

Abstract
We previously identified and characterized a homozygous LMNA nonsense mutation leading to the absence of A-type lamins in a premature neonate who died at birth. We show here that the absence of A-type lamins is due to degradation of the aberrant mRNA transcript with a premature termination codon. In cultured fibroblasts from the subject with the homozygous LMNA nonsense mutation, there was a decreased steady-state expression of the integral inner nuclear membrane proteins emerin and nesprin-1α associated with their mislocalization to the bulk endoplasmic reticulum and a hyperphosphorylation of emerin. To determine if decreased emerin expression occurred post-translationally, we treated cells with a selective proteasome inhibitor and observed an increase in expression. Our results show that mislocalization of integral inner nuclear membrane proteins to the endoplasmic reticulum in human cells lacking A-type lamins leads to their degradation and provides the first evidence that their degradation is mediated by the proteasome.
Keywords: Nuclear envelope, Lamin, Emerin, Nesprin, Proteasome, Muscular dystrophy
Lamins are intermediate filament proteins that form the nuclear lamina, a fibrous meshwork underlying the inner nuclear membrane of higher eukaryotic cells [1]. In human, two types of lamins are expressed in somatic cells according to their sequences and biochemical properties, A-type and B-type. Lamins A and C, encoded by the LMNA gene, are the predominant A-type lamins in mammalian somatic cells and arise from alternative RNA splicing. Two genes, LMNB1 and LMNB2, respectively, encode the somatic mammalian B-type lamins B1 and B2 [1]. While lamins B1 and B2 are expressed in most of all somatic cells, the expression of A-type lamins is developmentally regulated and generally restricted to differentiated cells [2,3]. The nuclear lamina is attached to the inner nuclear membrane via interactions with integral proteins, among them emerin, nesprin-1α, LBR, and LAPs [4]. Lamins A and C also interact with chromatin [5] and DNA [6]. The nuclear lamina functions in maintenance of nuclear architecture, DNA replication, and the synthesis of mRNA [4].
Mutations in LMNA cause several human diseases affecting different tissues, which are sometimes referred to as “laminopathies” [4]. One of these diseases is limb-girdle muscular dystrophy type1B (LGMD1B), an autosomal dominant disorder characterized by symmetrical weakness starting in the proximal lower limb muscles and gradually affecting the upper limb muscles [7,8]. It is associated with cardiac conduction disturbances and dilated cardiomyopathy, similar to autosomal dominant Emery–Dreifuss muscular dystrophy and isolated cardiomyopathy with conduction defect 1A, which are also caused by LMNA mutations.
The molecular mechanisms underlying the various disease phenotypes caused by LMNA mutations are unknown. However, several investigators in the field have proposed two main hypotheses to explain the tissue-specific effects observed in laminopathies [4]. The “mechanical stress” hypothesis proposes that abnormalities in the nuclear lamins caused by LMNA mutations lead to structural defects and increased nuclear fragility, eventually resulting in nuclear disruption in mechanically stressed cells. The “gene expression” hypothesis proposes a tissue-specific role of lamins in transcription and abnormalities in expression of genes involved in cell differentiation or maintenance of the differentiated state of certain tissues if there are LMNA mutations. These two hypotheses are not necessarily mutually exclusive, as, for example, defective nuclear mechanics have been linked to altered expression of stress-activated genes in fibroblasts lacking A-type lamins [9].
We previously investigated the molecular and cellular alterations in cultured skin fibroblasts from two members of a family with LGMD1B carrying a LMNA nonsense mutation (Y259X) [10]. A heterozygous 66-year-old woman had a typical LGMD1B phenotype, whereas her grandchild carrying two mutated LMNA alleles, leading to absence of A-type lamins, died at birth after preterm delivery by cesarean section [11]. Fibroblasts from the homozygous neonate had nuclei with marked structural defects and mislocalization of emerin and nesprin-1α to the endoplasmic reticulum [10]. In this report, we further examine cells from the subject with the homozygous LMNA Y259X mutation and present evidence that emerin “escaped” from the inner nuclear membrane to the bulk endoplasmic reticulum in these cells lacking A-type lamins undergoes protea-some-mediated degradation.
