

Abstract
Background: Renin-angiotensin system (RAS) components have been identified in human ciliary body and aqueous humour, pointing to a role for the RAS in the regulation of aqueous humour dynamics. Here, the authors examine the functional expression of a RAS and the effects of angiotensin II (AII) on a signal transduction pathway and ion secretion mechanism in cultured human ciliary body non-pigmented epithelium (HNPE).
Methods: RAS expression was examined in cultured HNPE cells using polymerase chain reaction (PCR) analysis. Secretory function was determined using spectrofluorescence imaging microscopy to measure cell calcium (Ca2+I) and volume responses. Single channel patch clamp techniques were employed to investigate ion channel activity.
Results: PCR analysis demonstrated the expression of angiotensinogen and the AT1b receptor in HNPE cells. A large conductance potassium (BK) channel (mean 190 (SEM 5.6) pS, n = 22 cells), was observed in plasma membrane patches. This channel was calcium sensitive with channel open probability (Po) increasing with increasing Ca2+I (K0.5 10.79 (0.44) μM Ca2+, Hill coefficient of 1.04 (0.04)). AII (100 nM) increased the number (N) of active BK channels in HNPE cells and also the probability of channel opening (Po). N.Po increased from 0.008 (0.002) to 1.38 (0.4) following the addition of AII (p=0.0064). AII also induced a rapid rise in Ca2+I from resting values of 112 (14) nM to a peak of 992 (106) nM (p<10−4). A simultaneous cell volume reduction of 24.70% (3.34%) (p<10−4) occurred during this calcium signal. Losartan (1 μM) significantly blocked the AII induced BK channel activation (p=0.0131), the Ca2+I response (p<10−4), and the AII induced volume effect (p=0.0046).
Conclusion: It was demonstrated that AII activates a Ca2+I signalling system which subsequently increases potassium ion channel activity. These effects are accompanied simultaneously by cell volume loss, indicating that AII acts as receptor operated secretagogue in HNPE cells. The ability of an AT1 receptor antagonist to inhibit these processes may thus offer a new family of pharmaceutical agents to the current armamentarium in the treatment of glaucoma.
Keywords: renin-angiotensin system, secretory function, ciliary body, epithelium
The circulating renin-angiotensin system (RAS), through the production of its biological effector angiotensin II (AII), has an important role in the control of electrolyte homeostasis, renal haemodynamics, and blood pressure.1 An increasing amount of evidence now suggests that local RAS exist in peripheral tissue, including the eye.2,3 Recently, renin mRNA, angiotensin converting enzyme mRNA, and angiotensinogen mRNA have been demonstrated in human ocular tissues,4 strongly supporting the concept of a local intraocular synthesis of AII. In humans, prorenin, the inactive precursor of renin, has been detected in ciliary body tissue5,6 and the presence of angiotensin converting enzyme has been shown in ciliary body, vitreous, and aqueous humour.7–9 In addition, AII has been identified in human anterior uveal tract and aqueous humour.10,11 Currently, direct investigation of the effects of locally generated AII are not possible in vivo and studies defining the role of AII in aqueous humour formation are limited. A human non-pigmented ciliary body epithelial cell line (HNPE) has been established by transfection with simian virus 40.12 This HNPE cell line has been found to retain the electrophysiological properties found in vivo or in primary cultured non-pigmented cells.13,14 Here we have determined the functional expression of a RAS in these cultured cells and the effects of AII on cell calcium, an intracellular mediator of secretion. The ability of AII to trigger secretion is indicated through its effects on potassium ion channel activation and cell volume reduction.
METHODS
Cell culture
The cell clone (ODM Cl-2) was established from a primary culture of human non-pigmented ciliary epithelium. The cells were grown in polystyrene culture flasks (25 cm2, Falcon 3801 Primaria) containing Dulbecco's modified Eagle's medium (DMEM catalogue No 320–1965, Gibco Laboratories, Life Technologies, Inc) with 10% fetal calf serum (FCS) and 50 mg per ml gentamicin sulphate (garamycin, Schering corporation). The medium contained 4500 mg per litre, d-glucose and l-glutamine. NaHCO3 was added to the medium to adjust the pH to between 7.3 and 7.4 and the final osmolality was 328 mOsm. The medium was sterilised by membrane filtration with 0.2 μm Corning pore filters. The cells were grown at 37°C in 5% carbon dioxide in incubators. Cells were passaged every 6–7 days at a split ratio of 1:2 or 1:3 and the medium changed three times a week. All of the cells studied experimentally were grown on filter cups (Millipore, area 0.6 cm2, Corning, Bedford, MA, USA), with a base composed of collagen (types I, III, and IV). Experiments were performed on single cells isolated from monolayers between passages 6 to 16. HNPE cells were enzymatically isolated 4–5 days after attaining monolayer confluency using 0.05% trypsin/0.02% EDTA.
