuscine-like fluorescence in the retinal pigment epithelium in response to protease inhibitor treatment. Mech Ageing Dev 1989;49:23–40 [DOI] [PubMed] [Google Scholar]
- 35.Abrahamson M, Grubb A. Increased body temperature accelerates aggregation of the Leu-68→Gln mutant cystatin C, the amyloid-forming protein in hereditary cystatin C amyloid angiopathy. Proc Natl Acad Sci 1994;91:1416–20. [DOI] [PMC free article] [PubMed] [Google Scholar]