Materials and methods
Reagents
Lactacystine and cycloheximide (CHX) were purchased from Sigma–Aldrich. Lactacystine was prepared as a 10 mM stock solution in dimethyl sulfoxide and stored at −80 °C until use. CHX was dissolved in ethanol as a stock solution of 100 μg/ml.
Cells
Skin fibroblasts were obtained from a male child homozygous for the Y259X LMNA mutation who died at birth after delivery at 30 gestational weeks [10,11]. Fibroblasts from a 12-week-old fetus with no LMNA mutation were used as control. Fibroblasts were obtained from skin biopsies obtained after informed consent of the parents and complied with ethical guidelines approved by the Institutional Review Board. Fibroblasts were maintained at 37 °C in a humidified incubator containing 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf bovine serum (Invitrogen) and 0.1% gentamycin.
RNA extraction
At 80% confluence, media were removed from cultures and cells immediately lysed by addition of 2 ml RNAplus (Biop-robes) directly to culture plate. Total RNA was prepared as described by the manufacturer. Adequacy and integrity of extracted RNA were assessed by Agilent analysis (Agilent Technologies) and concentrations measured by ultraviolet absorbance spectroscopy.
Indirect immunofluorescence microscopy
Fibroblasts were grown on coverslips and washed with phosphate-buffered saline (PBS) then fixed as previously described [10,12]. Fibroblasts were then incubated with the primary antibodies for 1 h at room temperature. Primary antibodies used were anti-lamin A/C monoclonal MANLAC (1:10, provided by G.E. Morris), anti-lamin B1 polyclonal (1:250, a gift from J.C. Courvalin), anti-emerin polyclonal (1:100, provided by G. Morris), and anti-nesprin-1α (1:200, provided by E.M. McNally). After washes with PBS, fibroblasts were incubated with secondary antibodies (1:500). Secondary antibodies used were Alexa fluor 568 goat anti-mouse immunoglobulin G (Molecular Probes) and fluorescein isothiocyanate-conjugated goat anti-mouse immunoglobulin G (Chemicon). Cells were then washed with PBS and slides mounted in Mowiol (Santa Cruz Biotechnologies) with 0.1 μg/ml 4′,6-diamidino-2-phenylindole (dapi). Immunofluorescence microscopy was performed using an Axiophot microscope (Carl Zeiss). Micrographs were processed using Adobe Photoshop 5.5 (Adobe Systems).
Real-time reverse transcription-polymerase chain reaction (RT-PCR)
Reverse transcription was performed on 5 μg total RNA using the SuperSript First-Strand Synthesis System (Invitrogen). Quantitative PCR was performed using a LightCycler (Roche Diagnostics). PCR mix (20 μl) included 2 μl of each diluted reverse transcription product, 4 mmol/l MgCl2, and 300 nmol/l of each primer in 1× LightCycler DNA Master SYBR Green. Specific primers for cDNAs were chosen with the Light-Cycler program according to following Genbank Accession Nos.: LMNA (Accession No. NM_170707), LMNB1 (Accession No. NM_005573), EMD (Accession No. NM_000117), SYNE1 (Accession No. NM_022027), LBR (Accession No. NM_002296), and NUP153 (Accession No. NM_005124). For LMNA, the primers were common to both lamin A and lamin C mRNA transcripts. PCR was performed using the following conditions: denaturation and enzyme activation at 95 °C/15 min and cycling at 95 °C/30 s, 64 °C/10 s, and 72 °C/10 s. Analysis was carried out with the LightCycler 3.5 software (Roche). Relative levels of mRNA expression were calculated according to the ΔΔCT method [13]. Individual expression values were normalized by comparison with β-actin mRNA.