Intracellular calcium imaging
The isolated cells were placed in glass bottomed perfusion chambers containing DMEM. The cells were loaded with Fura-2/AM (Molecular Probes) in DMSO (final concentration <0.01%) at a concentration of 5 μM and incubated in 5% carbon dioxide and 95% air at 37°C for 35 minutes. Cells were washed to remove the medium and Fura-2 completely, in the following solution containing (mM): 138 NaCl, 4.7 KCl, 1.2 MgCl2, 1.2 KH2PO4, 6 HEPES, 10 glucose, and 2.5 CaCl2, pH 7.4 with NaOH, pCa 3, and 297 mOsm. All experiments were performed at room temperature 23–25°C to minimise dye leakage. Microfluorimetric measurements of intracellular calcium were made using an inverted epifluorescence microscope (Diaphot 200, Nikon) equipped with ×40 and ×100 oil immersion lens. The light from a 100 W xenon lamp (Nikon) was filtered through alternating 340 nm and 380 nm interference filters (10 nm band width, Nikon). A custom built chopper circuit controlled the motorised filter wheel, and allowed an electronic adjustment of the duration of the exposure at each wave length. The resultant fluorescence was passed through a 400 nm dichroic mirror, filtered at 510 nm, and then collected using an intensified CCD camera system (Darkstar, Photonic Science). Video fluorescence images were digitised and analysed using the Starwise, Fluo system (Imstar, Paris, France). Fluorescence signals were calibrated in vivo over a range of calcium concentrations in the presence of the calcium ionophone 4-bromo-A23187 as previously described in detail.15
Cell volume measurements
Fluorometric analysis
An estimate of cell volume was obtained by analysing the fluorescent intensity of Fura-2 at the “isosbestic” wavelength. At this wavelength the fluorescent intensity is independent of intracellular calcium levels and dependent on dye concentration. Thus relative changes in cell volume can be determined from changes in fluorescent intensity at the isosbestic wavelength according to the following equation: volume/volumeo=((380* −Bk) + (340* × −Bk))/2, where 380* represents the fluorescent intensity at 380 nm, 340* is the value of the signal at 340 nm, and Bk is the value of the backround fluorescence signal. Three dimensional images of cell volume calculated by this method were generated by the computer and the ratiometric cell calcium level could be superimposed on this volume image, yielding simultaneous spatiotemporal measurements of cell calcium and volume.
Morphometric analysis
Isolated HNPE cells were seeded on glass bottomed petri dishes in NaCl Ringer solution and placed on the inverted stage of a microscope equipped with Nomarski optics. (Nikon Diaphot TMD). Video images of the cells were captured at ×100 magnification with a camera (WAT 902, Watec, Japan) connected to a video frame grabber (model LG3, Scion Corporation). An image of the cell was stored on a computer each second and later analysed with edge detection software on a Macintosh computer using the public domain NIH Image program (v1.57ppc), developed at the US National Institutes of Health (available on floppy disk from the National Technical Information Service, Springfield, Virginia, part number PB95–500195GEI). Assuming a spherical shape of isolated cells, the cell volume was calculated as 2π3.
Single channel patch clamp recording
The cell attached and inside out configurations of the patch clamp technique were used for single channel studies.16 Patch pipettes (tip resistance 4–10 MW) were prepared from borosilicate glass capillaries (Vitrex, Modulohm, Herlev, Denmark), pulled and polished on a programmable puller (DMZ, Zeitz Instrumente, Augsburg, Germany). The patch pipettes were filled with either standard NaCl Ringer or an intracellular-like K+ solution (“Kint” composition given in Table 1). All solutions were filtered through 0.2 μm Millipore cellulose discs. Current and voltage recordings from patch clamp electrodes were amplified (Biologic RK 300 patch clamp amplifer, Echirolles, France), passed through a 30 KHz filter, and stored on video tape (Betamax Sony, Tokyo, Japan) after pulse code modulation (PCM model 501ES, Sony, Tokyo, Japan). The single channel current records were digitised and displayed in real time using a digital oscilloscope (Model 310 Nicolet, Madison, WI, USA). Single channel current records for illustrations were filtered at 1 KHz using a Tchebycheff filter and replayed on a chart recorder (Model RS3200 Gould Instruments, Valley View, OH, USA). Current and voltage signals were digitised using a 16 bit A/D converter (CED 1401, Cambridge Electronic Design, Cambridge) after low pass (−3dB) filtering at 5 KHz (8 pole Bessel filter, Model 902, Frequency Devices Inc, Haverhill, MA, USA) and analysed with an 80486–50 MHz Dell PC. Steady state current voltage relations were run and analysed using VGEN and PAT programs (J Dempster, Strathclyde Electrophysiology Software version 6.1, University of Strathclyde, Scotland) and instantaneous I-V trials were generated using pulsestim and vclamp programs (Cambridge Electronic Design, release EPC 5.0). Single channel current amplitude histograms, open probability, and open and closed dwell times were analysed from data segments of 20–60 second duration using the pat program. In cell attached configuration cation movement across the membrane from the cytoplasm to the external side is defined as outward current and shown as upward deflections in single channel records. Channel activity was determined from the product of the number of channels active in the patch (N) and the channel open probability (Po).
Table 1.
Composition (in mmol/l) of saline solutions used in bath and patch pipette
| NaCl | KCl* | MgCl2 | CaCl2 | HEPES | EGTA | pH* | |
| Extracellular | |||||||
| NaCl Ringer | 110 | 3.7 | 1 | 2 | 6 | – | 7.4 |
| Kext pCa3 | – | 107 | 1 | 1 | 10 | – | 7.4 |
| Internal Kint | |||||||
| pCa8 | – | 107 | 1.22 | 1.27 | 10 | 10 | 7.2 |
| pCa7 | – | 107 | 1.15 | 5.92 | 10 | 10 | 7.2 |
| pCa6 | – | 107 | 1.03 | 9.36 | 10 | 10 | 7.2 |
| pCa5 | – | 107 | 1.00 | 9.94 | 10 | 10 | 7.2 |
| Kgluc | KCl | MgCl2 | CaCl2 | NaAc | NaPyr | Asp | GTPgS | EGTA | HEPES | |
| *pH of Kint solutions was adjusted with KOH which increased the K+ concentration to 120 mmol/l. | ||||||||||
| Kgluc = potassium gluconate, NaAc = sodium acetate, NaPyr = sodium pyruvate, Asp = aspartic acid. | ||||||||||
| Note: In potassium gluconate solutions Ca2+ activity is reduced by 90% due to a chelating effect of gluconate (measured with a Ca2+ selective macroelectrode). To compensate for this, free calcium was present at 10-fold higher concentrations. | ||||||||||
| Whole cell pipette solutions | ||||||||||
| pH 7.2 | ||||||||||
| pCa8 | ||||||||||
| 100 | 20 | 1.22 | 2.96 | 3 | 5 | 5 | 0.01 | 5 | 10 | |
Whole cell current voltage analysis
The whole cell current voltage configuration was obtained from the cell attached mode after breaking the membrane patch by applying a brief negative pressure pulse (−200 mbar) in the pipette. Whole cell currents were amplified (RK300 Biologic), digitised at 20 KHz (CED 1401), and sampled in real time on hard disk at an acquisition rate of 5 KHz. Current voltage relations were analysed using CED vclamp software. The “outside out” configuration was obtained from whole cell patches by withdrawing the electrode from the cell. This resulted in the formation of a membrane patch at the tip of the electrode with the extracellular surface facing the bathing solution.