Western blot analysis
Fibroblasts (3 × 106) were harvested from each culture at the appropriate time interval, washed with ice-cold PBS, and total protein extracted as previously described [10,12]. For Western blot analysis, equal amounts of protein (10 μg) were electrophoresed on 10% SDS–polyacrylamide gels, transferred to nitrocellulose membranes (Invitrogen) by electroblotting at 4 °C for 1 h at 400 mA, and stained with Ponceau S. Primary antibodies used were anti-lamin A/C Jol5 monoclonal antibody (1:250, provided by provided by C. Hutchison), anti-lamin B1 (1:2000, provided by J.C. Courvalin), anti-emerin (1:500, Novocastra), anti-nesprin-1α (1:500, provided by E.M. McNally), and anti-β-actin polyclonal antibody (1:2000, Sigma). Secondary antibodies were horse-radish peroxidase-conjugated rabbit anti-mouse and goat anti-rabbit antibodies (1:2000, DAKO). Protein detection was performed using a SuperSignal West Pico Chemiluminescent Substrate (Pierce) and visualized using Hyperfilm ECL (Amersham).
Results and discussion
Expression of mRNAs encoding nuclear envelope proteins in cells homozygous for the LMNA Y259X mutation
Our initial aim was to determine whether the presence of homozygous Y259X LMNA nonsense mutation affects the expression of mRNA encoding lamins A and C in cultured skin fibroblasts. Total RNA was extracted from skin fibro-blasts cultured from unaffected fetal tissue and from the subject with the homozygous mutation (fibroblastsY259X/Y259X). Although similar amounts of total mRNA were analyzed as revealed by β-actin mRNA detection, real-time quantitative RT-PCR revealed a significant decrease of the two different lamin A/C mRNA transcripts encoded by LMNA in fibroblastsY259X/Y259X compared to controls (Fig. 1A). Expression of mRNAs encoded by LMNB1 (lamin B1), EMD (emerin), SYNE1 (nesprin-1α), LBR (integral inner nuclear membrane protein LBR), and NUP153 (nucleoporin NUP153) was not significantly affected in fibroblastsY259X/Y259X compared to controls (Fig. 1A).
Fig. 1.
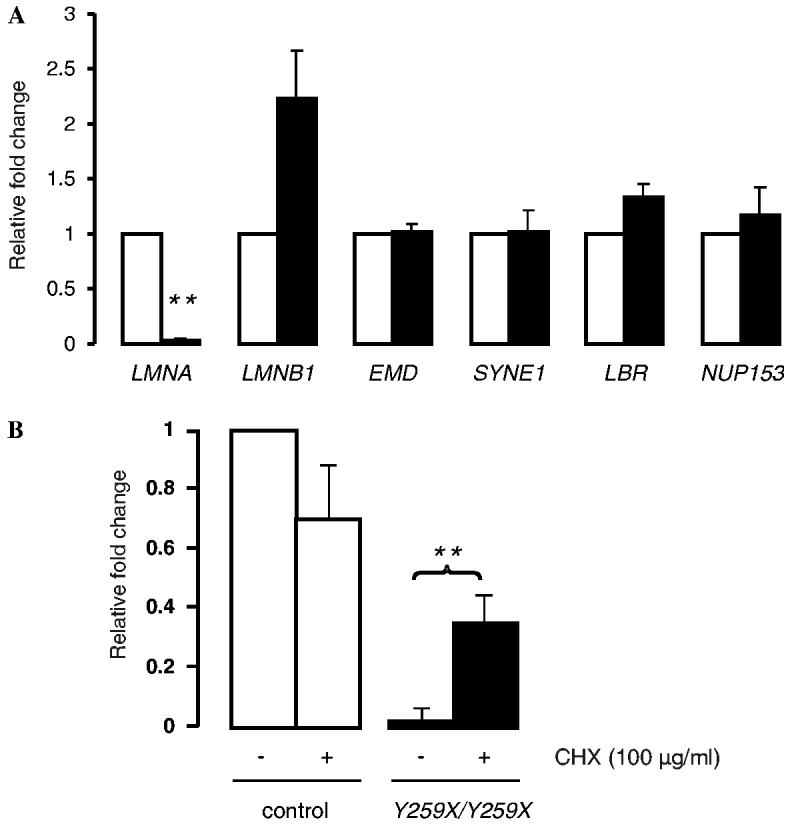
(A) Expression of mRNA encoded by LMNA, LMNB1, EMD, SYNE1, LBR, and NUP153 in cultured skin fibroblasts from control (open bars) and LMNAY259X/Y259X (solid bar) subjects using real-time quantitative RT-PCR. Bars indicate fold overexpression of the indicated mRNA. Values are means ± standard deviations. n = 3 (**p < 0.005). (B) Effect of CHX on the expression of mRNA encoded by LMNA gene. CHX (100 μg/ml) was added to cultured skin fibroblasts from control (open bars) and LMNAY259X/Y259X (solid bars) subjects using real-time quantitative RT-PCR. Bars indicate fold overexpression of the indicated mRNA. Values are means ± standard deviations. n = 3 (**p < 0.005).
The nonsense RNA-mediated decay (NMD) process might be the molecular mechanism involved in the degradation of mRNA carrying Y259X LMNA mutation. The c.1865T > A, p.Y259X mutation lies in exon 4 and generates a premature termination codon. NMD is a mRNA surveillance mechanism that leads to selective degradation or elimination of mRNA transcripts containing premature termination codons arising from somatic mutations or errors in gene expression, leading to decreased translation in cells [14]. In mammalian NMD, an intron apparently functions as a signal for triggering degradation by leaving a mark on the mRNA at the exon–exon junction as a consequence of the splicing event. This mark enables the NMD surveillance pathway to differentiate between premature termination codons and normal stop codons present in the last exon, hence ensuring the degradation only of transcripts containing nonsense codons that are followed by an intron [15,16]. In this mechanism, when the ribosome encounters a premature termination codon, the final exon–exon tag(s) are not removed and this marks the defective mRNA for destruction [14]. We investigated the activity of this mechanism on lamin A/C mRNA in fibro-blastsY259X/Y259X by using CHX. The addition of CHX to control fibroblasts induced a decrease of 30% of lamin A/C mRNA (Fig. 1B). This could be explained by a degradation of normal lamin A/C mRNA, which is not translated due to blocking translation by ribosomes. In contrast, the addition of CHX increased lamin A/C mRNA in fibro-blastsY259X/Y259X (8-fold increase) compared to untreated fibroblastsY259X/Y259X (Fig. 1B).
These results suggest that Y259X LMNA nonsense mutation leads to lack of expression A-type lamins through the RNA NMD process. Degradation of mRNA from the allele bearing mutation Y259X appears to occur by NMD, even thought it does not fulfill the canonical requirements for NMD (generation of a premature termination codon that lies more than 50 nucleotides 5′ to the last exon–exon junction). However, there are several genes whose mRNA degradation appears to show exceptions to this rule, including those encoding hexoseaminidase A [17], myelin protein zero [18], and the hepatocyte nuclear factor-1β [19]. The failure of some genes to follow the “55 nucleotides boundary rule” suggests that either alternative or additional signals may exist to activate the NMD mechanism.
Effect of lack of A-type lamins in cells homozygous for the LMNA Y259X mutation on expression inner nuclear membrane proteins
To determine if lack of A-type lamins in cells homozygous for the LMNA Y259X mutation leads to alterations in the expression and localization of lamin-associated integral proteins of the inner nuclear membrane, we analyzed the subcellular localizations and steady-state amounts of selected proteins. As previously described [10], nuclei from the fibroblasts have morphological abnormalities with an uneven localization of lamin B1, which is not detectable at “bleb-like” structures of the nuclear envelope (Fig. 2A, arrowheads). Emerin and nesprin-1α were not exclusively localized to the nuclear envelope but showed a cytosolic-appearing distribution, likely in the endoplasmic reticulum membranes (Fig. 2A), as we have also shown previously [10].