PCR analysis of angiotensinogen
The set of oligonucleotide primers to amplify the human angiotensinogen gene by the polymerase chain reaction (PCR) was selected based on the published cDNA nucleotide sequence (GenBank accession number X15324) and with the aid of a primer select program of Dnastar (Dnastar Inc, Madison, WI, USA). The forward primer: 5'-GCGGAAGCGAGCACCCCAGTC-3' (location 28 to 48); and the reverse primer: 5'-GGTCCAAGGCTCCCAGATAGA-3' (location 482 to 462) were synthesised in the DNA Synthesis Facility at Yale University. PCR conditions were determined according to the primer select program (Dnastar Inc, Madison, WI, USA). The PCR method of Saiki et al17 was used to anneal the above primers to DNA from 108 pfu of a cDNA library of the human non-pigmented ciliary epithelial cell line ODM-2 NPE18 and from a cDNA library of the human ciliary body.19 Reaction mixtures in 100 μl in MicroAmp tubes (Perkin Elmer, Emeryville, CA, USA) contained, 10 mM TRIS-HCl (pH 9), 50 mM KCl, 0.1% Triton X-100, and 1.5 mM MgCl2 as buffering components, and DNA from 108 pfu of λUni-ZAP, 200 mM (each) deoxynucleoside triphosphates, each oligonucleotide primer at 1 μM, and 5U of Taq polymerase (Promega, Co) as reaction components. PCR was done in a Perkin Elmer DNA thermal cycler 480 (Perkin Elmer Cetus, Norwalk, CT, USA). Samples were inserted into the machine during an initial incubation of the lambda phage particles at 94°C for 1 minute. Each cycle consisted of a denaturation step at 94°C for 1 minute, 1 minute of annealing at the optimal temperature 63°C, and 1 minute of polymerisation at 72°C. This cycle was repeated 35 cycles. The final polymerisation step was extended an additional 5 minutes. Five per cent of the resulting PCR product was size fractionated by electrophoresis on 1% agarose-SeaPlaque gels (FMC BioProducts, Rockland, ME, USA). As internal controls in PCR amplifications we included in the reaction mixture: (1) not template, and/or (2) RNA instead of cDNA as template. In both cases no PCR products were resolved on agarose gel, indicating that RNA samples were not contaminated by genomic DNA. The gel purified DNA fragment was sequenced directly using the method based on a Sequenase PCR sequencing kit (United States Biochemical, Cleveland, OH, USA).
RT-PCR analysis of AII receptors
Total RNA was extracted from HNPE cells grown on petri discs, and from adrenal gland and kidney using the acid guanidinium thiocyanate-phenol-chloroform method.20 The total RNA was analysed by gel electrophoresis and quantified by spectrophotometry. Total RNA (5 μg) was subjected to first strand cDNA synthesis using random hexamer primers and Superscript II transcriptase (Gibco-BRL, USA) in a final volume of 20 μl system. After incubation at 42°C for 1 hour, the reaction mixture was treated with RNase H before proceeding to PCR analysis. The final mixture (2 μl) was directly used for PCR amplification. Different sets of oligonucleotide primers based on the corresponding AII receptor subtypes were synthesised (Gibco-BRL, USA) for PCR analysis. AT1a or AT1b was amplified with the oligonucleotide primers specific for their respective 3'-non-coding sequences as previously reported.21 Sense and antisense for AT1a are 5'-GCACACTGGCAATGTAATGC-3' and 5'-GTTGAACAGAACAAGTG ACC-3', respectively. Sense and antisense for AT1b are 5'-GCCTGCAAGTGAAG TGATTT-3' and 5'-TTTAACAGTGGCTTTGC TCC-3', respectively. The PCR products were 385 bp for AT1a and 204 bp for AT1b. Control experiments were carried out with the glyceraldehydes-3- phosphate dehydrogenase (GADPH) sequence.22 Sense and antisense for GADPH are 5'-TCCCTCAAGAT TGTCAGCAA-3' and 5'-AGATCCACAACGGATACATT-3' respectively. The RT-PCR product for GADPH was 309 bp. PCR reactions were carried out in a volume of 50 μl system containing the corresponding sense and antisense primer sequences using the PCR reagent system (Gibco-BRL, USA). The PCR condition is denaturing: 94°C, 1 minute; annealing: 58°C, 1 minute; and elongating: 72°C, 2 minutes. The reaction is repeated for 40 cycles. The amplified mixture (10 μl) is finally analysed on 2% agarose gel electrophoresis and the amplified DNA bands were detected using ethidium bromide staining.