Fig. 2.

(A) Localization of lamins, emerin, and nesprin-1α in cultured skin fibroblasts from control and LMNAY259X/Y259X subjects. Mouse monoclonal antibodies directed against lamin A/C and rabbit polyclonal antibodies directed against lamin B1, emerin, and nesprin-1α were used. Arrowheads show nuclear bleb-like structures without lamin B1 labeling. Bar = 10 μm. (B) Expression of lamin A/C, lamin B1, nesprin-1α, and emerin in cultured skin fibroblasts from control and LMNAY259X/Y259X subjects was determined by Western blotting.
We next examined the steady-state expression levels of lamin B1, emerin, and nesprin-1α. On Western blotting, although similar amounts of total cellular proteins were loaded as revealed by β-actin detection, lamins A and C were not detectable in fibroblastsY259X/Y259X (Fig. 2B [10]). However, the expression of lamin B1 was not increased in fibroblastsY259X/Y259X (Fig. 2B), showing that the nuclear lamina in fibroblastsY259X/Y259X has the same quantity of lamin B1 as control fibroblasts. Hence, there does not appear to be a compensatory mechanism increasing the quantity of other lamins. However, there were detectable decreases in the steady-state expression levels of nesprin-1α and emerin (Fig. 2B). For emerin, this decrease is accompanied by the appearance of a more slowly migrating isoform with an apparent molecular mass of approximately 36 kDa (Fig. 2B). These observations demonstrated that down-regulation of emerin and nesprin-1α occurred post-translationally because expression of their mRNAs was not affected. Hence, the loss of A-type lamins in fibroblasts leads to mislocalization of emerin and nesprin-1α and an associated degradation of the proteins.
It has been previously demonstrated that emerin is phosphorylated; hence, the higher molecular weight isoform we detected in fibroblastsY259X/Y259X may be phosphorylated [20-24]. To assess whether the protein with an apparent molecular mass of 36 kDa recognized by anti-emerin antibodies was a phosphorylated form of emerin, we treated fibroblastsY259X/Y259X with the phosphatase inhibitor okadaic acid (OA). Western blotting carried out using an anti-emerin antibody demonstrated that 10 μM OA induced an increase in the emerin isoform with an apparent molecular mass of 36 kDa (Fig. 3A). This suggests that this is a phosphorylated form of emerin. Treatment of control fibroblasts with OA (0.01 and 1 μM) and subsequent Western blot analysis using an anti-emerin antibody also showed an appearance of the isoform with an apparent molecular mass of 36 kDa observed in fibroblastsY259X/Y259X (Fig. 3B).
Fig. 3.

(A) Effect of OA on expression of emerin in cultured skin fibroblasts from LMNAY259X/Y259X subject. Expression of a protein with an apparent molecular mass of 36 kDa that is recognized by anti-emerin antibody is increased by treatment with 10 μM OA, as determined by Western blot analysis. (B) Effect of OA on the expression of emerin in cultured skin fibroblasts from control subject. Addition of OA (0.01 and 1 μM) to cultured skin fibroblasts resulted in expression of a protein recognized by anti-emerin antibodies with an apparent molecular mass of 36 kDa, which is of the same molecular mass as that expressed in cultured skin fibroblasts from LMNAY259X/Y259X subject. (C) Treatment of cultured skin fibroblasts with lactacystine. The addition of lactacystin (50 μM) to cultured skin fibroblasts from control subject does not affect the expression of emerin, whereas in cultured skin fibroblasts from LMNAY259X/Y259X subject it induces a diminution of the protein recognized by anti-emerin antibody with an apparent molecular mass of 36 kDa and an increase of the recognized protein with an apparent molecular mass of 34 kDa. Bars from control (open bars) and LMNAY259X/Y259X subjects (solid bars) on the histogram indicate relative emerin fold change. Values are means ± standard deviations. n = 3 (*p < 0.005).