RESULTS
BK channels
A large conductance K+ channel (BK channel) was observed in “cell attached” plasma membrane patches when the pipette contained Kint solution and the cells bathed in NaCl Ringer solution. Out of 100 patches from single HNPE cells analysed in this study, the BK channel was found in 77 membrane patches and the number of active BK channels per patch varied between one and five. The channel activity is voltage dependent. Single channel conductance (γ) and probability of opening (Po) were increased at more negative pipette voltages (membrane depolarisation). Only outward currents could be recorded through the BK channel and the channel thus behaves as a pure “outward rectifier.” This channel property is apparent from the single channel current voltage (I/Vp) relation (Fig 1A). In the cell attached patch configuration the mean conductance for outward current through the channel was 190 (5.6) pS (n=12) with a reversal potential of −35.66 (6.02) mV (n=7). This should correspond to the membrane potential of the HNPE cell since the chemical potential for K+ across the cell membrane patch is close to zero. Voltage clamping the cell plasma membrane to potentials which should drive K+ into the cell failed to activate the BK channel. In addition, the open probability of the channel increases when the pipette voltage (Vp) is clamped at negative values favouring K+ efflux (Fig 1B). Thus, in physiological terms, the BK channel acts a secretory ion channel. High conductance BK channels are typically inhibited by quaternary ammonium compounds such as tetrapentylammonium (TPeA), which are known to evoke a drastic reduction in the BK channel conductance and open state probability when acting from outside of the cell membrane.23 This drug is useful for identifying BK channels as other K+ channels are insensitive to external TPeA at concentrations less than 10 μM.24 The effect of TPeA on BK channel activity in the “outside out” patch configuration is shown in Figure 1D. The addition of TPeA (1 μM) to the bath resulted in a significant reduction in control single channel outward current from 14.06 (0.28) pA to 2.8 (0.9) pA, (p<10−4, n=4) and in channel activity (Po) from 0.58 (0.074) to 0. 033 (0.06) (p=0.0012, n=4), at holding voltage Vp of +80 mV.
Figure 1.

(A). Demonstrates the voltage dependency of BK channel activity. The single channel current as a function of the patch membrane potential is shown. NaCl Ringer is in the bath and a high K+ solution (Kint, pCa 3) in the pipette. The channel activity is voltage dependent with single channel current increasing on membrane depolarisation. Only outward currents could be recorded through this K+ channel, as voltage clamping the patch to potentials greater than –40 VP, which should drive K+ into the cell, failed to produce channel activation. (B) Illustrates the open probability voltage (N.Po/V) relation of the BK channel. Control (solid circles): the open probability of the channel (N.Po) is seen to increase when the pipette voltage (VP) is clamped at more negative values (membrane depolarisation). Angiotensin II (open squares): N.Po is found to be significantly higher at each level of VP tested in the presence of AII (100 nM). (C) Control whole cell current voltage relation showing outward rectification (solid squares). The channel underlying the whole cell membrane conductance was inhibited by 1 μM tetra-pentyl ammonium (TPeA), which significantly reduces whole cell conductance (G) at all membrane voltages tested between −100 to +100 mV (open circles). (D) (Upper trace) Single channel current recording of BK channels from an excised (outside out) membrane patch with Kint in the bath and pipette (VP +80 mV). The open state (outward current) is seen as upward deflections on the trace and the closed channel state is indicated by the broken line. (Lower trace) Demonstrates the significant reduction in channel activity when (1 μM) TpeA is added to the bathing solution.
Whole cell current recording
Whole cell K+ currents showed outward recification when the pipette contained a K+ rich solution at pCa 8 and the cells exposed to a similar K+ (Kext) solution (Fig 1C). The reversal potential was 0 mV in agreement with the Nernst equilibrium potential for K+ under these symmetrical K+ solutions. Whole cell conductance (G) for outward current at (Vp +100 mV) was 19 nS and for inward G was 5.5 nS. Thus the activity of a large number of channels favours K+ efflux over K+ influx in the whole cell configuration as predicted from the single BK channel I/Vp relation. The channel underlying the whole cell membrane conductance was inhibited by tetrapentylammonium. TPeA (1 μM) significantly reduced G at all membrane voltages tested between −100 and +100 mV (Fig 1C, p<10−4, n=4), indicating the BK channel is the predominant conductance pathway for K+ under these conditions.
Ca2+ sensitivity of BK channels
Large conductance K+ channels are often found to be Ca2+ sensitive—that is, an increase in Ca2+ on the cytosolic side of the ion channel increases the probability and duration of channel opening.25 In order to examine if this was a property of BK channels in HNPE cells, active patches containing the channel were excised from the cell into an “inside out” configuration where the cytosolic side of the channel faces the bathing solution. The bath was then perfused with solutions of increasing levels of Ca2+ from pCa 9 to pCa 3 (10−9 to 10−3 M) and the corresponding changes in BK channel activity recorded. An increase in the level of Ca2+ in the bathing solution facing the cytosolic side of the cell membrane increased the number of BK channels active in the patch membrane and the open probability of these channels (Fig 2D). The relation N.Po versus Ca2+i was fitted by the Hill equation:
Figure 2.

The effect of AII (100 nM) on BK channels in HNPE cells. (A) Illustrates a current amplitude recording of ion channel activity in a membrane patch in cell attached mode at VP −100 mV, NaCl Ringer in the bath, and Kint in the pipette. The open state (outward current) is seen as upward deflections on the trace and the closed channel state is indicated by the broken line. (B) Demonstrates the increase in channel activity following the addition of AII to the bathing solution. AII increases the number of active BK channels in the membrane patch and also the probability of opening of these channels. (C) Shows the effect of losartan on this response. The addition of AII, holding at the same pipette voltage, in the presence of losartan (1 μM) fails to produce a significant increase in BK channel activity. (D) Illustrates the product of the number of BK channels active in a patch of plasma membrane (N) and the probability of opening of these channels (Po), against increasing Ca2+ levels facing the cytosolic aspect of the plasma membrane patch. (Inside out configuration, Kint pCa 3–9 in the bathing solution, Kint pCa 6 in the pipette, n=6, VP −50 mV). The lines through the data points are calculated from the best fit to the Hill equation given in the text.
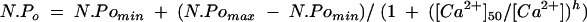 |
Open probability (Po) increased with increasing Ca2+i and the best curve fit gave a half maximal activation (K0.5) of the channel at cytosolic Ca2+ = 10.79 (0.44) μM ([Ca2+]50) at Vp −50 mV, Hill coefficient of 1.04 (0.04), reaching a maximum Po of ∼0.99 for Ca2+i at 100 μM. Increasing Ca2+i further to 1 mM had little additional effect on Po.