Emerin is degraded by proteasome when mislocalized in the endoplasmic reticulum
The decrease in steady-state emerin expression in fibroblastsY259X/Y259X lacking A-type lamins suggested that an integral protein that has “escaped” from the inner nuclear membrane to the endoplasmic reticulum is subjected to degradation. One potential mechanism of protein degradation is by the proteasome. To determine if the observed decrease in emerin in fibroblasts lacking A-type lamins could result from proteasome-mediated degradation, we used lactacystin, a specific inhibitor of the 20S-proteasome [25]. Treatment of the cells with 50 μM lactacystin for 24 h and subsequent Western blot analysis using an anti-emerin antibody showed an approximate 2-fold increased expression of emerin and a decrease of the phosphorylated form (Fig. 3C). These results suggest that emerin, including the phosphorylated form of approximately 36 kDa, is degraded by the proteasome.
Conclusion
We have shown that NMD of lamin A/C mRNA transcripts with a Y259X nonsense mutation leads to lack of A-type lamins. As reported previously by several groups, lack of A-type lamins in fibroblasts leads to a mislocalization of emerin, which is normally localized to the inner nuclear membrane, to the bulk endoplasmic reticulum [10,26,27]. A similar mislocalization is observed for nesprin-1α [10]. We have now demonstrated an increase in a phosphorylated form of emerin in human fibroblasts lacking A-type lamins. Associated with emerin's mislocalization and increased phosphorylation is a decrease in its steady-state cellular concentration, which may be mediated by the proteasomal degradation (Fig. 4). Abnormal folding of inner nuclear membrane proteins that have “escaped” to the bulk endoplasmic reticulum, perhaps induced by phosphorylation, may lead to conjugation to ubiquitins and subsequent proteasomal degradation. This mechanism would allow the cell to eliminate abnormally localized proteins and could also lead to alterations in physiological functions related to these proteins.
Fig. 4.

Schematic representation of the degradation of integral inner nuclear membrane proteins in fibroblasts from LMNAY259X/Y259X subject. Activation of NMD mechanism by the LMNA Y259X nonsense mutation leads to the degradation of lamin A/C mRNA. Subsequent cytosolic-appearing distribution of integral inner nuclear membrane protein (emerin and nesprin-1α) occurs. Emerin is in addition partially phosphorylated. This cytosolic mislocalization leads to degradation of emerin and nesprin-1α, likely through a proteasome-mediated process.
Our results in fibroblasts from human subjects lacking A-type lamins differ from those previously reported in fibroblasts from Lmna null mice. In mouse fibroblasts lacking A-type lamins, emerin is mislocalized to the bulk endoplasmic reticulum but the steady-state concentration appears to be unaffected [26]. The apparent conflicting results between human and mice cells may be secondary to species-specific factors, isolation or culture conditions or other factors. Other differences between mouse and humans resulting from deletion of the genes encoding Atype lamins have also been observed, including the grossly normal phenotype of Lmna +/− mice compared to the disease phenotype in heterozygous human subjects with LMNA mutations that cause muscular dystrophy and cardiomyopathy.
Acknowledgments
We are grateful to J.-C. Courvalin, C. Hutchison, E.M. McNally, and G.E. Morris for providing antibodies and P. Bausero and N. Vignier for assistance with real-time quantitative RT-PCR. This study was supported by grants from the National Institutes of Health (AR048997), European Union Fifth Framework (Euro-Laminopathies Contract 018690), Association Française contre les Myopathies (Grant 11057), and Human Frontiers Science Program (RGP0057/2001-M101). A.M. was supported in part by fellowship grants from Association Française contre les Myopathies and Fondation pour la Recherche Médicale.