Angiotensin II activation of BK channels
We have demonstrated that the BK channel is a predominant ion channel in the HNPE cell with high conductance for K+ secretion. This potassium channel may therefore play an important part in the formation of aqueous humour. Inhibitors of renin and angiotensin converting enzyme (ACE) have been shown to reduce intraocular pressure in humans.26,27 If AII is indeed a secretagogue, then a likely ion transport target is the BK channel. Figure 2 demonstrates the effect of AII (100 nM) on single BK channels in HNPE cells. The addition of AII to the control bathing solution (Fig 2A) increased the number of active BK channels in the membrane patch from one to three and also the probability of opening of these channels (Fig 2B). The N.Po significantly increased from 0.008 (0.002) in the control recordings to 1.38 (0.4) within 10 seconds after the addition of AII (n=5, p=0.0064, Vp −100 mV) in the example illustrated. The addition of AII to the bathing solution significantly increased N.Po at all voltages tested, at 10 mV intervals, between Vp −40 mV to −120 mV as demonstrated in Fig 1B (p<0.01, n=5 at each 10 mV step). The fact that the patch pipette was in the “cell attached” configuration means that the channels are isolated from the bathing solution, thus preventing contact between the peptide and the channel. The activation of BK channels following AII addition must therefore act via a receptor operated mechanism. The specific AT1 receptor antagonist, losartan, was used to further examine the role of the AII receptor in this response. The addition of AII (100 nM) to the bathing solution in the presence of losartan (1 μM) failed to produce an increase in BK channel activity. (Control-N.Po: 0.008 (0.006), AII + losartan-N.Po: 0.016 (0.010), n=5, p=0.5121, Fig 2C) This response differed significantly from that of the control-AII response for membrane potentials tested with Vp holding between −40 mV to −120 mV (p<0.01, n=5 at each 10 mV step).
Angiotensin II effects on intracellular calcium and cell volume
The BK channel is activated by calcium, a known intracellular mediator of secretion.28 Angiotensin II activation of these channels may be mediated via intracellular calcium signalling. This hypothesis was tested in HNPE cells loaded with the calcium fluoroprobe, Fura-2. The extracellular addition of AII (100 nM) to single HNPE cells induced a rapid and significant rise in intracellular free calcium from mean basal values of 112 (14) nM to a mean peak of 992 (106) nM (p<10−4, n=7) which was followed by an oscillatory decline back to basal levels (Fig 3) A normal Ca2+i response to AII was also elicited when the peptide was added following the reduction of external calcium. Under conditions of low external calcium, AII (100 nM) produced oscillations in Ca2+i of similar magnitude and pattern as in the control experiments. The mean basal and peak Ca2+i response to AII from HNPE cells exposed acutely (<20 seconds) to 1 nM extracellular calcium was 100 (8) nM and 966 (168) nM, respectively (n=6). Under these conditions, the Ca2+i response to AII was not significantly different from that observed in normal extracellular calcium (p=0.97). These results demonstrate that AII can activate release of calcium from intracellular stores and the initiation of the calcium response to AII does not require entry of calcium across the plasma membrane.
Figure 3.

(Lower) Changes in intracellular free Ca2+ levels in a single HNPE cell in response to angiotensin II (100nM). Intracellular calcium levels increased from 83 nM to a peak of 1179 nM which is followed by a gradual oscillatory decline back to basal levels. (Upper) This graph also illustrates the simultaneous change in cell volume in the same cell. The volume changes are represented as V/Vo, where Vo is the original mean cell volume. Cell volume decreases from a resting cell volume of 1.0 to a minimum of 0.66 in response to AII, followed by a restorative oscillatory increase toward the resting state. (Right) Upper and lower columns indicating mean (SEM) volume and Ca2+i responses, respectively, illustrated to the same scale as the single cell response (n=7; AII: angiotensin II, n=7; Lsn+AII: losartan + angiotensin II, n=7).
The process of secretion involves the net efflux of osmotically active ions with a concomitant reduction in cell volume. Secretion in HNPE cells may therefore be examined by monitoring acute fluctuations in cell volume. Using spectrofluorescence imaging single HNPE cell volume was measured in parallel with the calcium signal (Fig 3). The addition of AII (100 nM) to HNPE cells induced a significant peak volume loss of 24.70% (3.34%) (n=7, p<10−4). Figure 3 demonstrates that changes in cell volume coincide with alterations in the Ca2+i signal; however, these volume changes lag slightly out of phase. The oscillatory cycles in decreasing cell volume, or secretory events, are observed to follow several seconds after the cycles in increasing Ca2+i. In order to confirm the role of AII receptors in this process the AT1 receptor antagonist, losartan, was used. Exposure of HNPE cells to losartan (1 μM) had no effect on basal intracellular calcium (p=0.8, n=7). In the presence of losartan (1 μM) the addition of AII (100 nM) to the bathing solution failed to produce a significant increase in Ca2+i (Control-Ca2+i: 103 (8) nM, AII+losartan-Ca2+i: 141 (23) nM, n=7, p=0.1446). In addition, under these conditions cell volume did not significantly change from a mean resting cell volume of 1.0 (0.024) to 0.959 (0.043) in the presence of AII (p=0.42, n=7). This response differed significantly from the volume response in the absence of losartan (p=0.0046). Taken together, these results provide evidence for AII induced secretion in HNPE cells via calcium activation of BK channels. These findings were also supported by direct morphometric measurement of cell volume. The addition of AII (100 nM) to the bathing solution resulted in a similar and significant reduction in cell volume from resting levels of 100% (3.6%) to 82% (5.4%) (n=7, p=0.017). However, AII (100 nM) in the presence of losartan (1 μM), failed to produce a reduction in cell volume (resting volume: 100% (4%), AII+losartan: 98% (4.2%), n=6, p=0.74) and this differed significantly from the control AII cell volume effect (p=0.04)
The expression of AII receptor subtypes and angiotensinogen in HNPE cells
RT-PCR coupled with specific primers based on the corresponding AII receptor subtypes genes—namely, AT1a and AT1b, was performed to study the mRNA expression of their respective genes in the HNPE cells. Results showed that AII receptor subtypes in the cells appeared to be differentially expressed. RT-PCR analysis showed that mRNA from AT1a (Fig 4) was not detected, whereas mRNA from AT1b was found to be expressed in the cells. On the other hand, AT1a and AT1b mRNAs were detected consistently in the rat tissues of kidney and adrenal gland, which were known to express these receptor subtypes and were employed as positive controls for the experiments. in parallel by PCR using oligonucleotide primers complementary to the human angiotensinogen gene, as described in the Methods section. PCR analysis demonstrated the predicted 455 nt fragment in the human non-pigmented epithelium (ODM-2), as shown in Figure 5, confirming the presence of the human angiotensinogen gene in this cell type.
Figure 4.

RT-PCR analysis of mRNA from angiotensin II receptor subtypes in HNPE cells. (AT1a) Lanes 1 and 2 show AT1a mRNA expression in HNPE cells and kidney respectively. (AT1b) Lanes 1 and 2 show AT1b mRNA expression in HNPE cells and adrenal gland respectively. The arrows 385 bp, 204 bp, and 309 bp indicate the expected size of amplified products from AT1a, AT1b, and GAPDH respectively. Lane M indicates the DNA marker (φ X174 RF/Hae III fragments). Lanes 3 and 4 indicate GAPDH mRNA expression in cells and kidney as well as adrenal gland respectively.
Figure 5.

PCR of angiotensinogen from human cDNA libraries of the ciliary body (CB) and the HNPE cell line (ODM-2) of the ciliary epithelium. Equivalent lambda particle forming units (pfu) particles from libraries constructed in λUni-Zap XR vector were amplified in parallel by PCR using oligonucleotide primers complementary to the human angiotensinogen gene, as described in Methods. The predicted 455 nt fragment amplified from the libraries was resolved on a 1% agarose gel with ethidium bromide stain, and its migration position was marked at right. Lane M = 1 kb ladder DNA marker; CB = human ciliary body library; ODM-2 = human NPE cell line cDNA library.
DISCUSSION
Evidence now suggests the presence of a local renin-angiotensin system in the human eye4,7,11 and, specifically, RAS components have been identified in human ciliary body and aqueous humour.5–11 In addition, corneal application of ACE and renin inhibitors has been shown to lower intraocular pressure in patients with ocular hypertension and primary open angle glaucoma.26,27 These findings point to a role for the RAS in the regulation of aqueous humour dynamics. It has previously been hypothesised that the effect of the RAS is mediated predominantly by its ability to alter vascular tone at this site.3 However, in this report we demonstrate direct effects of AII on a receptor activated signal transduction pathway and ion secretion mechanism in human ciliary body non-pigmented epithelium.
Our data support previous work by Barros et al,29 who first identified the large conductance potassium channel (BK or “Maxi-K” channel) in this cell type. BK channels have been well characterised over the past decade in fluid secreting epithelia and have been confirmed to be a regulatory driving force for secretion in many cell types.30–32 In this study, we have demonstrated, from single channel and whole cell current analysis, that the BK channel dominates the overall conductance of the HNPE cell membrane. In addition, we have confirmed the sensitivity of this channel to small changes in the level of cytosolic free Ca2+, a known ubiquitous mediator of secretion.28,33 The Ca2+ dependency of the HNPE large conductance potassium channel was found to be similar to that of BK channels in other cell types.34,35 When activated by an increase in intracellular Ca2+, BK channels respond with increased open probability and concomitantly increased ion flux. These potassium channels therefore can serve as a link between cellular processes which involve intracellular Ca2+ elevation and those which involve membrane excitability and thus ion transport or solute secretion.36
Angiotensin II, through AT1 receptor associated Gq protein activation, is known to release intracellular store calcium via the phosphoinositol/phospholipase C pathway.37 In this study we show that extracellular AII increases intracellular calcium in HNPE cells. This process is independent of external calcium indicating that this effect is predominately mediated through intracellular calcium store mobilisation rather than transmembrane Ca2+ flux. In addition, we demonstrate that extracellular AII activates the predominant ion channel in this cell, the calcium sensitive BK channel. Channel activation is likely to be indirectly mediated by the intracellular calcium mobilising effects of AII. This is in keeping with previous studies demonstrating the modulation of BK channels by AII induced increases in Ca2+i.38–40 A direct activation of the channel by AII is ruled out as the cell attached pipette, which is measuring channel activity, isolates the channel from the external bathing solution.
BK channels, in addition to permitting K+ flux, are thought to set the membrane potential of various secretory cell types.41,42 Activation of these channels, therefore, may result in cell membrane potential change leading to concomitant activation of other voltage sensitive ion channels.28 Cell solute loss secondary to AII activation of BK channels would thus be envisaged. In this study we demonstrate that that mobilisation of Ca2+i by AII is sufficient to cause cell volume loss or secretion in the HNPE cell and that subsequent alterations in cell volume are tightly coupled with changes in Ca2+i. These data are in keeping with previous volume studies in secretory cells which have observed that the magnitude of agonist induced cell volume loss is generally between 15–25% and have also reported a similar coupling response between Ca2+i and cell volume.28,43,44 Our results thus demonstrate a direct link between the activity of an intracellular messenger and secretory cell volume loss in the HNPE cell. immunohistological studies and those of Lin et al46 who first demonstrated the presence of receptors for AII on cultured HNPE cells using radioligand binding studies. These findings suggest an autocrine role for AII in this cell type. Thus, the non-pigmented ciliary epithelial cells are the site of angiotensin production, angiotensin receptor expression and angiotensin induced secretion.