References
- 1.Worman HJ, Courvalin JC. The inner nuclear membrane. J. Membr. Biol. 2000;177:1–11. doi: 10.1007/s002320001096. [DOI] [PubMed] [Google Scholar]
- 2.Stewart C, Burke B. Teratocarcinoma stem cells and early mouse embryos contain only a single major lamin polypeptide closely resembling lamin B. Cell. 1987;51:383–392. doi: 10.1016/0092-8674(87)90634-9. [DOI] [PubMed] [Google Scholar]
- 3.Guilly MN, Kolb JP, Gosti F, Godeau F, Courvalin JC. Lamins A and C are not expressed at early stages of human lymphocyte differentiation. Exp. Cell Res. 1990;189:145–147. doi: 10.1016/0014-4827(90)90267-e. [DOI] [PubMed] [Google Scholar]
- 4.Muchir A, Worman HJ. The nuclear envelope and human disease. Physiology. 2004;19:309–314. doi: 10.1152/physiol.00022.2004. [DOI] [PubMed] [Google Scholar]
- 5.Glass CA, Glass JR, Taniura H, Hasel KW, Blevitt JM, Gerace L. The alpha-helical rod domain of human lamins A and C contains a chromatin binding site. EMBO J. 1993;12:4413–4424. doi: 10.1002/j.1460-2075.1993.tb06126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stierle VV, Couprie J, Ostlund C, Krimm I, Zinn-Justin S, Hossenlopp P, Worman HJ, Courvalin JC, Duband-Goulet I. The carboxyl-terminal region common to lamins A and C contains a DNA binding domain. Biochemistry. 2003;42:4819–4828. doi: 10.1021/bi020704g. [DOI] [PubMed] [Google Scholar]
- 7.van der Kooi AJ, Ledderhof TM, de Voogt WG, Res CJ, Bouwsma G, Troost D, Busch HF, Becker AE, de Visser M. A newly recognized autosomal dominant limb girdle muscular dystrophy with cardiac involvement. Ann. Neurol. 1996;39:636–642. doi: 10.1002/ana.410390513. [DOI] [PubMed] [Google Scholar]
- 8.Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, de Visser M, Schwartz K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B) Hum. Mol. Genet. 2000;9:1453–1459. doi: 10.1093/hmg/9.9.1453. [DOI] [PubMed] [Google Scholar]
- 9.Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Invest. 2004;113:370–378. doi: 10.1172/JCI19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muchir A, van Engelen BGM, Lammens M, Mislow JM, McNally E, Schwartz K, Bonne G. Nuclear envelope alterations in fibroblasts from LGMD1B patients carrying nonsense Y259X heterozygous or homozygous mutation in lamin A/C gene. Exp. Cell Res. 2003;291:352–362. doi: 10.1016/j.yexcr.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 11.van Engelen BGM, Muchir A, Hutchison CJ, van der Kooi AJ, Bonne G, Lammens M. The lethal phenotype of a homozygous nonsense mutation in the lamin A/C gene. Neurology. 2005;64:374–376. doi: 10.1212/01.WNL.0000149763.15180.00. [DOI] [PubMed] [Google Scholar]
- 12.Muchir A, Medioni J, Laluc M, Massart C, Arimura T, Van Der Kooi AJ, Desguerre I, Mayer M, Ferrer X, Briault S, Hirano M, Worman HJ, Mallet A, Wehnert M, Schwartz K, Bonne G. Nuclear envelope alterations in fibroblasts from patients with muscular dystrophy, cardiomyopathy, and partial lipodystrophy carrying lamin A/C gene mutations. Muscle Nerve. 2004;30:444–450. doi: 10.1002/mus.20122. [DOI] [PubMed] [Google Scholar]
- 13.Ponchel F, Toomes C, Bransfield K, Leong FT, Douglas SH, Field SL, Bell SM, Combaret V, Puisieux A, Mighell AJ, Robinson PA, Inglehearn CF, Isaacs JD, Markham AF. Real-time PCR based on SYBR-Green I fluorescence: An alternative to the TaqMan assay for a replicative quantification of gene rearrangements, gene amplifications and micro gene deletions. BMC Biotechnol. 2003;3:18. doi: 10.1186/1472-6750-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amrani N, Sachs MS, Jacobson A. Early nonsense, mRNA decay solves a translational problem. Nat. Rev. Mol. Cell. Biol. 2006;7:415–425. doi: 10.1038/nrm1942. [DOI] [PubMed] [Google Scholar]
- 15.Carter MS, Li S, Wilkinson MF. A splicing-dependent regulatory mechanism that detects translation signals. EMBO J. 1996;15:5965–5975. [PMC free article] [PubMed] [Google Scholar]
- 16.Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem. Sci. 1998;23:198–199. doi: 10.1016/s0968-0004(98)01208-0. [DOI] [PubMed] [Google Scholar]
- 17.Rajavel KS, Neufeld EF. Nonsense-mediated decay of human HEXA mRNA. Mol. Cell. Biol. 2001;21:5512–5519. doi: 10.1128/MCB.21.16.5512-5519.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoue K, Khajavi M, Ohyama T, Hirabayashi S, Wilson J, Reggin JD, Mancias P, Butler IJ, Wilkinson MF, Wegner M, Lupski JR. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 2004;36:361–369. doi: 10.1038/ng1322. [DOI] [PubMed] [Google Scholar]
- 19.Harries LW, Bingham C, Bellanne-Chantelot C, Hattersley AT, Ellard S. The position of premature termination codons in the hepatocyte nuclear factor-1 beta gene determines susceptibility to nonsense-mediated decay. Hum. Genet. 2005;118:214–224. doi: 10.1007/s00439-005-0023-y. [DOI] [PubMed] [Google Scholar]
- 20.Ellis JA, Craxton M, Yates JR, Kendrick-Jones J. Aberrant intracellular targeting and cell cycle-dependent phosphorylation of emerin contribute to the Emery–Dreifuss muscular dystrophy phenotype. J. Cell Sci. 1998;111:781–792. doi: 10.1242/jcs.111.6.781. [DOI] [PubMed] [Google Scholar]
- 21.Roberts RC, Sutherland-Smith AJ, Wheeler MA, Jensen ON, Emerson LJ, Spiliotis II, Tate CG, Kendrick-Jones J, Ellis JA. The Emery–Dreifuss muscular dystrophy associated-protein emerin is phosphorylated on serine 49 by protein kinase A. FEBS J. 2006;273:4375–4578. doi: 10.1111/j.1742-4658.2006.05464.x. [DOI] [PubMed] [Google Scholar]
- 22.Cartegni L, di Barletta RM, Barresi R, Squarzoni S, Sabatelli P, Maraldi NM, Mora M, Di Blasi C, Cornelio F, Merlini L, Villa A, Cobianchi F, Toniolo D. Heart specific localization of emerin: new insights into Emery–Dreifuss muscular dystrophy. Hum. Mol. Genet. 1997;6:2257–2264. doi: 10.1093/hmg/6.13.2257. [DOI] [PubMed] [Google Scholar]
- 23.Hirano Y, Segawa M, Ouchi FS, Yamakawa Y, Furukawa K, Takeyasu K, Horigome T. Dissociation of emerin from BAF is regulated through mitotic phosphorylation of emerin in a Xenopus egg cell-free system. J. Biol. Chem. 2005;280:39925–39933. doi: 10.1074/jbc.M503214200. [DOI] [PubMed] [Google Scholar]
- 24.Brill LM, Salomon AR, Ficarro SB, Mukherji M, Steller-Gill M, Peters EC. Robust phosphoproteomic profiling of tyrosine phosphorylation sites from human T cells using immobilized metal affinity chromatography and tandem mass spectrometry. Anal. Chem. 2004;76:2763–2772. doi: 10.1021/ac035352d. [DOI] [PubMed] [Google Scholar]
- 25.Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8:397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 26.Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 1999;147:913–920. doi: 10.1083/jcb.147.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J. Cell Sci. 2001;114:4557–4565. doi: 10.1242/jcs.114.24.4557. [DOI] [PubMed] [Google Scholar]