In order to further examine the role of the AII receptor in this secretory process the specific AT1 receptor surmountable antagonist, losartan, was used.37 In the presence of losartan, the addition of AII failed to significantly increase Ca2+i or reduce cell volume. On a molecular level, losartan also significantly blocked the AII induced increase in K+ channel activity normally observed under control conditions. As the cell attached pipette prevents exposure of the channel to the drug, this effect must be mediated via AT1 receptor antagonism rather than by direct interaction with the potassium channel.
In conclusion, we have established that a calcium sensitive BK channel dominates the overall characteristics of the HNPE cell membrane. We have confirmed using RT-PCR, the presence of the AII receptor on the HNPE cell. We have shown that external AII activates an intracellular calcium signalling system which subsequently increases potassium channel activity in the plasma membrane. These effects are accompanied simultaneously by cell volume loss, indicating that AII acts as receptor operated secretagogue in HNPE cells. The ability of an AT1 receptor antagonist to inhibit these processes may thus offer a new family of pharmaceutical agents to the current armamentarium in the treatment of glaucoma.
Acknowledgments
This work was supported by the Health Research Board of Ireland and the National Institute of Health National Eye Institute (EY-04873).
Abbreviations
ACE, angiotensin converting enzyme
AII, angiotensin II
DMEM, Dulbecco's modified Eagle's medium
FCS, fetal calf serum
HNPE, human ciliary body non-pigmented epithelium
PCR, polymerase chain reaction
RAS, renin-angiotensin system
REFERENCES
- 1.Oparil S, Haber E. The renin-angiotensin system. N Engl J Med 1974;291:446–57. [DOI] [PubMed] [Google Scholar]
- 2.Re RN. Cellular biology of the renin-angiotensin systems. Arch Intern Med 1984;144:2037–41. [PubMed] [Google Scholar]
- 3.Van Haeringen NJ. The renin-angiotensin system in the human eye. Br J Ophthalmol 1996;80:99–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wagner J, Jan Danser AH, Derkx FH, et al. Demonstration of renin mRNA, angiotensinogen mRNA, and angiotensin converting enzyme mRNA expression in the human eye: evidence for an intraocular renin-angiotensin system. Br J Ophthalmol 1996;80:159–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sramek SJ, Wallow IH, Day RP, et al. Ocular renin-angiotensin: immunohistochemical evidence for the presence of prorenin in eye tissue. Invest Ophthalmol Vis Sci 1988;29:1749–52. [PubMed] [Google Scholar]
- 6.Wallow IH, Sramek SJ, Bindley CD, et al. Ocular renin angiotensin: EM immunocytochemical localization of prorenin. Curr Eye Res 1993;12:945–50. [DOI] [PubMed] [Google Scholar]
- 7.Weinreb RN, Sandman R, Ryder MI, et al. Angiotensin-converting enzyme activity in human aqueous humor. Arch Ophthalmol 1985;103:34–6. [DOI] [PubMed] [Google Scholar]
- 8.Igic R, Kojovic V. Angiotensin I converting enzyme (kininase II) in ocular tissues. Exp Eye Res 1980;30:299–303. [DOI] [PubMed] [Google Scholar]
- 9.Ferrari-Dileo G, Ryan JW, Rockwood EJ, et al. Angiotensin-converting enzyme in bovine, feline, and human ocular tissues. Invest Ophthalmol Vis Sci 1988;29:876–81. [PubMed] [Google Scholar]
- 10.Danser AH, Derkx FH, Admiraal PJ, et al. Angiotensin levels in the eye. Invest Ophthalmol Vis Sci 1994;35:1008–18. [PubMed] [Google Scholar]
- 11.Osusky R, Nussberger J, Amstutz C, et al. Individual measurements of angiotensin II concentrations in aqueous humor of the eye. Eur J Ophthalmol 1994;4:228–33. [DOI] [PubMed] [Google Scholar]
- 12.Coca-Prados M, Wax MB. Transformation of human ciliary epithelial cells by simion virus 40: induction of cell proliferation and retention of β2-adrenergic receptors. Proc Natl Acad Sci USA 1986;83:8754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Helbig H, Korbmacher C, Wohlfarth J, et al. Electrical membrane properties of a cell clone derived from human non-pigmented ciliary epithelium. Invest Ophthalmol Vis Sci 1989;30:882–9. [PubMed] [Google Scholar]
- 14.Yantorno RE, Coca-Prados M, Krupin T, et al. Volume regulation of cultured, transformed, non-pigmented epithelial cells from human ciliary body. Exp Eye Res 1989;49:423–37. [DOI] [PubMed] [Google Scholar]
- 15.Donnadieu E, Cefai D, Tan YP, et al. Imaging early steps of human T cell: activation by antigen-presenting cells. J Immunol 1992;148:2643–53. [PubMed] [Google Scholar]
- 16.Hamill OP, Marty A, Neher E, et al. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch 1981;391:85–100. [DOI] [PubMed] [Google Scholar]
- 17.Saiki RK, Scharf S, Faloona F, et al. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 1985;230:1350–4. [DOI] [PubMed] [Google Scholar]
- 18.Escribano J, Hernando N, Ghosh S, et al. cDNA from human ocular ciliary epithelium homologous to βig-h3 is preferentially expressed as an extracellular protein in the corneal epithelium. J Cell Physiol 1994;160:511–21. [DOI] [PubMed] [Google Scholar]
- 19.Escribano J, Ortego J, Coca-Prados M. Isolation and characterization of cell-specific cDNA clones from a subtractive library of the ocular ciliary body of a single normal human donor: transcription and synthesis of plasma proteins.J Biochem 1995;118: 921– 31. [DOI] [PubMed] [Google Scholar]
- 20.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987;162:156–9. [DOI] [PubMed] [Google Scholar]
- 21.Kitami Y, Okura T, Marumoto K, et al. Differential gene expression and regulation of type-1 angiotensin II receptor subtypes in the rat. Biochem Biophys Res Commun 1992;188:446–52. [DOI] [PubMed] [Google Scholar]
- 22.Fort P, Marty L, Piechaczyk M, et al. Various rat adult tissues express only one major mRNA species from the glyceraldehyde-3-phosphatedehydrogenase multigenic family. Nucleic Acids Res 1985;13:1431–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwatsuki N, Petersen OH. Inhibition of Ca2+ - activated K+ channels in pig pancreatic ancinar cells by Ba2+, Ca2+ guanine and guanidine. Biochem Biophys Acta 1985;819:249–57. [DOI] [PubMed] [Google Scholar]
- 24.Findlay I, Dunne MJ, Wallheim CB, et al. Quinine inhibits Ca2+ independent K+ channels whereas tetraethylammonium inhibits Ca2+ activated K+ channels in insulin-secreting cells. FEBS Lett 1985;185:4–8. [DOI] [PubMed] [Google Scholar]
- 25.Kaczorowski GJ, Knaus HG, Leonard RJ, et al. High-conductance calcium-activated potassium channels; structure, pharmacology, and function. J Bioenerg Biomembr 1996;28:255–67. [DOI] [PubMed] [Google Scholar]
- 26.Constad WH, Fiore P, Samson C, et al. Use of an angiotensin converting enzyme inhibitor in ocular hypertension and primary open-angle glaucoma. Am J Ophthalmol 1988;105:674–7. [DOI] [PubMed] [Google Scholar]
- 27.Denis P, Nordmann JP, Elena PP, et al. One study to evaluate safety and efficacy of SR 43845, a rennin inhibitor, in healthy volunteers. Invest Ophthalmol Vis Sci 1995;36:S734. [Google Scholar]
- 28.Petersen OH. Stimulus-secretion coupling: cytoplasmic calcium signals and the control of ion channels in exocrine acinar cells. J Physiol 1992;448:1–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barros F, López-Briones LG, Coca-Prados M, et al. Detection and characterization of Ca2+-activated K+ channels in transformed cells of human non-pigmented ciliary epithelium. Curr Eye Res 1991;10:731–8. [DOI] [PubMed] [Google Scholar]
- 30.Maruyama Y, Gallacher DV, Petersen OH. Voltage and Ca2+ - activated K+ channel in basolateral acinar cell membranes of mammalian salivary glands. Nature 1983;302:827–9. [DOI] [PubMed] [Google Scholar]
- 31.Petersen OH, Marvyama Y. Calcium activated potassium channels and their role in secretion. Nature 1984;307:693–6. [DOI] [PubMed] [Google Scholar]
- 32.Petersen OH. Calcium-activated potassium channels and fluid secretion by exocrine glands. Am J Physiol 1986;251:G1–13. [DOI] [PubMed] [Google Scholar]
- 33.Tepikin AV, Petersen OH. Mechanisms of cellular calcium oscillations in secretory cells. Biochim Biophys Acta 1992;1137:197–207. [DOI] [PubMed] [Google Scholar]
- 34.Rothberg BS, Magleby KL. Gating kinetics of single large-conductance Ca2+-activated K+ channels in high Ca2+ suggest a two-tiered allosteric gating mechanism. J Gen Physiol 1999;114:93–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vergara C, Latorre R, Marrion NV, et al. Calcium-activated potassium channels. Curr Opin Neurobiol 1998;8:321–9. [DOI] [PubMed] [Google Scholar]
- 36.Cui J, Cox DH, Aldrich RW. Intrinsic voltage dependence and Ca2+ regulation of mslo large conductance Ca-activated K+ channels. J Gen Physiol 1997;109:647–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Gasparo M, Catt KJ, Inagami T, et al. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev 2000;52:415–72. [PubMed] [Google Scholar]
- 38.Sansom SC, Ma R, Carmines PK, et al. Regulation of Ca(2+)-activated K(+) channels by multifunctional Ca(2+)/calmodulin-dependent protein kinase. Am J Physiol Renal Physiol 2000;279:F283–8 [DOI] [PubMed] [Google Scholar]
- 39.Romero F, Silva BA, Nouailhetas VL, et al. Activation of Ca(2+)-activated K+ (maxi-K+) channel by angiotensin II in myocytes of the guinea pig ileum. Am J Physiol 1998;274:C983–91 [DOI] [PubMed] [Google Scholar]
- 40.So SC, Wu WL, Grima J, et al. Functional expression of sperm angiotensin II type I receptor in Xenopus oocyte: modulation of a sperm Ca2+-activated K+ channel. Biochim Biophys Acta 1998;1415:261–5. [DOI] [PubMed] [Google Scholar]
- 41.Rudy B. Diversity and ubiquity of K channels. Neuroscience 1988;25:729–49. [DOI] [PubMed] [Google Scholar]
- 42.Wang W, Sackin H, Giebisch G. Renal potassium channels and their regulation. Annu Rev Physiol 1992;54:81–96. [DOI] [PubMed] [Google Scholar]
- 43.Foskett JK, Melvin J. Activation of salivary secretion: coupling of cell volume and [Ca2+]i in single cells. Science 1989;244:1582–5. [DOI] [PubMed] [Google Scholar]
- 44.Foskett JK. (Ca2+) modulation of C1-content controls cell volume in single salivary acinar cells during fluid secretion. Am J Physiol 1990;259:C998–1004. [DOI] [PubMed] [Google Scholar]
- 45.Sramek SJ, Wallow IH, Tewksbury DA, et al. An ocular renin-angiotensin system. Immunohistochemistry of angiotensinogen. Invest Ophthalmol Vis Sci 1992;33:1627–32. [PubMed] [Google Scholar]
- 46.Lin C, Stone RA, Wax MB Angiotensin binding sites in rabbit anterior uvea and human ciliary epithelial cells. Invest Ophthalmol Vis Sci 1990;31:147–52. [PubMed] [Google Scholar]